
Abstract
Oncolytic viruses (OVs) are novel cancer gene therapies that are moving toward the forefront of modern medicines. However, their full therapeutic potential is hindered by the lack of convenient and reliable strategies to visualize and quantify OV growth kinetics and therapeutic efficacy in live cells. In this study, we present an innovative imaging approach for single-cell real-time analysis of OV replication and efficacy in cancer cells. We selected SG33 as a prototypic new OV that derives from wild-type Myxoma virus (MYXV). Lausanne Toulouse 1 (T1) was used as control. We equipped SG33 and T1 genomes with the ANCHOR system and infected a panel of cell lines. The ANCHOR system is composed of a fusion protein (OR-GFP) that specifically binds to a short nonrepetitive DNA target sequence (ANCH) and spreads onto neighboring sequences by protein oligomerization. Its accumulation on the tagged viral DNA results in the creation of fluorescent foci. We found that (1) SG33 and T1-ANCHOR DNA can be readily detected and quantified by live imaging, (2) both OVs generate perinuclear replication foci after infection clustering into horse-shoe shape replication centers, and (3) SG33 replicates to higher levels as compared with T1. Lastly, as a translational proof of concept, we benchmarked SG33 replication and oncolytic efficacy in primary cancer cells derived from pancreatic adenocarcinoma (PDAC) both at the population and at the single-cell levels. In vivo, SG33 significantly replicates in experimental tumors to inhibit tumor growth. Collectively, we provide herein for the first time a novel strategy to quantify each step of OV infection in live cells and in real time by tracking viral DNA and provide first evidence of theranostic strategies for PDAC patients. Thus, this approach has the potential to rationalize the use of OVs for the benefit of patients with incurable diseases.
Introduction
Since the dawn of time, viruses have been responsible of pandemics, epidemics, and a variety of infectious diseases. They were first discovered at the end of the 19th century, accountable for multiple pathologies yet unidentified. Since it was reported that a 42-year-old female patient with myelogenous leukemia underwent tumor remission after an influenza infection, the attempt to use viruses to eliminate tumors has never stopped. 1
Employing therapeutic oncolytic viruses (OVs) against tumor cells has grown exponentially since the 1950s/60s, with the first OV derived from Herpes Simplex Virus 1 (HSV-1, Imlygic©) 2 having received its product license in 2015 for advanced melanoma; multiple other viral oncolytic strains are currently investigated in (pre)clinical studies for the treatment of diverse neoplasms. 3 With OV finally reaching patients' bed, several limitations and challenges remain to be tackled to broader the use of such innovative therapies in oncology programs.
One of the main hurdles so far in virotherapy programs is to achieve real-time noninvasive monitoring of viral replication, propagation, and oncolysis in live cells to address OV biodistribution, pharmacokinetics, and to identify potential detrimental side effects after infection of normal cells. Thus, novel technological approaches are needed for the in-depth understanding of OV biology and therapeutic efficacy.
Methods studying viral infection and replication include enzyme-linked immunosorbent assay (ELISA) and antigen-specific immunofluorescence, quantitative PCR (qPCR), and reverse transcriptase-qPCR, 4 and transmission electron microscopy 5 —a laborious and cumbersome technique. These are usually end-point analyses that necessitate fixed or lysed biological samples and are unable to discern all the various stages of a viral life cycle. Furthermore, these techniques provide at best multiple timepoint analyses and, consequently, cannot be used in longitudinal studies.
Nowadays, fluorescent reporter proteins associated with early or late viral promoters are the most common method used in preclinical programs to track viral infection. 6 For in vivo preclinical and clinical studies, the sodium iodide symporter (NIS) expressed by viral genomes provides noninvasive imaging information on viral infection in cancer models 7 and NCT03456908. A limit to this technique, however, is the necessity of administering a radiomarker (125I) to ensure detection. Collectively, these methods provide an understanding of the viral replication cycle and viral progeny assembly but are missing early steps in viral infection or replication.
Viral oncolytic activity, often simultaneously studied in OV research, is often defined through cell viability assays such as tetrazolium colorimetric assays (MTT/MTS), which prove to have the same end-point analyses limits.
To follow viral infection and propagation in real time to address every step of the viral cycle would be essential to gain basic knowledge on OV behavior and efficacy in cancer cells. This, in turn, will help accelerate the progress of virotherapy toward the clinic by providing priceless biodistribution and pharmacokinetic data.
The ANCHOR system is a novel technique based on genetic engineering that allows real-time monitoring of DNA localization. In brief, the ANCHOR system is composed of a fusion protein (OR-GFP) that specifically binds to a short nonrepetitive DNA target sequence (ANCH) and spreads onto neighboring sequences by protein oligomerization. Its accumulation results in the creation of a fluorescent focus that allows DNA tracking in live cells. In recent studies, the ANCHOR system was successfully used for the real-time visualization and quantification of human cytomegalovirus replication in living cells, 8 the in vivo labeling of adenovirus DNA, 9 to trace baculovirus infection 10 and to shed light on critical early steps characterizing HIV-1 infection. 11
During this study, we developed for the first time in the field an OV genome equipped with the ANCHOR system to detect and quantify viral DNA and oncolytic activity in live cells. We selected prototypic Myxoma virus (MYXV) as a large enveloped DNA virus that is pathogenic for lagomorphs but not for humans. MYXV has already demonstrated oncolytic potential against diverse tumoral cells, while remaining innocuous for healthy cells. 12 In this study, we characterize for the first time the oncolytic properties of the vaccinal strain SG33, derived from parental MYXV. Using the ANCHOR system, we found that SG33 surpasses MYXV replication and we precisely defined SG33 viral cycle in model cells, both at single-cell and population levels.
Pancreatic adenocarcinoma (PDAC) is a disease with no cure that will soon rank second worldwide in death related to cancer. 13,14 PDACs are heterogeneous by essence, both at the molecular and cellular levels, and such heterogeneity strongly contributes to resistance to therapy. 14 –16
Several groups including ours demonstrated that OVs have therapeutic potential for this disease, 17,18 but to our knowledge, the importance of interpatient tumor heterogeneity on virotherapy efficacy has been scarcely explored so far. As a proof of concept, we characterized the efficacy of replication and oncolysis of SG33 in primary samples from patients with PDAC, both in vitro and in vivo in experimental models of tumors. We found that SG33 spares normal pancreatic cells and replicates in PDAC primary cultures. In vivo, SG33 infects and spreads into tumors to inhibit cancer growth. Collectively, our study represents a significant step toward precision medicine for virotherapy of cancer.
MATERIALS AND METHODS
Cell lines and culture conditions
Experimental procedures performed on mice were approved by the ethical committee of INSERM CREFRE US006 animal facility and authorized by the French Ministry of Research: APAFIS#3600-2015121608386111v3.
RK13 (rabbit kidney cells), Hela (human cervix cancer cells), and WM266-4 (human melanoma cancer cells) were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM; ThermoFisher Scientific, Illkirch, France) 4.5 g/L glucose (for MiaPACA2) containing 10% fetal bovine serum (FBS; ThermoFisher Scientific), 100 IU/mL penicillin (ThermoFisher Scientific), 100 μg/mL streptomycin (ThermoFisher Scientific), and 250 ng/mL fungizone (ThermoFisher Scientific), 2 mM glutamine (Sigma), and 100 IU/mL plasmocin (Invivogen) (hereafter referred to as complete medium). Pancreatic cancer patient-derived primary cells PDAC012T, PDAC087T, and PDAC072T were obtained and cultured as previously described. 19 Cells were incubated at 37°C with 5% CO2.
Viruses cloning, viral production, and titration
SG33-ANCHOR and T1-ANCHOR were obtained by homologous recombination of lacZ gene from the SG33-LacZ and T1-LacZ viruses with the donor plasmid pVmyxlac-ANCHORGFP-GPT. SG33-LacZ and T1-LacZ were derived from the SG33-attenuated vaccine strain and Lausanne-like Toulouse 1 strain (T1) of MYXV, respectively. 20 In these recombinant viruses, the lacZ gene under the control of the poxvirus late p11 promoter is inserted between the M009L and M010L MYXV genes. The donor plasmid pVmyxlac-ANCHORGFP-GPT includes the lacZ sequence in which three sequences were inserted through appropriate restriction sites; the ANCHOR system is composed of a sequence coding for a fusion protein ORGFP and a nonrepetitive DNA target sequence ANCH, and finally the EcoGPT selection cassette. ORGFP and EcoGPT genes were each under the control of a poxviral promoter p7.5. In the SG33-ANCH virus, the ORGFP expression cassette is lacking to generate a nonfluorescent viral DNA genome.
MYXV-ANCHOR recombinant viruses were selected by resistance to mycophenolic acid 21 and GFP positive criteria by fluorescence microscopy. MYXVs were produced in RK13 cells. In brief, RK13 cells were infected until 80% of cytopathic effect. The supernatant and cell lysate were harvested, and were either frozen and thawed three times (crude virus), or purified through two 36% sucrose cushion as previously described. 22 MYXV quantification was performed by plaque assay and the virus titer was determined as plaque forming units (PFUs). 23
Real-time viral replication analysis at the cell population level
Cells were seeded in 96-well plates (Corning, Boulogne-Billancourt, France) at 10 × 103 cells per well in 100 μL of complete medium. After 24 h, cells were washed with phosphate buffered saline (PBS) and infected at corresponding multiplicity of infection (MOI) during 1 h at 37°C in 100 μL of phenol-free complete (see above) culture medium, so as to avoid viral green fluorescence quenching. Infection medium was then replaced with phenol-free complete culture medium. After infection, cells were imaged in real time at 37°C, 5% CO2 with the IncuCyte Zoom (Sartorius). Images were acquired at 10 × magnification every 3 h using brightfield and green filters (Ex: 440–480 nm, Em: 504–544 nm) to determine cell confluence and viral replication, respectively, and were analyzed using the Zoom2016B software.
Real-time viral cytolytic activity assay
Cells were seeded in 96-well plates (Corning) at 10 × 103 cells per well in 100 μL of complete medium. After 24 h, cells were washed with PBS and infected with SG33-ANCH lacking the ORGFP protein at corresponding MOIs during 1 h at 37°C in 100 μL of phenol-free complete culture medium. Infection medium was then replaced with phenol-free complete culture medium supplemented with 250 nM of cytotox green (Ex: 491 nm, Em: 509 nm), a reagent for real-time quantification of cell death (Sartorius). Cells treated by 0.5 μM of Staurosporine were used as a positive control. After infection, cells were imaged in real time at 37°C, 5% CO2 with the IncuCyte Zoom (Sartorius). Images were acquired at 10 × every 3 h using brightfield and green filters (Ex: 440–480 nm, Em: 504–544 nm) to determine cell confluence and cell death, respectively, and were analyzed using the Zoom2016B software.
Viral genome replication quantitation
Cells were seeded in 12-well plates (Corning) at 100 × 103 cells per well in 1 mL of complete medium. After 24 h, cells were washed with PBS and infected at corresponding MOIs during 1 h at 37°C in 200 μL of phenol-free complete (see above) culture medium. Infection medium was then replaced with phenol-free complete culture medium. Viral replication was measured with the IncuCyte Zoom (Sartorius) at 10 × using a green filter (Ex: 440–480 nm, Em: 504–544 nm) and analyzed using the Zoom2016B software.
Cells were recovered and DNA was extracted using the One-4-All Genomic DNA Mini-Prep kit (Bio Basic) following manufacturer's instructions. Samples were diluted in sterile water before viral DNA was quantified by qPCR, as described previously, 24 using QuantiNova Probe directed toward the M071lq gene of MYXV with the LightCycler 96 (Roche, Boulogne-Billancourt, France) system. Data were analyzed using the LightCycler 480 1.5 software.
Viral DNA quantification in live cells
Cells were seeded and infected in 96-well plates as previously described. After 48 to 72 h of infection, cells were fixed with formalin (Sigma) according to manufacturer's instructions and imaged at 10 × magnification using the CellInsight CX7 High-Content Screening (Cell Insight CX7 HCS; ThermoFisher, Illkirch, France), with light emitting diode (LED) 10 × (numerical aperture, NA, 0.30) nonconfocal with the following light sources (Ex/Em, in nm): 438/480 (blue), 485/521 (green), and 549/600 (red). Number of cells was determined after nuclei staining with Hoechst 33342 (ThermoFisher). Fluorescence intensity was quantified using HCS Studio software for integrated data collection and analysis (ThermoFisher).
Single-cell analysis of viral infection
Cells were seeded in 96-well plates (PerkinElmer) at 10 × 103 cells per well in 100 μL of complete medium. After 24 h, cells were washed with PBS and infected at corresponding MOIs during 1 h at 37°C in 100 μL of phenol-free medium. Infection medium was then replaced with 100 μL of phenol-free complete culture medium. Forty-five whole-well images were acquired every 15 min for 25 h using the Operetta CLS High-Content Analysis System (PerkinElmer), with LED 5 × Air (NA 0.4) confocal with eGFP filter (Ex: 460–490 nm, Em: 500–550 nm). Images were exported and analyzed using the Columbus program (PerkinElmer).
Viral propagation assay in three-dimensional spheroid model
RK13 and PDAC087T cells were seeded in 96-well ultralow attachment bottom-rounded plates (Corning) at 5.103 cells per well and centrifuged 400 g for 10 min. After 5 to 7 days of culture, spheroids were formed and infected at corresponding PFU of SG33-ANCHOR (10 × 103 to 500 × 103) and incubated at 37°C, 5% CO2. Four (RK13 cells) or 7 (PDAC087T cells) days later, spheroids were imaged using the Operetta CLS High-Content Analysis System (PerkinElmer), with LED 5 × Air (NA 0.16) nonconfocal with brightfield and eGFP filter (Ex: 460–490 nm, Em: 500–550 nm). Images were acquired using Harmony 4.9, exported and analyzed using the Columbus (PerkinElmer) 2.8.2 program. PDAC087T cells were also imaged using the LSM 880 AxioObserver (Zeiss), laser 10 × plnApo (0.45NA) confocal with brightfield, and FITC filter (Ex: 470/40 nm, Em: 525/50 nm). Images were acquired using Zeiss black 2.3 and analyzed using Zeiss blue 2.3.
Experimental tumors
PDAC087T cells were engrafted subcutaneously into the flank of SCID CB17 mice (2 × 106 cells per mouse, n = 5 mice per group). Two weeks later, when tumors became palpable, 5 × 105 PFUs of SG33 were diluted in 50 μL of PBS and injected intratumorally. Control tumors received 50 μL of PBS (placebo). Tumor size was measured using a caliper and monitored using the Aixplorer echography system. Mice were killed 2 weeks after virus injection and tumors were collected and imaged for fluorescence using the Ivis Spectrum (PerkinElmer). Tumors were sampled and minced in liquid nitrogen, and centrifugated for 20 min at 11,000 g and 4°C. Cleared supernatants were used to infect RK13 cells, and viral replication was analyzed as previously described.
Quantification and statistical analysis
Unless otherwise indicated, data represent mean ± standard error of the mean of at least three biological replicates. Statistical significance was calculated by unpaired student test with Welch's correction, using Graphpad Prism 8.0 software (Graphpad). p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.005.
RESULTS
We generated the MYXV Lausanne T1 and SG33 viral genomes equipped with ANCHOR-GFP molecular beacon (Material and Methods and Fig. 1A, B) to address viral replication in real time in live cells. RK13, which are highly permissive to MYXV, were infected with MOI = 0.1 to MOI = 5 of either Lausanne T1 or SG33. SG33-ANCHOR infection resulted in significantly larger viral plaques and perinuclear viral replisomes characteristic of Poxvirus infection (Fig. 2A, top panel), as compared with cells infected with T1-ANCHOR (Fig. 2A, bottom panel).
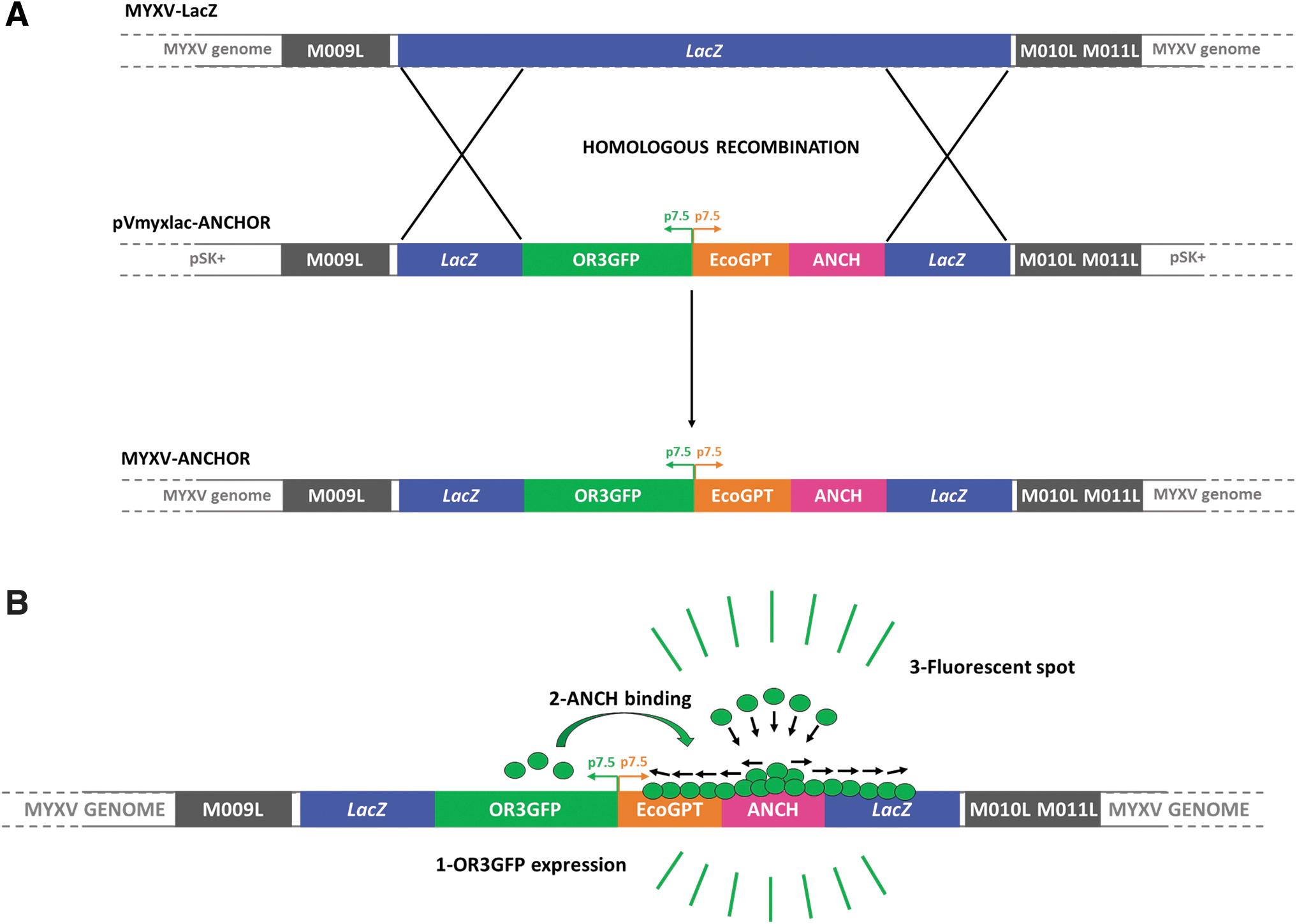
MYXV-ANCHOR construction.
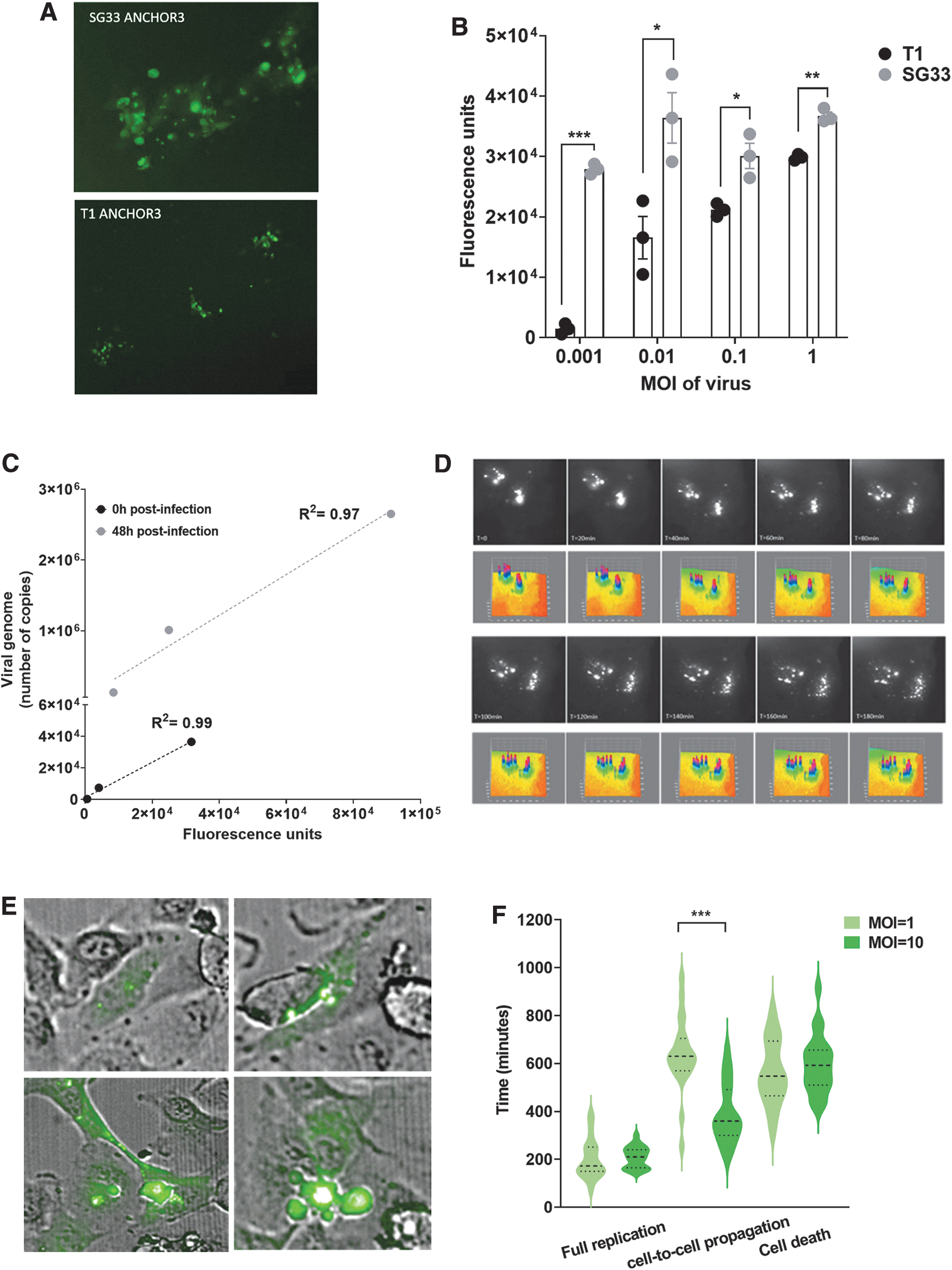
Analysis of SG33-ANCHOR viral cycle in model cells.
Forty-eight hours later, replication level in infected cells was quantified by live imaging. RK13 cells infected with vaccinal SG33 demonstrated higher viral genome replication, as compared with cells infected with parental T1 Lausanne strain (Fig. 2B), with a 2- to 19-fold increase in fluorescence intensity, and the highest difference in replication was measured when using the lowest dose of virus for infection. These data demonstrate that in model cells, SG33 replicates to a larger extent than parental T1 Lausanne MYXV strain. Additional data demonstrated that SG33 genome replication was readily detected after infection of WM266-4 (melanoma) and Hela (cervix) cancer cells (Supplementary Fig. S1A).
To confirm the correlation between fluorescence intensity and viral genome replication, we infected RK13 cells with increasing levels of SG33-ANCHOR (MOI 0.1, 1, 5) and measured total fluorescence intensity directly after infection, and 48 h postinfection using the Incucyte Zoom system (Supplementary Fig. S1B). Similarly, we quantified viral genome copies in these RK13 cells at both timepoints. We found a strong correlation between the two approaches at both timepoints (Fig. 2C), with R 2 = 0.99 at T = 0 and R 2 = 0.97 at T = 48 h postinfection, demonstrating that ANCHOR and qPCR share the same performance for viral genome quantification.
We next performed time lapse studies to better characterize the viral cycle of SG33 in permissive cells (Fig. 2D and Supplementary Movie S1). Using high-definition microscopy and three-dimensional (3D) surface plots of viral replication foci, we detected viral input (T = 0, Fig. 2D) that progressively disaggregate to form viral DNA factories in infected cells (T = 180, Fig. 2D).
We next performed longer noninvasive viral genome tracking studies to determine cell-to-cell viral propagation and virus-induced cell death. Thus, RK13 cells were infected with MOI = 1 and MOI = 10 of SG33-ANCHOR. Uninfected RK13 cells were used as control to address cell autofluorescence. SG33-ANCHOR genomes were readily detectable in living cells, at the single-cell level, either as viral input (Fig. 2E top left panel) or viral DNA factories (Fig. 2E top right). Time monitoring indicates that SG33 reaches full replication 202 ± 19 min and 202 ± 10 min after infection with MOI = 1 and MOI = 10, respectively (Fig. 2F). Remarkably, we confirm that SG33 MYXV traffics from cell to cell using cytoplasmic bridges 25 (Fig. 2E, bottom left panel). Notably, we identified that higher dose of virus significantly accelerates viral propagation from cell to cell (390 ± 29 min vs. 638 ± 36 min after infection with MOI = 10 and MOI = 1 of SG33, respectively). We show that SG33-ANCHOR is lytic (Fig. 2D, bottom right panel), and that once infected, RK13 cells die within 563 ± 31 min (MOI = 1) and 598 ± 28 min (MOI = 10), respectively.
Next, we aimed to monitor SG33 viral spread and oncolytic activity at the cell population level. Thus, RK13 cells were infected with increasing MOI of SG33-ANCHOR. Figure 3A and Supplementary Movie S2 depict viral progression as imaged noninvasively using the Incucyte Zoom. We detected and quantified viral replication down to MOI = 0.01 of SG33-ANCHOR, which increased dose dependently to reach a peak of replication as early as 24 h after infection with MOI = 10 of virus, and after 60 h of infection of cells with MOI = 1 of virus (Fig. 3B). Linear regression analysis indicates a positive correlation between the viral dose and the increase in viral replication. In parallel, we analyzed cell confluence and found that SG33 replication correlates with dose-dependent inhibition of cell proliferation (Fig. 3C).
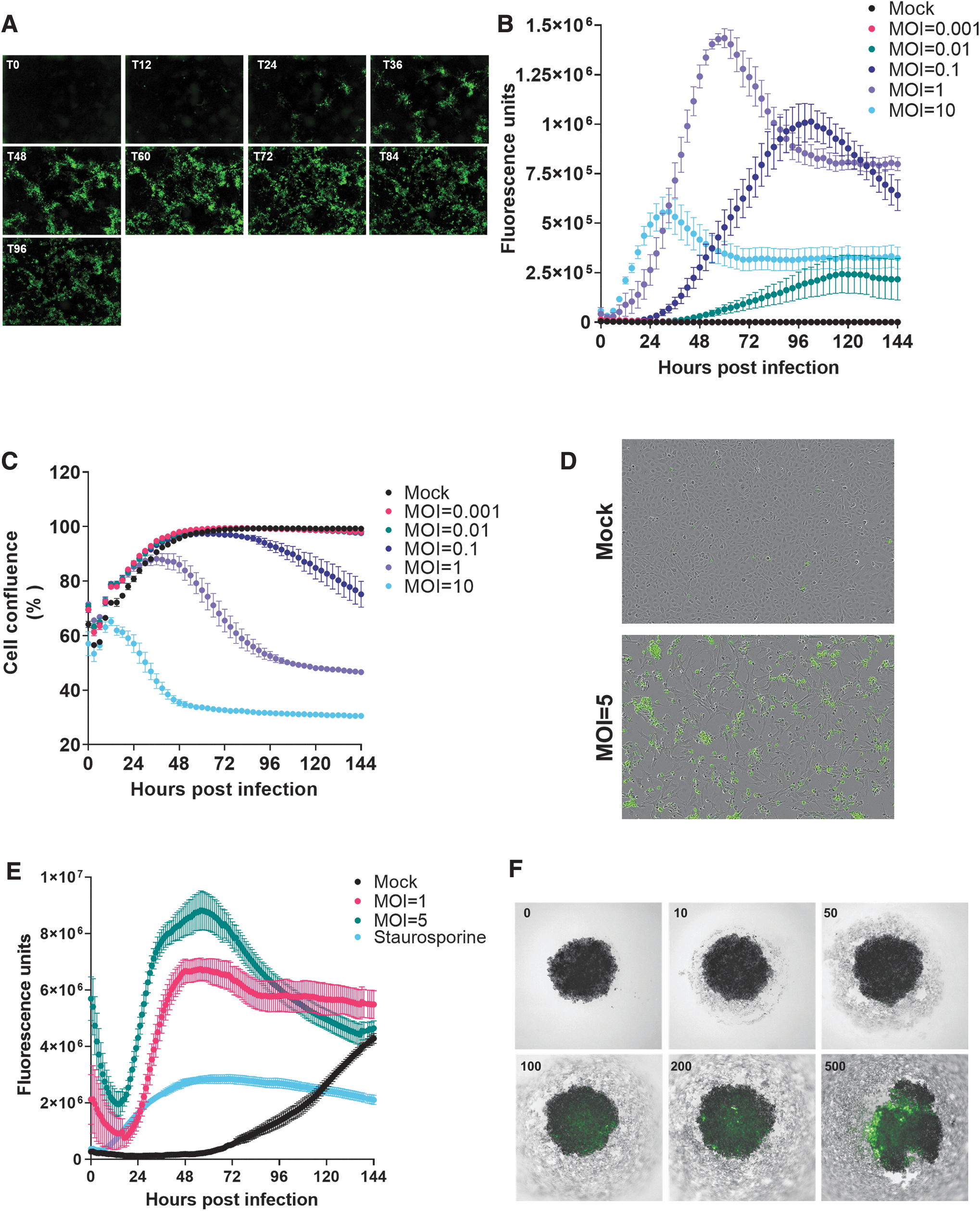
SG33 cytolytic activity in model cells. RK13 cells were infected with SG33-ANCHOR and imaged every 3 h using the Incucyte Zoom.
To determine whether the decrease in cell confluence was due to virus-induced cell death, we performed the same experiment with SG33-ANCH virus lacking the ORGFP protein, and a green fluorescent cytotoxicity marker to characterize cell death (Fig. 3D). Cells treated with staurosporine were used as control. Figure 3E shows that SG33 increases cytolysis in RK13 cells in a dose-dependent manner. Collectively, we demonstrate here for the first time the oncolytic potential of SG33-ANCHOR in live infected cells.
Three-dimensional cell culture systems are becoming increasingly important in preclinical cancer research, as they recapitulate microtumors, metastases, and the tumor microenvironment much better than monolayer culture systems could. Although 3D tumor cell cultures are increasingly employed in virotherapy research, 26 live monitoring of OV replication in spheroids has not be achieved so far. Accordingly, we generated spheroids from RK13 cells. Five days later, 3D cultures were infected with increasing PFUs of SG33-ANCHOR (10 × 103 to 500 × 103). Cultures were monitored noninvasively for GFP fluorescence using the Operetta microscope 4 days postinfection. As shown in Fig. 3H, we identified a dose–response increase in fluorescence demonstrating infection of 3D models by SG33-ANCHOR. Remarkably, we found that SG33 infection strongly alters spheroids structure and increases cancer cell death, as monitored by detection of cellular debris.
As a pilot study of applying SG33-ANCHOR as oncolytic virotherapy in PDAC, we selected a panel of patient-derived primary cells of basal-like and classical PDAC subtypes. 19 We first addressed SG33-ANCHOR specificity and found that the virus failed to replicate into and spared normal pancreatic HPDE cells (Fig. 4A and Supplementary Fig. S1C).
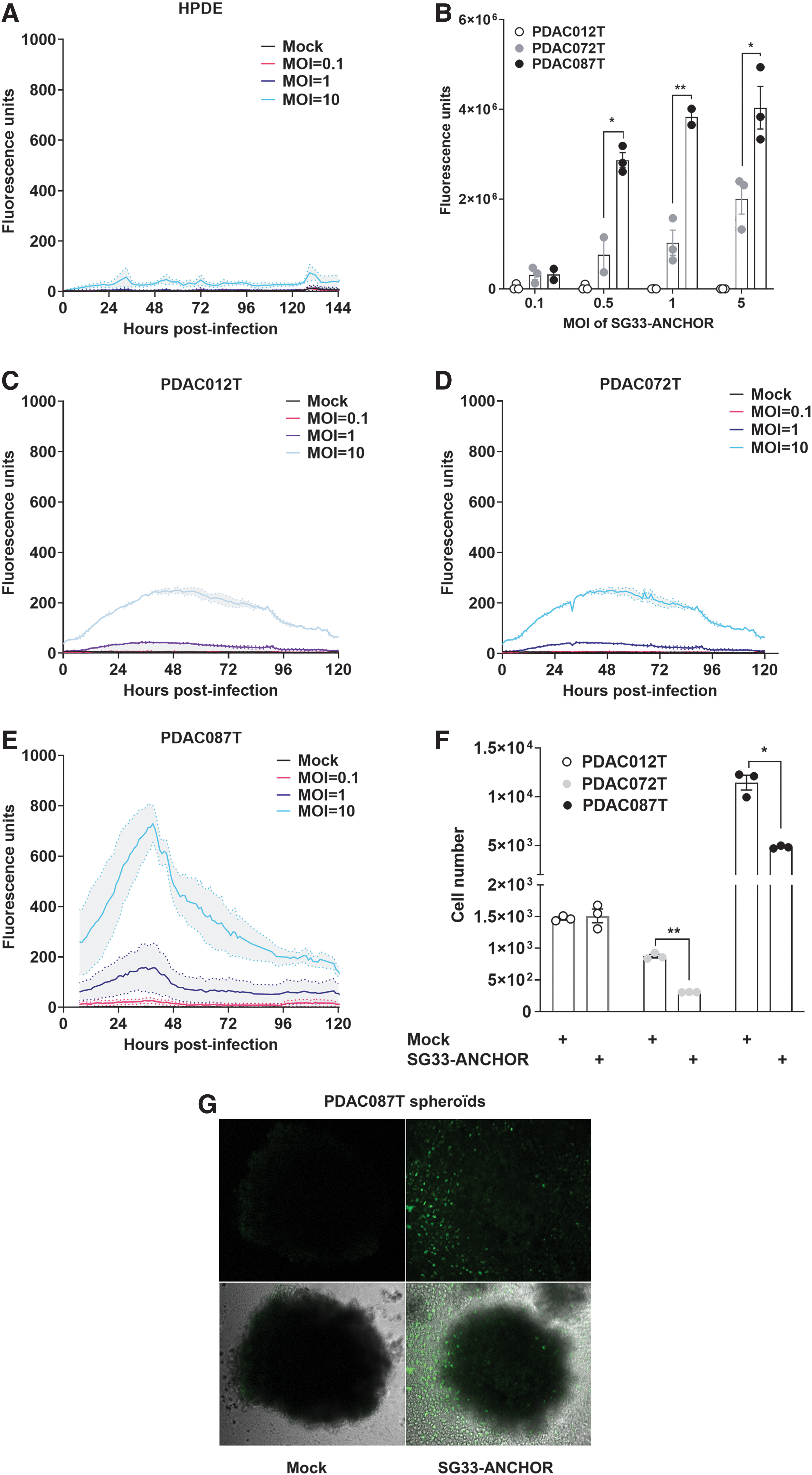
Characterization of SG33 replication and oncolytic activity in primary cells derived from pancreatic cancer.
We next addressed SG33-ANCHOR replication at the single-cell level and quantified viral DNA content in infected PDAC primary cell lines by fluorescence using the CellInsight CX7 HCS platform. As shown in Fig. 4B, we identified that PDAC012T primary cells poorly replicate SG33-ANCHOR; in contrast, we identified a dose-dependent increase in SG33 viral DNA production in PDAC087T cells, and, to a lesser extent, in PDAC072T cells.
Next, we monitored SG33-ANCHOR replication in PDAC012T, PDAC087T, and PDAC072T live cultures at the population level (Fig. 4C–E). PDAC cells were infected with increasing MOI of SG33-ANCHOR, ranging from MOI = 0.1 to MOI = 10, and viral replication and cell death were monitored noninvasively. We found that SG33-ANCHOR replicates in all primary cancer cells tested, although at different levels. In PDAC087T, SG33-ANCHOR replication is higher than that of PDAC012T and PDAC072T. Interestingly, SG33-ANCHOR is oncolytic for PDAC072T cells, despite low replication levels, and PDAC087T cells, while PDAC012T cells resisted to SG33-ANCHOR infection (Fig. 4F). These data indicate for the first time that SG33-ANCHOR replicates specifically in human cancer cells and is oncolytic for PDAC primary cells but could also be affected by cellular heterogeneity found in the tumors.
Next, we determined whether viral replication and progression could be followed in 3D cultures of PDAC primary cells. For this, we formed tumoroids of PDAC087T cells and infected them with 500 × 103 PFU of SG33-ANCHOR, with noninfected tumoroids as control. Seven days later, viral replication was detected and, similarly to what was seen in RK13 spheroids, SG33-ANCHOR altered spheroid composition with a significant increase of cellular debris (Fig. 4G). In addition, viral particles can be detected in detached dying/dead cells, as seen by fluorescent signals in cell debris around the spheroid. Overall, we managed to detect SG33 replication and propagation within 3D tumor models, to induce cancer cell death.
We next investigated whether SG33-ANCHOR replication and antitumoral activity could be characterized in experimental tumors, in vivo. We selected PDAC087T cells that were engrafted subcutaneously in athymic mice. When tumors were palpable, 5 × 105 PFUs of SG33-ANCHOR were injected intratumorally. Control tumors received PBS. Tumor size was measured using a caliper. We found that SG33-ANCHOR infection impairs tumor burden (Fig. 5A) and delays tumor growth (−38% ± 6%, p < 0.001, Fig. 5B). We performed echography analyses at the end of the infection protocol and before autopsy and measured a mean 25% decrease in tumor size in the SG33-ANCHOR–treated tumor group, as compared with tumors receiving placebo (Fig. 5C). Tumors were sampled and analyzed for fluorescence using the Ivis Spectrum. Results in Fig. 5D illustrate the presence of infected cells within tumors treated with SG33-ANCHOR.
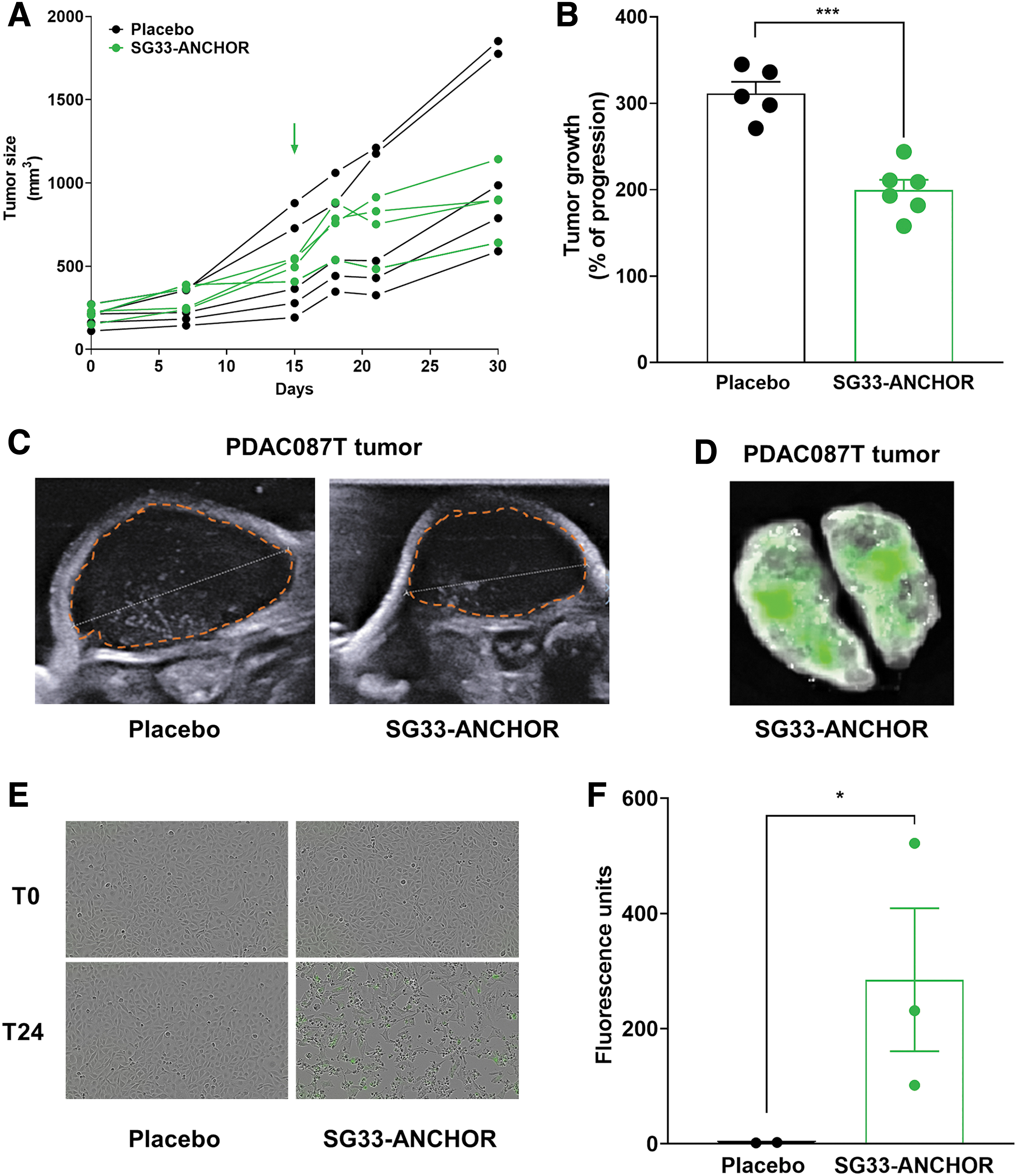
SG33-ANCHOR antitumoral efficacy and tracking in experimental pancreatic tumors. Experimental pancreatic tumors were generated following engraftment of PDAC087T cells in athymic mice. 5x105 p.f.u. of SG33-ANCHOR were injected intratumorally. Control mice received PBS (placebo, n = 5 mice per group).
Lastly, tumors from both groups were homogenized, and cell-free supernatant was used to infect reporter RK13 cells to quantify tumor viral load. We found that cells infected with cell-free supernatant from SG33-ANCHOR–treated tumors developed infection, as compared with cells treated with cell-free supernatant from control tumors (Fig. 5E, F). Collectively, these data demonstrate for the first time that SG33 is oncolytic for PDAC cells and that the ANCHOR system allows for rapid characterization of cancer cell susceptibility to OV infection and lysis, both in vitro and in vivo
DISCUSSION
OVs are innovative therapies that could be extremely beneficial within the treatment plan of patients with cancer. For this, however, a deep understanding of viral infection, replication, and spread as well as defining the oncolytic activity of these viruses are prerequisites before their use in a clinical context. The ANCHOR system is a novel tool of genetic engineering that allows for the noninvasive detection of DNA in live cells. In this study, we generated for the first time OV genomes equipped with the ANCHOR system to quantify OV replication and oncolytic activity in live cells, at single-cell and populations levels.
In proof-of-concept studies, we confirmed the infectious and lytic capacity of SG33, a new vaccinal strain of MYXV, 20,27 and demonstrate that SG33 outranks parental T1 Lausanne, in a dose-dependent manner. We also monitored the precise timing for SG33 to replicate, expanse to neighboring cells, notably confirming propagation using cytoplasmic bridges 25 to induce cell death, and demonstrated viral spreading within 2D and 3D cellular models. Undoubtedly, such discoveries were largely facilitated by the ANCHOR system, which demonstrate equivalent performance with end-point qPCR for viral DNA genome.
As stated before, the lytic potential of SG33 was previously shown 27 ; in this study, we demonstrate that the ANCHOR system does not hamper the replication and lysis capacities of this virus, paving the way for future theranostic strategies. However, we believe there is still room for improvement, notably using concomitant live detection of viral proteins fused to fluorescent proteins, when analyzing spheroid or whole tumor infection in animals.
We then demonstrated for the first time that SG33 has oncolytic potential for PDAC, a disease with no cure, that is eligible for virotherapy. 18 We infected primary human PDAC cells that recapitulate tumor phenotype and genotype diversity and defined different profiles of viral replication and cytopathic effects. Indeed, PDAC087T cells displayed the highest level of viral replication and subsequent cell death; PDAC012T and PDAC072T cells, however, showed comparable levels of SG33 replication but only PDAC072T cells were significantly lysed by the virus. Notably, we found that SG33-ANCHOR successfully infected and replicated into PDAC tumoroids, to induce cell death.
Hence, we demonstrated the antitumor capacity of SG33-ANCHOR in vivo since treated experimental tumors presented significant delays in tumor growth when compared with control tumors. As our study demonstrates heterogeneity in virotherapy efficacy in PDAC primary cells, it remains essential to identify cellular and molecular factors that govern SG33 efficacy in PDAC cells to accelerate clinical transfer. Importantly, SG33 originates from the in vivo recombination between a wild-type South American (T1 Lausanne) strain and a California MSD-derived strain, and carries a large deletion in the right open reading frame of its genome. 20 In greater details, SG33 genome lacks one copy of the viral m-t5 ankyrin coding sequence that coordinates with Akt cellular signaling to establish a new cellular environment more favorable for virus replication. 28 As this protein is partially lost in SG33, we obtained preliminary results indicating that AKT status is not fully predictive of PDAC cell infection and lysis by SG33 (data not shown).
We speculate that complemental high-throughput screening studies conveyed with the ANCHOR system following protocols established during this study are in reach to identify the molecular mechanisms involved in PDAC cells permissiveness to SG33 viral infection and oncolysis. Together with transcriptomic investigations, this will help define a molecular signature predictive of SG33 efficacy for patient stratification, and to devise new combinations to improve the therapeutic index of virotherapy.
To date, the human sodium/iodide symporter (hNIS) is the most prevalent reporter transgene to generate precise insights into the kinetics of gene expression, viral biodistribution, toxicity, and therapeutic outcomes in the field of virotherapy. hNIS promotes the accumulation of radiotracers at the site of transgene expression, 29 both in preclinical models and in patients. 30 Yet, this reporter system has several disadvantages, such as a limited intracellular retention capacity of radiotracers in hNIS-infected cells and potentially dangerous radiotracer uptake by both malignant and normal tissues. These include the thyroid gland and the digestive tract. 31
During this study, we demonstrate that ANCHOR “all-in-one system” allows for OV detection in tumors, up to 15 days after infection, without hindering viral replication or spread, and without the need of any supplemental tracers. Remarkably, the ANCHOR system is compatible with tumor viral load tittering ex vivo, which is mandatory to address the viral pharmacokinetics and to adapt treatment in patients. However, we failed to detect SG33 growth in tumors in live animals using GFP-labeled viral genomes, probably because of high-background autofluorescence due to animal hair bulbs and chlorophyll in food. Studies are currently undergoing to generate novel SG33 genomes equipped with red fluorescent proteins such as Santaka to allow for noninvasive in depth tissue fluorescent imaging of the virus.
CONCLUSION
Several oncolytic strains of viruses are now developed for the therapy of hematologic and solid tumors. 32 For all of them, novel strategies for the noninvasive monitoring of OV biodistribution and efficacy are key to overcome the current limitations of oncolytic therapy. Poxviruses such as MYXVs may take the stage as they are nonpathogenic for humans, are large virus with DNA genomes that can be equipped with therapeutic genes of interest, can be administered locally and in the blood to target distant metastasis, and can induce an immune response against the tumor. 33
By nature, SG33 holds the promise of increased specificity toward cancer cells and greater mobilization of immune cells, as already demonstrated for rabbit and ovine bone marrow-derived dendritic cells. 34 In this study, we demonstrate not only that SG33 replicates to higher levels than MYXV T1 and that SG33 is oncolytic for primary cancer cells, but also that the ANCHOR system provides sufficient specificity and efficacy necessary for longitudinal monitoring of SG33 viral infection in complex models without hampering viral fitness.
Considering that SG33 is a clinical-grade veterinary vaccine with limited manufacturing constraints that could be easily repurposed for human health, our study advocates for additional preclinical investigations to accelerate the clinical transfer of SG33-ANCHOR for the benefit of PDAC patients.
Footnotes
Authors' Contributions
Conceptualization was carried out by P.C., S.B., F.G., C.C., L.B., and N.D.; investigations were done by L.Q., S.T., S.K.-G., A.R., C.Q.-F., and H.L.; supervision was performed by P.C., S.B., and F.G.; writing—original draft—was by P.C. and L.Q.; writing—review and editing—was carried out by S.B., N.D., and F.G.; funding acquisition was taken care of P.C., S.B., and N.D.; and data analysis was carried out by P.C., S.B., F.G., L.Q., and A.R.
Acknowledgments
The authors acknowledge the imaging and the vectorology facilities of the CRCT technological cluster, and the technical assistance of Ms. Laetitia Ligat and Ms Emilie Martin.
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported by grants from CHU Toulouse (L.Q.) and Fonroga Fondation (A.R.). This study was supported from a BPI France funding (I-Lab) (S.T, SK-G) and TheradPox (EU).
Supplementary Material
Supplementary Figure S1
Supplementary Movie S1
Supplementary Movie S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.