
Abstract
Exon skipping therapies for Duchenne muscular dystrophy that restore an open reading frame can be induced by the use of noncoding U7 small nuclear RNA (U7snRNA) modified by an antisense exon-targeting sequence delivered by an adeno-associated virus (AAV) vector. We have developed an AAV vector (AAV9.U7-ACCA) containing four U7snRNAs targeting the splice donor and acceptor sites of dystrophin exon 2, resulting in highly efficient exclusion of DMD exon 2. We assessed the specificity of splice variation induced by AAV9.U7-ACCA delivery in the Dmd exon 2 duplication (Dup2) mouse model through an unbiased RNA-seq approach. Treatment-related effects on pre-mRNA splicing were quantified using local splicing variation (LSV) analysis. Filtering the transcriptome for differences in treatment-related splicing resulted in only 16 candidate off-target LSVs. Only a single candidate off-target LSV was found in both skeletal and cardiac muscle tissue and occurred at a known variable cassette exon. In contrast, four LSVs represented significant on-target correction of Dmd exon 2 splicing and transcriptome analysis showed correction of known dystrophin-deficient gene dysregulation. We conclude that the absence of off-target splicing induced by treatment with the U7-ACCA vector supports the continued clinical development of this approach.
Background
Exon 2 of the DMD gene, expressed in all 427 kilodalton (kDa) isoforms of the dystrophin protein, is the most commonly duplicated exon associated with Duchenne muscular dystrophy (DMD), accounting for ∼10% of all DMD duplications. 1,2 Single exon duplications are promising targets for molecular therapies, as skipping of a single copy of a duplicated exon would restore a wild-type (WT) reading frame and expression of full-length dystrophin, which may be expected to lead to better outcomes than existing therapies for DMD, 3,4 as well as other approaches currently in clinical trials. 5
As a platform for testing single exon skipping, we created a novel mouse model that contains a duplicated DMD exon 2 (the Dup2 mouse), which largely recapitulates the findings in the standard mdx mouse. 6 For any single exon duplication resulting in an out-of-frame transcript, highly efficient skipping of both copies of the exon would be predicted to result in an out-of-frame exon deletion transcript, with no improvement in phenotype. Exon 2, however, is a special case that presents a wide therapeutic window, as complete exclusion of the exon results in the expression of a highly functional dystrophin isoform that results from ribosome assembly on an internal ribosome entry site (IRES) within exon 5 and translational initiation from a methionine within exon 6 of DMD. 7,8
The extent of this therapeutic window is demonstrated by the exceedingly mild phenotype associated with the first North American founder allele demonstrated in DMD, a nonsense mutation within exon 1 (c.9G>A; p.Trp3X). 9 Despite the fact that a nonsense mutation would be predicted to result in expression of no dystrophin, most males who carry the mutation only have myalgias with exertion; the most severely affected individual stopped walking at age 62, whereas others had no reported symptoms into their eighth decade. 9 We demonstrated that this mild phenotype was due to the expression of significant amounts of an isoform of dystrophin resulting from utilization the glucocorticoid-sensitive exon 5 IRES. 8 The resulting isoform is ∼413 kDa in size and lacks the calponin homology domain 1 (CH1) that makes up the first half of the actin-binding domain 1 (ABD1) at the N-terminus of dystrophin. 7,8 Despite the canonical view that an intact ABD1 is required for dystrophin functionality, the human “experiment”—the presence of the founder allele in humans without significant symptoms, and clearly no effect on reproductive fitness—argues that the actin-binding domain 2 (ABD2), located within the dystrophin rod domain, is sufficient for significant preservation of muscle function. 8
We noted that duplications of exon 2 were nearly always associated with DMD, even though the duplication resulted in an altered reading frame and hence a premature termination codon in the second copy. At the same time, we noted that deletions of exon 2, which are similarly out of frame, had never been reported in either our large cohort or in the exhaustive dystrophinopathy mutational databases. 1,2,10 Assuming that such a mutation had never been ascertained because of IRES activity, we hypothesized that the IRES was active in the absence of exon 2 but not in the presence of a duplicated exon 2, a result confirmed by in vitro studies, and by the identification of an essentially asymptomatic patient with a deletion of exon 2. 8 The therapeutic implication of these findings is that there is a wide margin of safety for skipping of exon 2, as boys with exon 2 duplications cannot be “overtreated.” Efficient skipping will result in either the WT transcript containing one copy of exon 2, or in the complete exclusion of exon 2, resulting in IRES activation and the expression of the highly protective isoform lacking CH1.
To induce highly efficient exon 2 skipping, we have developed the
The purpose of this study is to identify and quantify potential off-target splicing effects induced by systemic delivery of the scAAV9.U7-ACCA vector in the Dup2 mouse. We delivered clinically relevant doses to P0 mice by a single systemic injection through the facial vein, and 3 months later examined RNA-seq data generated from skeletal muscle, heart, and liver from treated and control Dup2 mice, analyzing gene expression and splicing using an empiric and unbiased approach. We measured on and off-target splicing effects by quantifying “local splice variation” (LSV) at splice junctions defining the start or end of each exon. A single LSV measures the relative level of splice events detected by RNA-seq reads mapping across exon–exon, intron–exon, or exon–intron boundaries. An LSV quantifies the relative amount of splice junction reads on a “Percent Spliced In” (PSI) scale from 0 to 1. For example, an idealized exon skipping event at 20% skipping efficiency would be measured by counting splice junction reads at both the “source” and “target” LSVs. The source LSV is the splice donor site upstream of the skipped exon while the target LSV is the splice acceptor site of the exon downstream of the skipped exon. They will share a common set of RNA-seq junctions reads that skip the intervening exon but will differ in their junction reads to and from the skipped exon. Splicing viewed from the source LSV donor site will count 20% of its RNA-seq junction reads ( = 0.2 PSI value) mapping to the target LSV acceptor site, while 80% of its junction reads (0.8 PSI) will map to the skipped acceptor site. The same exon skipping event will be measured from the target LSV by counting the junction reads in common with the source LSV (0.2 PSI) and the 80% (0.8 PSI) junction reads that map from the skipped donor site to the target LSV acceptor site. Previous RNA-seq studies have shown that quantifying LSVs in this fashion identifies differentially spliced events with high accuracy. 17 LSV analysis shows no evidence for meaningful off-target splicing alteration, whereas significant on-target exon 2 splicing variation induced gene expression changes consistent with a reversal of known dystrophic gene expression signatures, results that provide support for the continued clinical development of this approach.
Methods
Mouse injection
As part of ongoing clinical development studies, male Dup2 mice 6 , were injected through facial vein at P0 with 1.8e14 vg/kg of scAAV9.U7.ACCA, and sacrificed at 3 months for evaluation of exon skipping and dystrophin expression. The full report of that study is under review elsewhere, but an outline of the study is included in Supplemental Data.
RNA isolation and sequencing
Frozen heart, gastrocnemius muscle, and liver tissue from 3 C57/Bl6 WT control, 3 untreated Dup2, and 3 treated Dup2 mice were sectioned on a cryostat and homogenized with a 26-gauge needle in 400 μL of 20 mM Tris*Cl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 1% Triton X-100. RNA was extracted using TRIzol® and precipitated with isopropanol according to the manufacturer's instruction (Life Technologies). Total RNA (∼1 μg) was depleted of mouse cytoplasmic and mitochondrial rRNA using tiling oligodeoxynucleotides and the RNase H digestion method. 18 The Illumina TruSeq Stranded Total RNA library Kits were used to prepare indexed libraries and paired-end, 2 × 125 bp reads were generated on an Illumina HiSeq instrument using v4 chemistry.
RNA-seq read alignment and differential gene expression
Paired-end FASTQ reads were preprocessed for quality trimming and adaptor removal using Trimmomatic
19
with the MAXINFO parameter set to 50:0.2. The effective read lengths of the libraries were extended using the FLASH
20
tool that merged overlapping paired-end reads. All reads (extended and nonoverlapping paired-end) were mapped by the STAR RNA-seq aligner
21
(version 2.6.0b) to the GRCm38/mm10 mouse reference genome with splice junction annotations built using the 1-pass mapping strategy with known junctions from the GENCODE comprehensive gene annotation GTF file (Release M17,
Gene-level read counts were extracted from the STAR output Binary Alignment/Map (BAM) files using featureCounts
22
with parameters set to strand-specific read counting, filtering secondary alignments, retaining only uniquely mapped reads and counting reads from multioverlapping gene features. Gene-level read count extraction was performed using featureCounts with Ensembl gene_id annotations from GENCODE release M17. Differential expression (DE) was performed using the edgeR Bioconductor package.
23
Read counts were normalized using the trimmed mean of M values method and filtered to remove low-count genes, requiring a count-per-million threshold value of 5 in at least 3 samples. Library size per sample averaged for heart 35.0 million read counts (range 30.2 to 40.3 million), for gastrocnemius muscle 29.1 million read counts (range 22.2 to 32.3 million), and for liver 8.9 million read counts (range 4.5 to 14.8 million). The design matrix and dispersion estimates were fit to a linear model using the edgeR glmFit function and DE was tested with the glmTreat function between treated and control samples relative to a fold change (FC) threshold of 2. Gene-level results were displayed as mean-difference plots, with log2 FC for each gene plotted against the average abundance in log2 counts per million mapped reads. Gene Ontology and pathway enrichment analysis was performed with the PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System (Version 16.0, released 2020-12-01;
LSV detection
The Modeling Alternative Junction Inclusion Quantification (MAJIQ/Voila v1.0.5) software
17
was used to detect and quantify splice variation from the filtered bam alignment files. MAJIQ Builder used the GENCODE comprehensive gene annotation file (
Thermodynamics of off-target interactions
To calculate RNA–RNA-binding energies, we used the ViennaRNA Package 2.0 server
25
running the RNAup algorithm (
Results
Splice junction quantification in treated versus untreated tissues using RNA-seq
To determine treatment-specific mRNA splicing and gene expression differences in the Dup2 mouse, total RNA was purified from gastrocnemius muscle, heart, and liver tissue, using three Dup2 mice from the treated condition (
The effect of
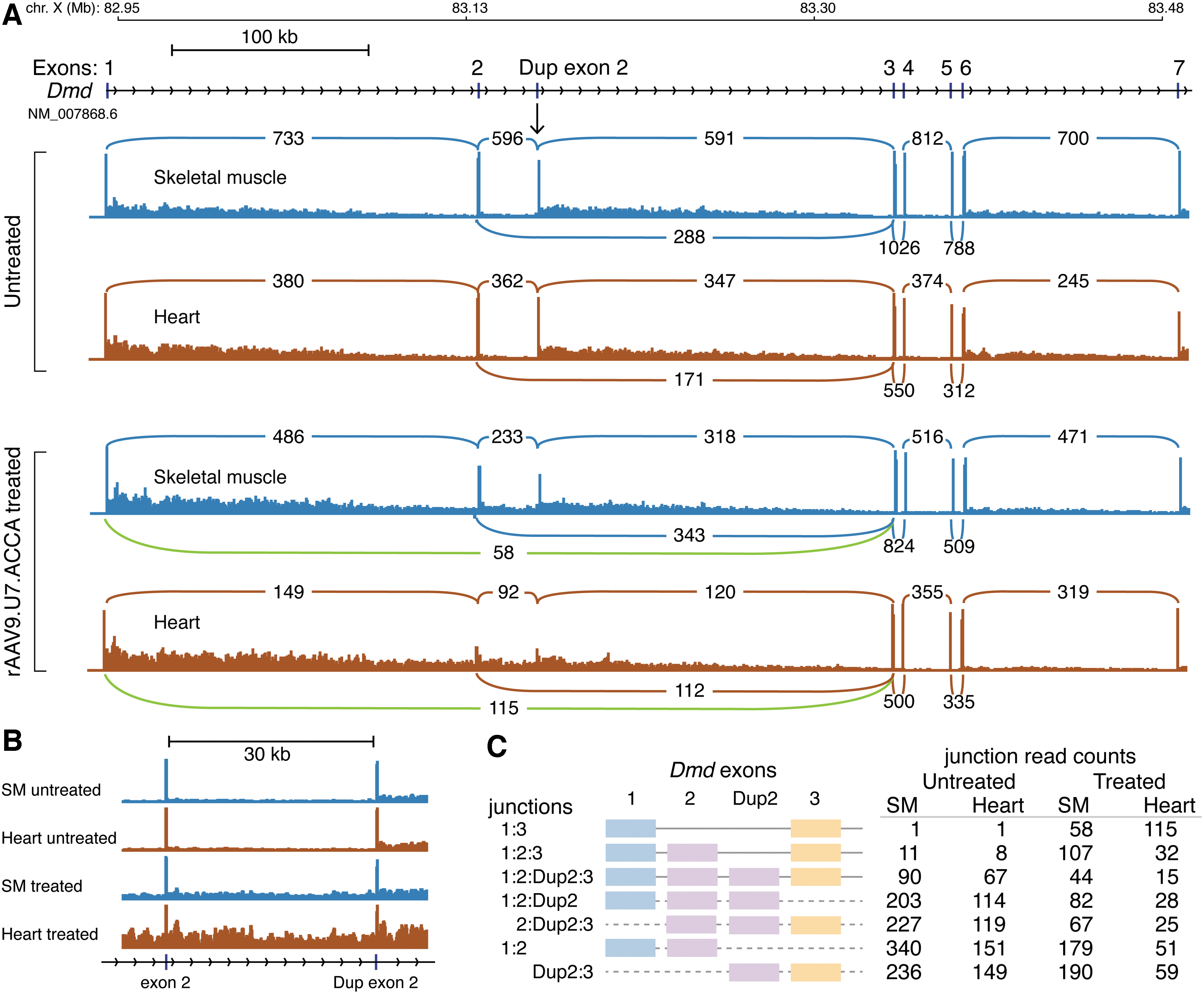
RNA-seq visualization of alternative spliced exons for Dmd transcripts in skeletal muscle and heart.
Since the genomic mapping of RNA-seq reads requires splitting of reads by the mapping algorithm, we extracted reads that mapped to Dmd exons 1, 2, or 3 and then realigned them directly to transcript sequences representing exon 1-3, exon 1-2-3 and exon 1-2-2-3 isoforms. Figure 1C shows the number of reads that completely span exon–exon junctions for exons 1-3 (skipping both copies of exon 2), exons 1-2-3 (skipping of a single exon 2), and exons 1-2-2-3 (the expected spliced transcript from the Dup2 mutation), plus reads that cannot be unambiguously assigned to an isoform but bridge the exon 1-2 or 2-3 junction. In both tissues, the level of exon 2 skipping indicated that the virally expressed modified U7snRNAs were active at their primary Dmd exon 2 targets and confirmed the general validity of quantifying treatment-induced alternative splicing from the mapped RNA-seq reads.
Global analysis of differential mRNA splicing associated with rAAV9.U7.ACCA treatment
To quantify on- versus off-target alternative splicing events from global RNA-seq mapping data, we used the MAJIQ software to detect differential splicing between groups of replicates from the

LSVs using MAJIQ.
In untreated skeletal muscle and heart tissue, PSI values >0.99 indicated that exon 1:2 and exon 2:3 junctions were constitutively spliced and Dmd exon 2 skipping (exon 1:3 junctions) was not detected (PSI value threshold <0.01) in the untreated condition from either source or target LSVs. The
Since the LSV methodology was capable of detecting and quantifying treatment-induced alternative splicing at Dmd exon 2, we cataloged transcriptome-wide LSVs in skeletal muscle, heart, and liver tissues. Differences in transcriptome complexity in each tissue led to variation in the total number of LSVs detected, where a candidate LSV was defined by two or more splice junctions emanating from a single splice donor site (source) or splice acceptor site (target). We used two sequential data filters to narrow the search for treatment-induced differences in constitutive splice junction usage. The first filter selected for constitutive splice sites in the untreated condition by requiring PSI values >0.99. This threshold is level of constitutive splicing observed for Dmd exon 2 in the untreated condition. In heart tissue, 35,880 constitutive LSVs were detected in 9,489 genes; in skeletal muscle, 29,606 constitutive LSVs were detected in 7,964 genes; and in liver, 8,084 constitutive LSVs were detected in 3,113 genes. The second filter required a minimum ΔPSI value of 0.10 between the treated versus untreated condition at these constitutive LSVs.
Only 20 candidate LSVs mapping to 12 genes exceeded a ΔPSI ≥0.10 threshold difference: 11 LSVs from heart, 8 LSVs from skeletal muscle, and 1 LSV from liver (Table 1). Four of these candidate LSVs were the on-target effects for Dmd exon 2 skipping. When ranked by highest ΔPSI values, the Dmd exon 2 source and target LSVs ranked 1 and 2 in heart tissue, and 5 and 6 in skeletal muscle. Five of these candidate LSVs were not splicing LSVs but instead represented alternative promoter LSVs associated with upregulation of promoter activity in the treated samples. Figure 3 shows two examples of upregulation of alternative promoter LSVs for the Tacc2 (transforming, acidic coiled-coil-containing protein 2) gene in heart tissue, and the Abacb (acetyl-Coenzyme A carboxylase beta) gene in liver tissue.
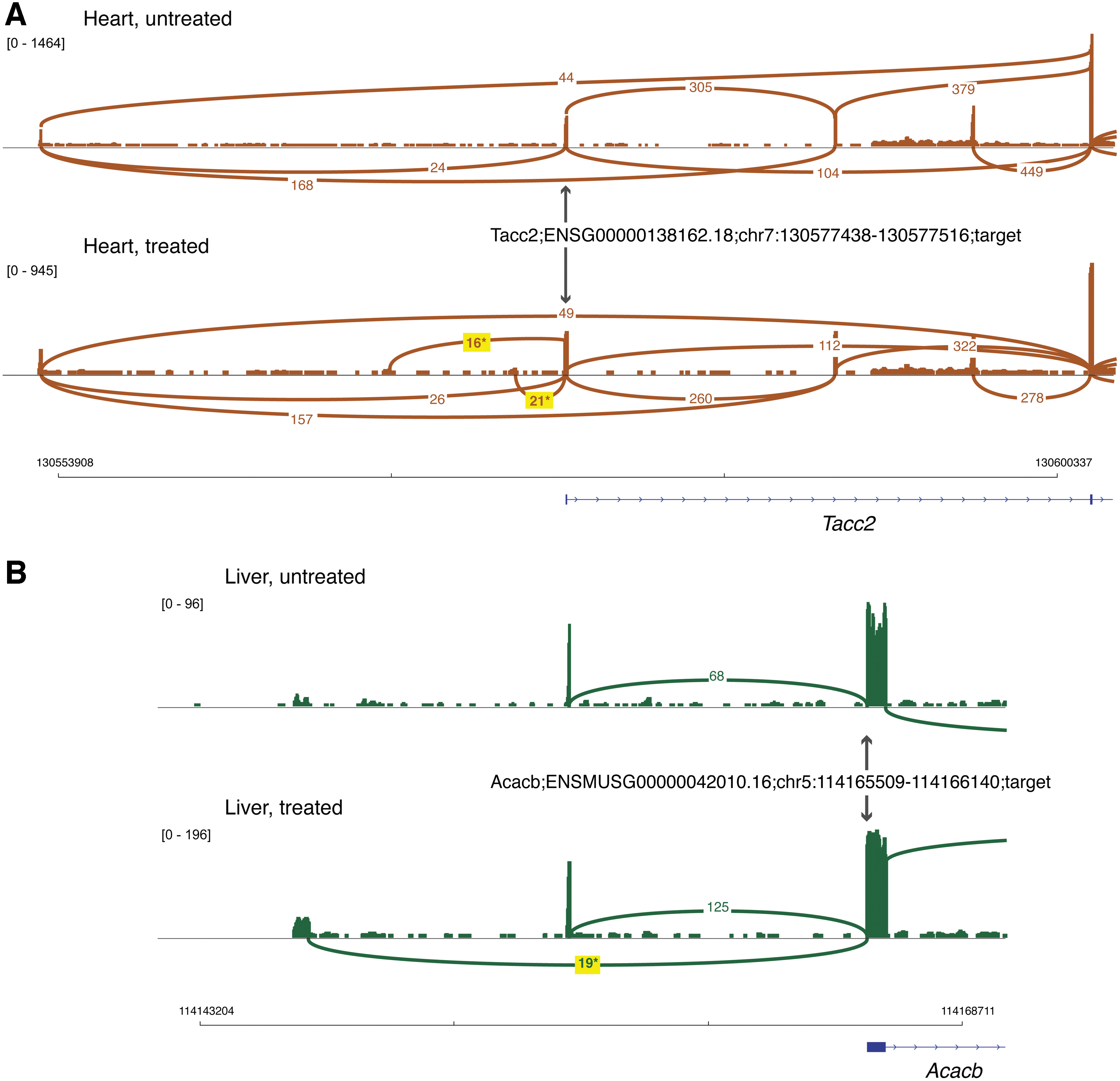
Differences in alternate promoter usage in candidate LSVs. Merged RNA-seq data from 3 animals is shown for the untreated/treated conditions as described in Figure 1, with the scale for RNA-seq read depth indicated by the bracketed numbers. The yellow boxes highlight the alternate promoter junction reads that are unique to the treated condition.
Treatment-induced differences in splice junction usage from constitutive exons
Bold marks targeted exon 2 sequence junctions. ΔPSI, delta percent spliced in; LSV, local splicing variation; Ssbp4, single-stranded DNA-binding protein 4; VastDB, Vertebrate Alternative Splicing and Transcription Database.
Analysis of off-target effects for the remaining candidate splicing LSVs
The 11 remaining candidate off-target LSVs mapped to 6 genes and were all known alternative splicing events annotated in the VastDB, a large annotated resource of alternative splicing events detected by RNA-seq from a diverse set of vertebrate cells and tissues.
10
Since we observed on-target exon 2 skipping of Dmd transcripts in both skeletal muscle and heart, we assumed that ΔPSI differences due to off-target
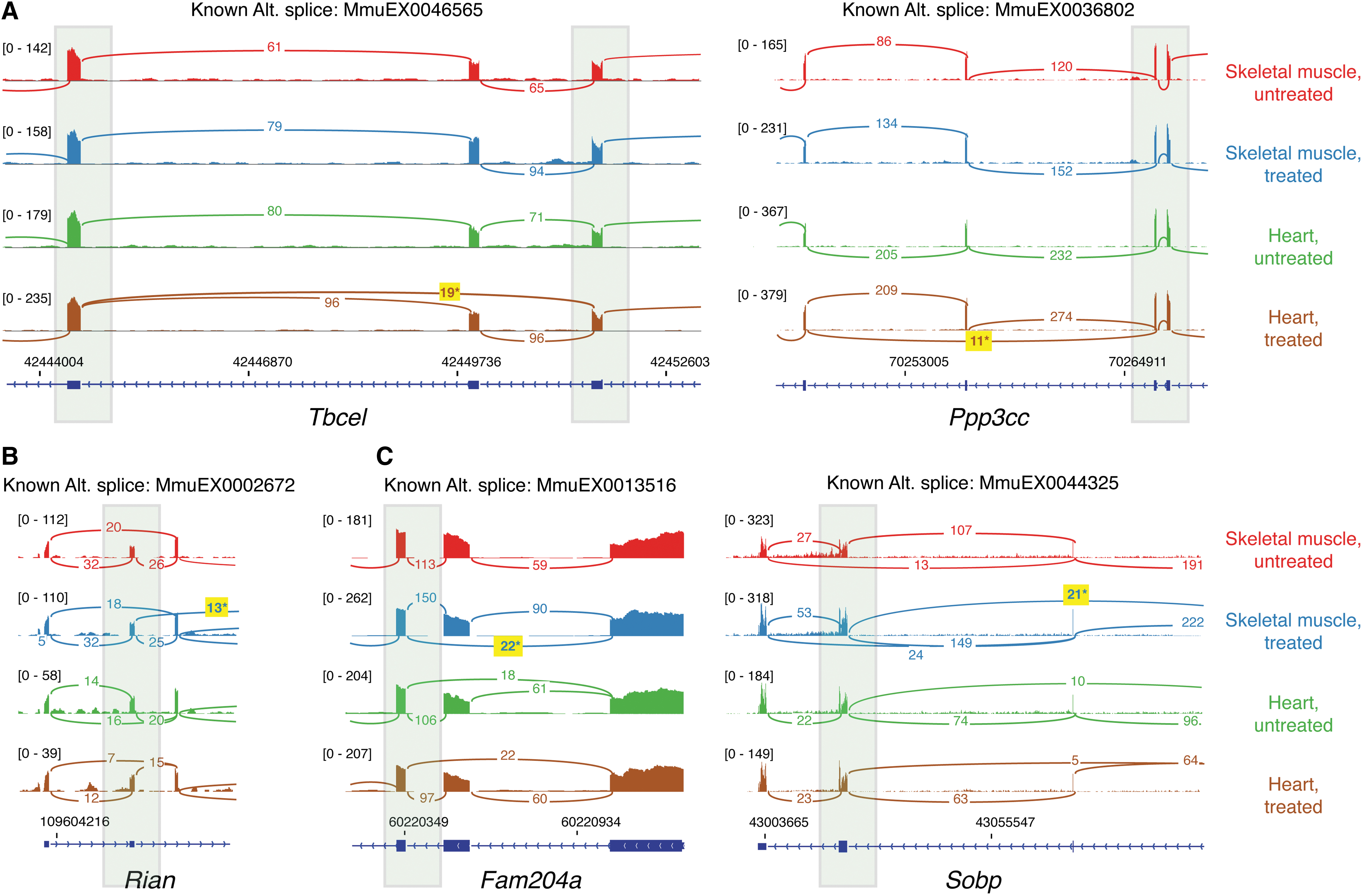
Tissue-specific AS candidate LSVs. Merged RNA-seq data from three animals are shown for the untreated/treated conditions as described in Figure 1, with the scale for RNA-seq read depth indicated by the bracketed numbers. The shaded green boxes indicate the location of candidate LSVs shown in Table 2 and the yellow boxes highlight the AS junction reads that are responsible for the detected LSVs.
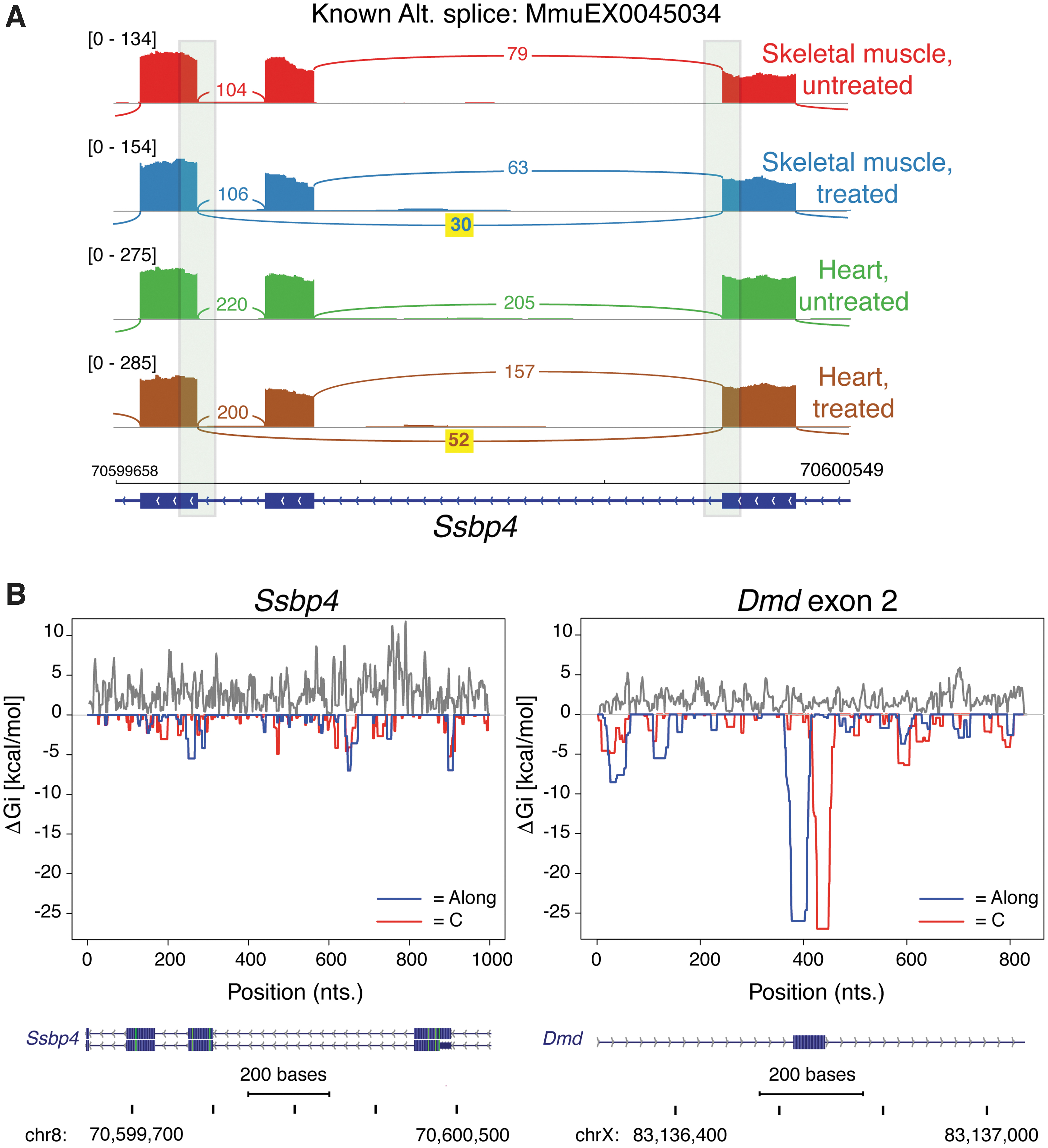
Off-target analysis for the Ssbp4 candidate LSVs.
In addition to Dmd exon 2 skipping, the remaining 4 LSVs in Ssbp4 were the only example of ΔPSI differences seen in both skeletal muscle and heart (Fig. 5A). In VastDB, these Ssbp4 LSVs mapped to a known mouse cassette exon (VastDB ID MmuEX0045034) whose inclusion/exclusion leads to alternative protein isoforms of single-stranded DNA-binding protein 4 (Ssbp4), which is alternatively spliced in multiple tissues, including heart, brain, lung, and adipose tissue. To identify putative off-target binding sites in Ssbp4 pre-mRNA, we computed the predicted binding energy between U7.A or C snRNA and the Ssbp4 pre-mRNA using the RNAup algorithm, 12 which computes the interaction energy of small RNA—target RNA hybrids combined with the probability that potential binding sites are accessible on the target pre-mRNA (not paired in secondary structure). Figure 5B shows the binding energy profiles for the U7.A or C snRNA interactions for every position along the target pre-mRNA sequences from the Ssbp4 MmuEX0045034 region and Dmd exon 2 region. While the RNA–RNA interaction scan detected an optimal binding energy overlapping the Dmd exon 2 splice sites, the Ssbp4 profile gave no indication of a strong attractive interaction between the U7.A or C snRNA and Ssbp4 pre-mRNA. This suggests that the Ssbp4 ΔPSI differences are an indirect effect of treatment on alternative splicing instead of a direct off-target effect due to modified U7 snRNA—target pre-mRNA binding.
Since treatment-induced Dmd exon 2 skipping was greater in heart versus skeletal muscle, we also evaluated off-target binding sites predicted by the RNAup algorithm for the heart-specific LSV events in Ppp3cc and Tbcel. The skipped Ppp3cc exon did not show putative binding sites for Along or C duplex formation, but the 140 nt. Tbcel skipped exon had an RNAup-predicted 13 base pair Along duplex at positions +59 to +71 within the exon (Supplementary Fig. S1A). The interaction-free energy (ΔGi) of this putative off-target site was −13.8, compared with a predicted ΔGi of −26.2 for the on-target Along Dmd exon 2 splice acceptor site. To evaluate how frequent putative binding sites below this interaction-free energy may occur in the transcriptome, we examined the distribution of putative Along-binding sites in 195,829 annotated exons from the mm10 reference mouse genome. For each exon plus 30 nts. of flanking intron sequence, we predicted the lowest interaction energy for that exon and then rank ordered this list of sites. The on-target Along Dmd exon 2 splice acceptor site was at the top of the list, while the ΔGi of the Tbcel site was seen at rank 1,581 or within the top 1% of sites. The putative off-target site in Tbcel maps to exon 3 of the gene, and reverse transcription PCR (RT-PCR) analysis of Tbcel mRNA from heart shows low-level skipping (0.8–2.1% of transcript) in WT BL6 mice, which increased to 20.6–23.0% in treated mice (Supplementary Fig. S2), comparable to results from RNA-seq.
Differential gene expression associated with rAAV9.U7.ACCA treatment
We also analyzed gene expression to evaluate whether restoration of dystrophin in skeletal muscle and heart led to specific transcriptome-wide changes. In skeletal muscle, there were 16 DE genes observed at a 5% false discovery rate (Fig. 6A and Supplementary Table S1). Upregulation of insulin-like growth factor 2 (Igf2) and sarcomeric components for the embryonic myosin heavy chain genes (Myh3 and Myh8) and troponin T2 (Tnnt2) are consistently observed in both mdx mouse 28 and human 29 studies of dystrophic skeletal muscle tissue. Expression of these four genes were restored in the treated condition to levels observed in skeletal muscle from C56BL/6J control mice (Fig. 6B). In a comparative protein and mRNA-level omics study using the mdx mouse, 30 the prune homolog 2 (Prune2) was among the 10 most upregulated genes in dystrophic muscle and Prune2 is downregulated in the treated condition. A similar but opposite effect was seen for the protein phosphatase 1, regulatory subunit 1A (Ppp1r1a) and the dual specificity phosphatase 26 (Dusp26) genes, which are downregulated in the mdx mouse, but upregulated in the treated condition (Fig. 6B).

Gene-level expression responses associated with treatment.
There were 30 DE genes observed in treated versus untreated heart (Fig. 6C and Supplementary Table S2) at a 1% false discovery rate, not including U7snRNA (Rnu7) reads that mapped to the precursor RNA from virally delivered
Discussion
The development and deployment of novel therapies directed toward alteration of mRNA splicing understandably requires studies to address the specificity of exon splicing effects. To address this question in the case of the rAAV9.U7ACCA vector, we chose an unbiased approach using a global RNA-seq analysis for several reasons. First, in silico analysis failed to identify regions of significant homology to the exon 2 target sequences that would serve as likely candidates for altered splicing amenable to a targeted RT-PCR approach. The second reason, a corollary to first, is that global RNA-seq analysis allows an unbiased assessment of all transcripts to search for unanticipated splicing events, or unannotated regions of target homology. Third, RNA-seq allows us to directly quantify the target DMD mRNA exon 2 junction sequences. Finally, RNA-seq allows characterization of expression of the entire transcriptome, confirming alterations known to be associated with muscle from DMD patients or DMD models and quantifying the degree of correction following delivery of the vector.
Among these, the most critical for assessing safety is identification of off-target and unanticipated splicing events. Using conservative quantitative thresholds to quantify the degree of LSV among all exon/exon junctions, our data confirmed altered splicing of the intended DMD exon 2 target junctions, which may be interpreted as a control for the sensitivity of the approach. We note that alteration of exon 2 splicing was more pronounced in the heart than in skeletal muscle, a finding consistent with greater vector tropism in heart than in skeletal muscle in non-human primates treated with the same vector. 33 At the same time, these data demonstrate no convincing evidence for off-target splicing alterations. The only gene that showed significant alteration of splicing in both cardiac and skeletal muscle was Ssbp4, where LSVs were found at a cassette exon location that is already known to be widely alternatively spliced. An increase in endogenous alternative splicing in Tbcel could be confirmed in heart by RT-PCR, but is unlikely to be of biological significance. The resulting Tbcel transcript isoform ablates the tubulin-specific chaperone cofactor E-like protein reading frame leading to a short N-terminal peptide, but ∼80% of the transcripts remained WT. While this study detected splice junctions in the large number of genes, a limitation is the detection of transcripts expressed at low levels, which will require substantially greater read depths for a more complete survey of transcriptomes in target tissues and cell types.
In evaluating the transcriptome, the pattern of DE of genes in the tissues studied suggested treatment-related, specific transcriptional responses as an indirect effect of dystrophin restoration. In dystrophic skeletal muscle from humans as well as animal models, the upregulation of the embryonic/developmental myosin heavy chain genes are robust markers of regenerating muscle fibers. The reduction in Myh3 and Myh8 transcript levels observed in skeletal muscle after
In contrast, the transcriptional signature in the heart was unique to the treated condition, differing from both untreated Dmd Dup2 and control C57BL/6J tissue. Many of the top 30 DE genes are direct targets for transcriptional regulation by the stress-induced activating transcription factors Atf4 and Atf5 in response to mitochondrial perturbations, including Fgf21, Asns, and Trib3. In a mouse model of mitochondrial myopathy, 35 deletion of mtDNA induces an integrated stress response and upregulation of Atf4 and Atf5, which in turn upregulate genes that have amino acid starvation response elements (AARE) in their upstream regulatory regions. We observed treatment-associated upregulation of both Atf5 and Atf4, and of genes known to be controlled by the AARE response, including: Fgf21, Mthfd2, Gdf15, Phgdh, and Psat1. Enrichment analysis of the treatment-induced upregulated genes in heart implicated the serine glycine biosynthesis (Phgdh and Psat1) and one-carbon metabolism pathways (Mthfd2, the mitochondrial folate-coupled dehydrogenase). Previous studies with mice overexpressing the CnAβ2 (Ppp3cb) catalytic subunit of the calcium-regulated phosphatase calcineurin have observed ATF4-dependent upregulation of Phgdh, Psat1, Mthfd2, and Aldhl2 specifically in cardiac tissue, and it has been proposed that this transcriptional response may have a protective effect on cardiac function mediated by increased antioxidant production. 36 The calcineurin gamma catalytic subunit encoded by Ppp3cc that we observed with a heart-specific exon skipping LSV event may be linked to this calcineurin-mediated upregulation of the serine glycine pathway genes. It is noteworthy that besides the four differentially spliced LSVs detected at the targeted Dmd exon 2, all the other 16 candidate LSVs occurred at known alternative splice junctions. It is possible that the observed differential splicing at these LSVs were also due to an indirect effect of dystrophin restoration.
Altogether, these results demonstrate that systemic delivery of the
Footnotes
Acknowledgments
The authors acknowledge the technical assistance of Hannah Huang.
Author Disclosure
Drs. K.M.F. and N.W. hold patents related to the scAAV9.U7 vector, and both they and Nationwide Children's Hospital receive royalties from the licensing of that patent to Audentes Therapeutics.
Funding Information
This work was supported by the Beauhawks Foundation (KMF).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.