
Abstract
Adeno-associated virus (AAV) is currently the most popular gene delivery vector for in vivo gene therapy. However, variability in titration methods between different laboratories affects the reproducibility of experiments and evaluation of safety and efficacy in clinical trials. We describe an optimized protocol for AAV titration, including quantitative PCR (qPCR) standard preparation and quantitation and treatment of AAV samples before qPCR and droplet digital PCR (ddPCR) titration. During the protocol development, we observed that quantitation of the qPCR standard was dependent on its conformation and that A260-based quantitation overestimated the plasmid copy numbers, introducing significant error. Linearized, free inverted terminal repeat (free-ITR), and supercoiled standards were compared with enhanced green fluorescent protein (EGFP), SV40p(A), and AAV2-ITR qPCR assays and we found that using the AAV2-ITR assay together with either linearized or supercoiled standard led to overestimation of the titers, while EGFP and SV40p(A) assays were more accurate with the linearized standard. Finally, we compared extraction of AAV1, AAV2, AAV5, AAV6, AAV8, and AAV9 genomes by heat denaturation, proteinase K treatment, and kit extraction. Kit extraction, which contained proteinase K treatment in denaturing buffer before spin-column purification, significantly increased the titers acquired for all the serotypes in both qPCR and ddPCR. These improvements resulted in an accurate quantitation of the ATCC reference standard and in a robust and reliable protocol for AAV titration.
Introduction
Adeno-associated virus (AAV)-based viral vectors are currently the number one choice for in vivo gene therapy, while also being a popular tool for in vitro studies. The first approved AAV-based therapy was Glybera, utilizing AAV serotype 1 (AAV1) vector for the treatment of lipoprotein lipase deficiency, followed by Luxturna (AAV2) for inherited retinal disease, and Zolgensma (AAV9) for spinal muscular dystrophy, with several others in clinical development. 1
One critical challenge in AAV gene therapy results from difficulties in equalizing dosing between different studies, as it is widely acknowledged that titration results between different laboratories can easily show 10-fold or larger variations in titers. 2,3 In extreme cases, this disparity can be the difference between therapeutic efficacy and toxicity, as was observed in response to in vivo short hairpin RNA delivery in mice, 4 highlighting how crucial it is to minimize such differences.
Among the first AAV titration approaches was dot blot hybridization of AAV genomes, which has now been largely replaced by the use of more accurate and less laborious quantitative PCR (qPCR). 5 In addition to genomic titers, capsid titers, transducing units, and different measurements of infectivity are commonly used to characterize AAV lots, 6 –9 although they are not widely used as the primary determinants of dosage. More recently, droplet digital PCR (ddPCR), which is capable of absolute quantitation without the need for a standard curve, has also gained popularity as an alternative to qPCR, although for now it remains more expensive and has a lower throughput (Table 1). 10
Comparison of quantitative PCR and droplet digital PCR
Adjusted to account for the wells required for a standard curve.
QX One can queue up to five plates, but does not offer parallel processing and reading.
Effect of ssDNA hybridization unproven, accurate quantitation of in vivo concatemers would require restriction digest.
ddPCR, droplet digital PCR; qPCR, quantitative PCR.
A major advancement toward more standardized titration across the field was the introduction of ATCC's AAV2 reference standard material (AAV2-RSM). 11 Both the U.S. Food and Drug Administration Agency (FDA) and the U.K. National Institute for Biological Standards and Control (NIBSC) recommend the utilization of reference standard material in confirming the correct calibration of in-house assays. 12,13 Unfortunately, due to the vast variety of different vector constructs and titration assays, the reference standard material is limited by the sequences included within. This prevents the direct validation of certain assays against the reference standard material and complicates the comparisons between vectors titrated using different assays.
In 2012, Aurnhammer et al. 14 published a near-universal AAV titration assay based on the detection of the inverted terminal repeat sequences of AAV2 (AAV2-ITRs), which has since become increasingly popular. However, D'Costa et al. 15 showed that correct quantitation with this method required digestion of the plasmid standard at the edge of the ITR regions, mimicking the vector genome structure and presumably facilitating the melting of the ITR hairpin structures during PCR amplification. This improved method was named free-ITR qPCR. ITRs can also suppress amplification of nearby sequences, 16 while several studies have noted the effects of plasmid supercoiling on amplification. 17 –19 To further complicate matters, plasmid size and conformation can also add bias to the original quantitation of the DNA standard. 18,20
While accurate quantitation of the standard is crucial for accurate titration by qPCR, other assumptions must also hold true. Namely, the sample must be free of plasmid DNA and unpackaged vector genomes, and the packaged AAV genomes must be available from the first PCR cycle. DNaseI digest is commonly used to address unpackaged DNA, while capsid opening is facilitated either by incubation at high temperature or proteinase K digest, sometimes in combination with spin-column purification. Currently, the field is still of mixed opinion regarding the need for proteinase K digestion. Lock et al. 10 noted that proteinase K digest increased qPCR titers on average by 2.4-fold but did not present their data. Werling et al. 13 looked at AAV2 and AAV8 vectors and concluded that proteinase K digest was necessary only for AAV8, with mixed results on spin-column purification. Furthermore, to our knowledge, no study has presented conclusive data on genome availability in ddPCR.
Here, we introduce a comprehensive strategy for the optimization of AAV genome titration, covering every step from standard preparation and choice of the assay for effective genome extraction. We also discuss possible caveats, highlight important considerations, and note recent significant advances in the field.
Materials and Methods
Standard preparation
Four micrograms of pAAV-d2EGFP (Supplementary Fig. S1) was digested with 4 U of single-cutter FastDigest PvuI (Fisher Scientific) or ITR-edge double-cutter (FastDigest PvuII, Fisher Scientific) for 20 min at 37°C in FastDigest Green Buffer (Fisher Scientific). The supercoiled standard was mock digested in the reaction buffer without an enzyme. The DNA was then purified with NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel), after which the concentration was measured three times with NanoDrop ND-1000 (Thermo Scientific), and diluted down to 109 N/μL in nuclease-free water to create the standard stock, which was then aliquoted and frozen at −20°C.
Standard quantitation
The linearized, free-ITR, and supercoiled plasmid standards were measured in three separate occasions. Spectrophotometric measurements were carried out with NanoDrop ND-1000 with appropriate blank solutions. Fluorometric measurements were performed using the Qubit dsDNA HS Assay kit (Invitrogen) and Qubit 2.0 Fluorometer (Life Technologies). Finally, concentration and conformation were determined with Fragment Analyzer 5200 using the Standard Sensitivity Large Fragment Analysis Kit (Agilent).
The standard concentrations were calculated from sequence-based molecular weight by SnapGene Viewer (GSL Biotech), adjusting for the two strands in dsDNA standards. The Qubit concentrations were used as the basis for all titers shown.
Vectors
The vectors were produced either by calcium phosphate or PEIpro (Polyplus-transfection) transfection of HEK293T cells as described previously (Supplementary Table S1). 21 Briefly, the vectors were harvested 48h post-transfection and purified by iodixanol gradient ultracentrifugation 22 (apart from AAV6-dEGFP-L, which did not undergo purification), followed by concentration and buffer exchange to Dulbecco's phosphate-buffered saline (DPBS) using the Amicon Ultra-15 Centrifugal Filter Unit (Millipore, Darmstadt, Germany). The AAV2-RSM (VR-1616) was obtained from ATCC.
AAV genome extraction
The AAV1, AAV2, AAV5, AAV6, AAV8, and AAV9 vectors were first diluted 1:10 in DPBS, and 5 μL was taken either for proteinase K treatment or extraction with the High Pure Viral Nucleic Acid Kit. The proteinase K treatment was carried out in total volume of 50 μL with 0.4 U of proteinase K (Thermo Scientific) with a 1-h incubation at 50°C, followed by a 10-min inactivation at 90°C. The kit extraction (including treatment with 1 mg of proteinase K) was carried out following the manufacturer's instructions. For the purposes of the standard conformation and assay comparison, 5 μL of undiluted vector was directly extracted with the High Pure Viral Nucleic Acid Kit.
Quantitative PCR
qPCR analysis was carried out with StepOne Plus Real-Time PCR System (Applied Biosystems) using TaqMan Fast Advanced Master Mix (Applied Biosystems). For standard comparisons, the default Fast TaqMan cycling conditions were used, whereas for genome extraction comparisons, a 15-min incubation at 95°C at the precycling stage was added to test the heat denaturation.
The standards used were diluted serially 1:10 to the range of 10–106 N/μL. For comparison of standard conformations and for the ITR, enhanced green fluorescent protein (EGFP), and SV40pA assays (Supplementary Table S2), the vectors were diluted in two independent dilution series to 1:104 and 1:105. For the genome extraction comparisons, the vectors were diluted to 1:104, 1:105, and 1:106.
Droplet digital PCR
The ddPCRs were set up in 1xSupermix for Probes (no dUTP; Bio-Rad). The AAV2-ITR assay was combined with either EGFP or SV40pA assay (Supplementary Table S2). The droplets were generated with the QX200 AutoDG droplet digital PCR system (Bio-Rad). The PCR contained a precycling incubation at 95°C for 10 min, followed by 40 cycles with 30 s at 95°C and 1 min at 60°C (2°C/s), and final enzyme inactivation at 98°C for 10 min. The droplets were stabilized at 4°C before reading with QX200 Droplet Reader (Bio-Rad).
For the treatment comparisons, the different AAV vectors were diluted 1:10 in DPBS and then either heated 15 min at 95°C, or, as for qPCR, treated with 0.4 U of proteinase K or extracted with the High Pure Viral Nucleic Acid kit. The vectors were diluted in 1:10 series in 0.1% Pluronic F-68 and analyzed at total dilutions of 1:105, 1:106, and 1:107 in duplicates.
For analysis of genome composition, the number of scored droplets was Poisson transformed into copy numbers with the following formula: c = −LN(1−(P/R))/Vd, where c denotes reaction concentration, P is the number of positive partitions, R the total number of partitions, and Vd the droplet volume (0.85 nL). 23
Results
DNA standard quantitation
Three different quantitation methods were used to measure the concentration of DNA standards in linearized, free-ITR, or supercoiled conformation. Out of the quantitation methods tested, the NanoDrop spectrophotometer gave the highest concentrations with the highest coefficient of variation of 23% (Fig. 1). As we were working at the very low end of the instrument range, we also compared the results at higher concentrations and found a similar overestimation (Supplementary Table S3).

Impact of plasmid forms on DNA quantitation using different methods.
There was no significant difference between the average values obtained by the Qubit Fluorometer and the Fragment Analyzer (Fig. 1 and Supplementary Table S4), both of which rely on the detection of fluorescent intercalating dyes. Interestingly, we observed much lower than expected values for the supercoiled DNA, suggesting that the nucleic acid binding dye-based methods tested here underestimated the concentration of DNA in the supercoiled conformation. Based on the Fragment Analyzer results, we verified that the DNA standards were in the expected conformations (Supplementary Fig. S2).
Choosing qPCR standard
To characterize the effects of standard conformation and the qPCR target sequence, five vectors were titrated with the different standards using AAV2-ITR, EGFP, and SV40pA assays. We observed a clear decrease in the cycle threshold (Ct) values when using the double-digested free-ITR standard with the AAV2-ITR assay as opposed to the linearized or supercoiled standards, correcting a clear overestimation of the titers (Fig. 2A). Unexpectedly, using the free-ITR standard with either EGFP or SV40pA assays showed a minor shift to the opposite direction, suggesting inhibition of amplification (Fig. 2B, C). Overall, the best agreement was observed when comparing the values obtained with the SV40pA and EGFP assays with the linearized standard with those obtained with the AAV2-ITR assay using the free-ITR standard (Fig. 2E and Supplementary Table S5). Importantly, the AAV2-RSM titer obtained with the free-ITR qPCR (3.76 × 1010 vgc/mL) was closest to the published reference standard value (3.28 × 1010 vgc/mL).

Effect of standard plasmid conformation on qPCR amplification. Serial dilutions of pAAV-d2EGFP in supercoiled, linearized, or free-ITR conformation were evaluated with three different titration assays:
AAV titration by ddPCR
Droplet digital PCR (ddPCR) is capable of absolute quantitation without the need for standard curve. We investigated how the values obtained using different qPCR assays and standards compared with those obtained from ddPCR. Consistent with the qPCR standard shifts, linearized standard for EGFP and SV40pA assays and free-ITR standard for the AAV2-ITR assay resulted in titers closest to the values obtained from ddPCR (Fig. 3 and Supplementary Table S6), with less than twofold difference for each. When comparing EGFP and SV40pA assay values obtained with the free-ITR standard, this difference stretched to ∼3.5-fold, confirming that this assay setup led to overestimation of titers in qPCR.
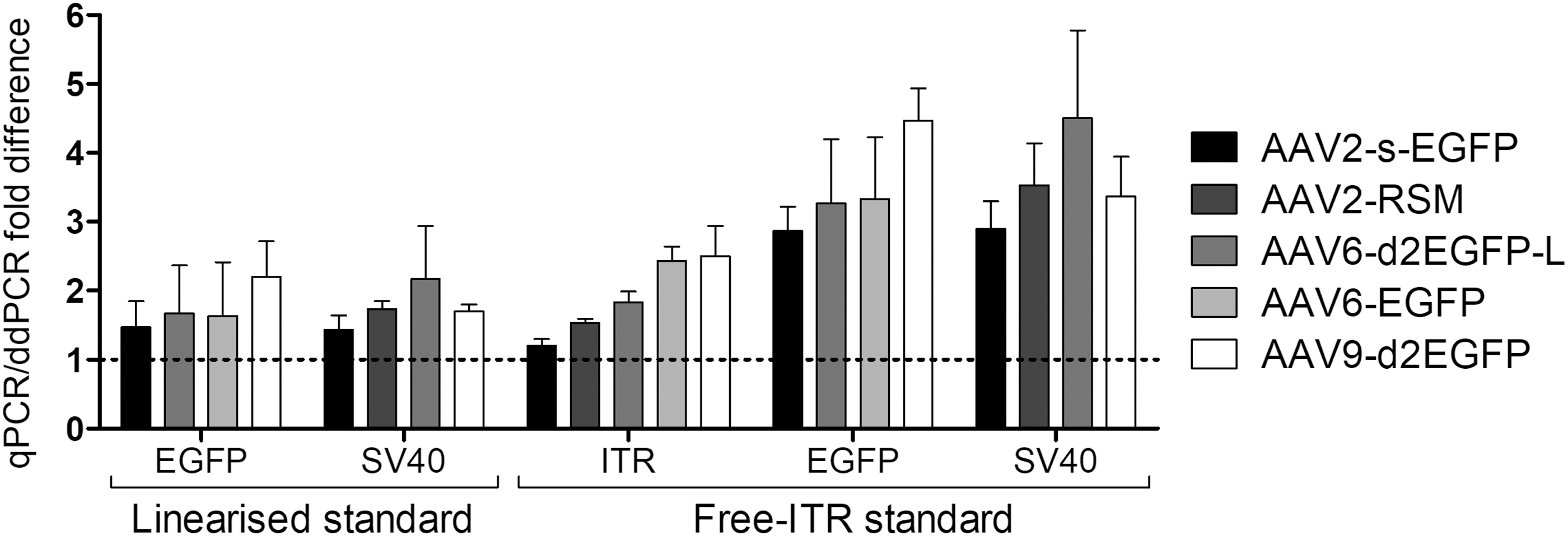
Comparison of qPCR and ddPCR titers. Fold differences in the titers of five vector lots, obtained with different assays in qPCR and ddPCR, were compared. The data are shown as mean ± SD (n = 3). ddPCR, droplet digital PCR.
Using ddPCR to study genome composition
In ddPCR, genomes are partitioned into droplets, each undergoing an independent PCR and endpoint scoring. This means that multiplexing in ddPCR can theoretically offer single-molecule resolution, which can be used to evaluate the genome composition of a vector lot. For this purpose, we Poisson transformed the droplet calls into genome copy numbers and used those to calculate the percentages of each target. We found that while the single-target titers were in most cases similar between all assays (Supplementary Table S6), there were striking differences when multiplexing two targets simultaneously. For simultaneous evaluation of ITR and EGFP targets, the range of double-positive population varied between 27.5% and 87.4%, and for ITR and SV40pA assays from 38.3% to 84.4% (Fig. 4). The two sets did also not necessarily correlate with each other; for AAV2-s-EGFP, 61.8% of the genome population was ITR-SV40pA double positive, but only 27.5% was ITR-EGFP double positive. We also compared our results for the AAV2-RSM with those obtained by Furuta-Hanawa et al. In our hands the double-positive population was markedly smaller (38.3% vs. 59.3%), which might be due to different dilutions used, as we found that the linear range for determining the genome composition was approximately only 10-fold (data not shown).
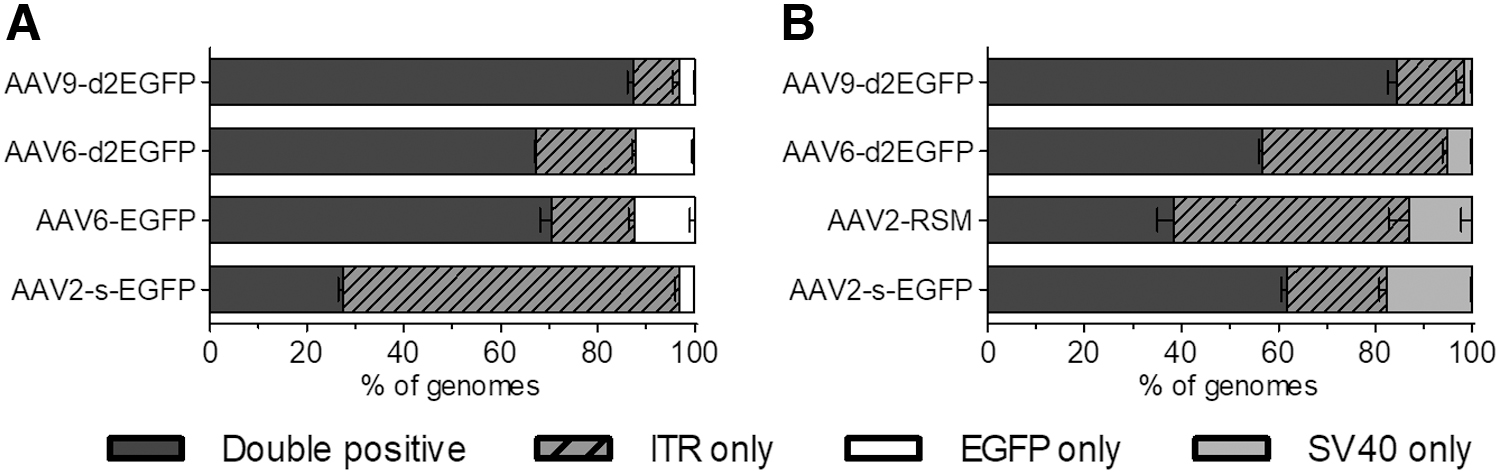
Composition of AAV genomes as evaluated by ddPCR. Fractions of
Pretitration treatment of AAV vectors
AAV is a nonenveloped virus with a highly stable icosahedral structure. Of the commonly used serotypes, AAV5 has the highest thermal stability, with reported melting temperature of 89.2°C. 24 It is thus reasonable that many laboratories use precycling incubation at 95°C for 15 min to denature capsids. We wanted to test heat denaturation against the standard proteinase K treatment and against genome extraction with the High Pure Viral Nucleic Acid Kit, which uses a higher concentration of proteinase K in a denaturing buffer combined with a spin-column purification.
We hypothesized that the benefit of the proteinase K treatment would likely be dependent on the thermal stability of the serotype. Unexpectedly, we found that the heat denaturation method consistently resulted in lower titers than the proteinase K treatment or kit extraction and that the fold improvement was not correlated with thermal stability (Supplementary Fig. S3). Furthermore, kit extraction consistently yielded higher titers than the proteinase K digest alone in both qPCR and ddPCR (Fig. 5 and Supplementary Table S7). Coefficient of variation was lowest for kit-extracted samples (14% and 11%, qPCR and ddPCR, respectively) and highest after heat denaturation (41% and 27%). Kit extraction also improved separation of the positive and negative droplet populations in ddPCR (data not shown).
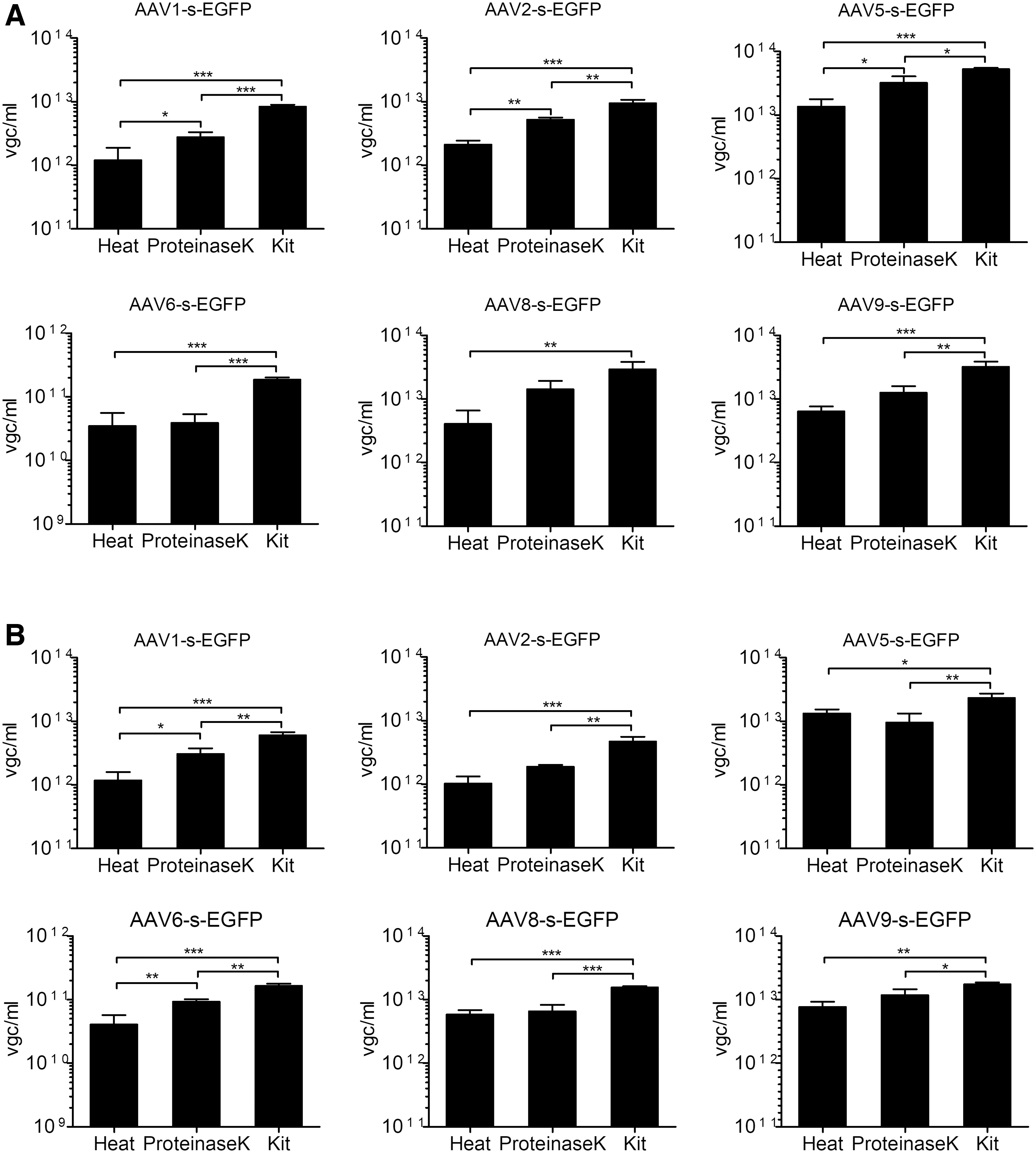
Titration of differently treated AAV vectors by qPCR. Six different serotypes were titrated after heat denaturation for 15 min at 95°C (heat), treatment with 0.4 U of proteinase K for 1h at 50°C (proteinase K), or genome extraction with the High Pure Nucleic Viral Acid Kit (kit).
We also tried the kit denaturation protocol without the spin-column purification, but found that the lysis buffer and higher concentrations of proteinase K resulted in PCR inhibition (data not shown). We further tested the recovery of the kit with three samples of previously purified genomes and obtained average recovery of 91% ± 6% (Supplementary Fig. S4). This implies that even the kit extraction method, which gives the highest values of the methods tested, is still underestimating the titers.
Discussion
High variability in AAV titers obtained in different laboratories continues to present challenges for accurate comparison and evaluation of findings. The availability of the standard materials from ATCC 2,3 has been an important step toward standardization, creating a tool for the calibration of AAV titration platforms. It is recommended that an in-house lot is titrated alongside the RSM and that this lot is then used as a consistent control for inter-run variation. However, before the platform can be validated using the RSM, it should first be optimized using all typical in-house samples, including different serotypes and sample types. Here, we have presented our optimized strategy for AAV titration, highlighting caveats during plasmid standard quantitation, choice of the assay, and preparation of vectors for titration. Our recommendations are summarized in Fig. 6.
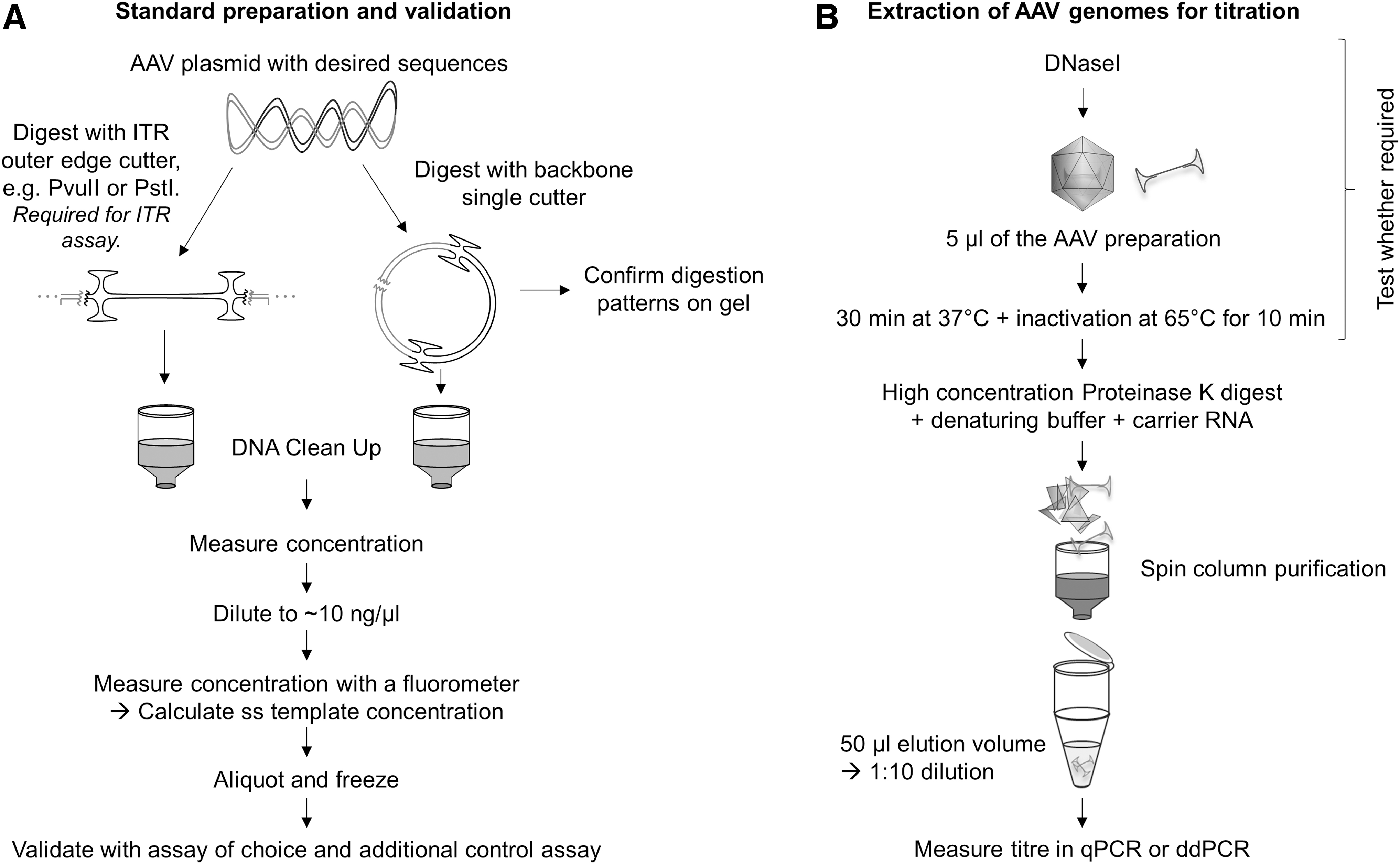
Guidelines for AAV titration.
Accurate quantitation of the DNA standard is a prerequisite for accurate qPCR titration. Comparison of different quantitation methods and the effect of DNA standard conformation revealed that both should be considered during standard preparation. We found that the NanoDrop ND-1000 produced higher concentrations with lower precision than the Qubit HS dsDNA Assay and the Fragment Analyzer SS Large Fragment Analysis Kit, which is not surprising considering the limited selectivity offered by spectrophotometry. If spectrophotometry is the only DNA quantitation method available, the range of the instrument being used should be considered, together with careful purification of the sample and choice of the final buffer. Furthermore, we want to highlight the importance of repeated measurements and comparison against another quantitation method whenever possible.
Linearization of qPCR plasmid standards has previously been recommended, as supercoiling can reportedly affect amplification of certain targets. 17,19 Lin et al. 18 also reported a method and conformation-based bias of measured DNA concentrations. Interestingly, they observed relatively increased overestimation of nonsupercoiled DNA by spectrophotometry, whereas our results show no such trend. Instead, both Qubit and Fragment Analyzer seemed to systematically underestimate the concentration of supercoiled DNA. Considering both the unpredictable effect of supercoiling on the amplification and on the concentration measurements, it is recommended to avoid the use of undigested plasmid DNA as a qPCR standard.
The AAV2-ITR titration assay enables the move away from using several different qPCR and ddPCR assays and standards, a practice that makes comparisons between different vectors more unreliable. D'Costa et al. showed that titration by AAV2-ITR assay required “freeing” the ITRs on the plasmid standard. While our results agreed with theirs, we also found that the free-ITR standard is not necessarily suitable for use with other assays, as it led to overestimation of both EGFP and SV40pA titers. This is perplexing as after denaturation, the free-ITR structure should result in a template essentially identical to the AAV genome. The effect was present regardless of whether the vectors being titrated were produced from the same plasmid as the standard or not.
It is known that AAV packages partial genomes with a 3′ end bias, 25 and this is likely to be the main reason for the differences that remained between the titers obtained from different assays in both qPCR and ddPCR. As AAV packages both [+] and [−] genomes, 26 this unfortunately cannot be overcome by the choice of a 5′ end primer set; instead, the choice of a primer set that is either central (such as transgene specific) or amplifies both ends (such as the ITRs) is likely to be more reliable than a set only amplifying one end of the genome. Only the former strategy seems compatible with ddPCR, as we were unable to distinguish between droplets that carried different amounts of the ITR amplicon. However, other methods, such as Southern blotting, alkaline gel electrophoresis, or single-molecule sequencing should be used as primary determinants of genome integrity, especially in the case of oversized or hairpin containing constructs. 25,27
A recent advancement made in AAV titration methodology is the two-dimensional ddPCR technology applied by Furuta-Hanawa et al. 28 This method uses an internal primer set in addition to ITR primers to better approximate the fraction of AAV particles carrying complete functional genomes. As each ddPCR droplet theoretically carries only one template, this allows for single-genome-based evaluation of both targets as opposed to qPCR multiplexing, which can only quantitate the presence of both targets in the genome pool. Unfortunately, this method still cannot fully distinguish between full-length and partial genome fragments, as one intact ITR is enough to score the droplet positive. In addition, we observed that the choice of internal primer set can have a major effect on the estimation of intact genomes; for one of our vectors, AAV2-s-EGFP, the difference was over twofold. The direction of the difference was also not consistent. An alternative strategy would be to combine a promoter-specific assay with a polyA assay to better approximate the presence of partial genomes. Interestingly, this observation also calls into question the current belief that AAV genome truncation happens randomly across the genome. 25
Just as important as a well-prepared standard and the assay choice is the preparation of the AAV samples for titration. Many laboratories start this process by DNaseI digestion, but we chose not to show our data here as variations in production, harvest, and purification methods between laboratories mean that the need for DNaseI treatment should always be validated individually. DNaseI digest may also be advisable when retitrating old vector lots, which might have undergone degradation, for example, due to several freeze/thaw cycles.
For successful amplification of the AAV target sequences in either qPCR or ddPCR, the capsids must be opened. Heat denaturation is often used due to its convenience, but we have unequivocally shown here that it is not sufficient to ensure release of the genomes even for the less thermally stable serotypes. Proteinase K treatment increased titers for five samples out of six in qPCR, but in ddPCR the trend was less clear, whereas the kit extraction consistently produced the highest titers. We hypothesize that this is due to a combination of factors: more efficient opening of capsids, elimination of aggregates, and removal of PCR inhibitors. Our results were more favorable toward kit extraction than those by Werling et al., 13 which is likely due to the fact that we used spin-column purification to allow for the use of higher concentrations of proteinase K in a PCR-incompatible digestion buffer. Our protocol also included addition of carrier RNA, which likely improved sample recovery. The effect of spin-column purification would likely be more pronounced for crude samples, as cell lysate is known to inhibit PCR. 29 For these reasons, we strongly recommend optimizing spin-column purification for both qPCR and ddPCR.
The biology of AAV vectors presents an additional challenge for ddPCR. As mentioned, AAV genomes are single stranded and both genome polarities are packaged. After extraction, these complementary genomes are no longer shielded by capsids and can anneal together, which in ddPCR would lead to partitioning of two genomes into one droplet and underestimation of titer. This is a probable explanation for the average 1.9-fold difference we observed between qPCR and ddPCR titers. Lock et al. attempted to bypass this problem by partitioning the virions without extracting the genomes, but this was not validated against other methods. 10
This work was carried out using single-stranded AAV (ssAAV) vectors, but self-complimentary AAVs (scAAVs) are becoming increasingly popular due to their favorable expression kinetics. 30 Several groups have reported additional issues with the titration of scAAVs, likely originating from the suppressive effects of the increased secondary structures. 16 The inhibition could be overcome with digestion of the vector genomes at or near the ITR hairpins. 16,31 The assay choice may also play an increasingly important role in the scAAV titration, and as such, titration assays optimized for ssAAV may not automatically be suitable for scAAV titration.
Conclusion
Careful optimization of the AAV titration is required to ensure comparable data both within and between laboratories. The conformation and quantitation method of the standard, extraction of AAV genomes, and assay of choice all affect titration. After proper optimization has been carried out, no large differences remain between the qPCR and ddPCR titers, although vigilance of possible sources of errors in both is important. It is also crucial that authors describe how their vectors have been titrated, preferably disclosing their value for the ATCC reference standard material, allowing the scientific community to better interpret their published work.
Footnotes
Acknowledgments
The authors thank Eila Korhonen for her excellent technical assistance, and Vesa Turkki and Lionel Galibert from Kuopio Center for Gene and Cell Therapy for insightful discussions regarding ddPCR.
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported by grants from the Academy of Finland, ERC, National Virus Vector Laboratory, Biocenter Finland, and the EU-EATRIS Infrastructure Network.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.