
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease that affects 1:5,000 live male births and is characterized by muscle wasting. By the age of 13 years, affected individuals are often wheelchair bound and suffer from respiratory and cardiac failure, which results in premature death. Although the administration of corticosteroids and ventilation can relieve the symptoms and extend the patients' lifespan, currently no cure exists for DMD. Among the different approaches under preclinical and clinical testing, gene therapy, using adeno-associated viral (AAV) vectors, is one of the most promising. In this study, we delivered intravenously AAV9 vectors expressing the microdystrophin MD1 (ΔR4-R23/ΔCT) under control of the synthetic muscle-specific promoter Spc5-12 and assessed the effect of adding a cardiac-specific cis-regulatory module (designated as CS-CRM4) on its expression profile in skeletal and cardiac muscles. Results show that Spc5-12 promoter, in combination with an AAV serotype that has high tropism for the heart, drives high MD1 expression levels in cardiac muscle in mdx mice. The additional regulatory element CS-CRM4 can further improve MD1 expression in cardiac muscles, but its effect is dose dependent and enhancement becomes evident only at lower vector doses.
Introduction
Duchenne muscular dystrophy (DMD), the most common inherited lethal childhood disease, is a severe muscle-wasting disorder caused by gene mutations in the DMD gene leading to premature protein truncation and loss of functional dystrophin protein from skeletal and cardiac muscle. 1,2 The lack of dystrophin results in a progressive muscle wasting, which ultimately leads to loss of independent ambulation and need of a wheelchair often by the age of 13 years. 3 Patients affected by DMD usually die in their mid-thirties and forties due to respiratory failure and cardiomyopathy. 4 The gene addition strategy based on delivering adeno-associated viral (AAV) vectors carrying recombinant cDNA-based micro- or mini-dystrophins into diseased tissues is a promising approach for the treatment of DMD. However, AAV vectors have a packaging size limitation, due to the viral capsid, of about 4.7 kb. Therefore, many forms of truncated dystrophins have been produced and tested for their efficacy in various models of dystrophin deficiency. 5 Over the last few years we have been working to optimize the delivery and expression of a microdystrophin called MD1. 6 This microdystrophin that is sequence optimized and expressed by a synthetic muscle-specific promoter called Spc5-12, 7 leads to normalization of muscle mass, specific force, and resistance to exercise-induced damage in mdx mice. 6,8 Furthermore, whole body systemic delivery of AAV2/8-Spc5-12-MD1 in canine (GRMD) model of DMD has recently shown to greatly reduce the dystrophic pathology 9 in skeletal muscles.
Obtaining high levels of dystrophin expression is crucial to provide a functional effect to the treated muscles and translate the experimental approach to a clinical benefit. 10 However, there is a number of factors that limits the amount of AAV vector injectable in vivo, such as manufacturing constraints and the risk of possible AAV-specific immune responses. 11 –13 A promising strategy to increase the transgene expression and therefore reduce the amount of vector to be delivered is optimizing the regulatory elements driving expression of the protein. In the case of DMD, the cardiac muscle is a key target for a gene therapy treatment as cardiac failure is among the main causes of death. 14 Transgene expression in cardiac muscle can be increased by using strong tissue-specific promoters and by including cardiac-specific enhancer sequences, 15 such as CS-CRM4. Spc5-12, a short (334 bp) synthetic promoter that allows high protein expression in skeletal and cardiac muscles, is made by random assembly of a combination of the myogenic regulatory elements E-box, MEF-2, TEF-1, and SRE. 7 In this study, we combined the CS-CRM4 enhancer module with the Spc5-12 promoter to drive MD1 expression after systemic delivery in both young and aged mdx mice. We show that the effect of the CS-CRM4 element in transgene expression is vector dose dependent and that an optimal vector dose could be identified (5E + 11 vg/mouse), whereby the addition of the CS-CRM4 enhancer significantly increased MD1 expression in cardiac muscles. In contrast, at higher dose (HD) of AAV9-Spc5-12-MD1 (1E + 12 vg/mouse) Spc5-12 already induces a significant MD1 overexpression in cardiac muscle that cannot be further improved by including the CS-CRM4 enhancer. At very low vector doses (1E + 11 vg/mouse), no MD1 is detected in cardiac muscle, regardless of the presence of the CS-CRM4 element. Our data provide novel insights into using cardiac-specific regulatory elements for enhanced but controlled dystrophin expression in the heart.
Results
Addition of a CS-CRM4 (CS) enhancer to the muscle-specific promoter Spc5-12 increases microdystrophin expression in both cardiac and tibialis anterior (TA) muscles
To test whether the addition of the cardiac-specific cis-regulatory module 4 (CS-CRM4, named “cs” here) to the promoter increased microdystrophin expression, AAV9-Spc5-12-MD1 and AAV9-csSpc5-12-MD1 constructs were generated and intravenously injected in 8-week-old male mdx mice at a dose of 5E+11 vg/mouse. Heart (H), Diaphragm (DIA), and TA were collected 12 weeks later. At this dose, the cardiac-specific enhancer significantly increased dystrophin expression in the Heart; immunofluorescence for dystrophin and laminin showed homogenous, intense staining after treatment with CS-CRM4-enhanced AAV-csSpc5-12-MD1, whereas a weaker staining was present in the cardiac muscles of mice treated with AAV-Spc5-12-MD1 (Fig. 1A and Supplementary Fig. S1A). Densitometric analysis of immunoblots detecting dystrophin expression confirmed this observation (1,211.0 ± 311.5 and 92.8 ± 72.3 U for cardiac muscles of AAV-csSpc5-12-MD1 and AAV-Spc5-12-MD1-treated mice, respectively, unpaired t-test, p = 0.006, n = 6; Fig. 1B, C). Immunostaining for dystrophin and laminin in TA muscles showed increased dystrophin expression in mice injected with AAV-csSpc5-12-MD1 compared with AAV-Spc5-12-MD1-injected mice (26.2% ± 2.9% compared with 8.9% ± 2.5% dystrophin-positive fibers, respectively, Bonferroni test, p < 0.01, n = 6) (Fig. 1D, E). Quantification of dystrophin expression by immunoblot showed a nonstatistically significant trend toward an increase in dystrophin expression after inclusion of the enhancer (232.0 ± 104.1 and 23.4 ± 2.6 U for TA muscles of AAV-csSpc5-12-MD1 and AAV-Spc5-12-MD1-treated mice, respectively, unpaired t-test, p = 0.07, n = 6; Fig. 1F, G). DIA muscles showed a similar amount of patches of dystrophin-positive fibers after injection with either vector. In these patches, a similar percentage of dystrophin-positive fibers were detected (11.8% ± 1.1% vs. 7.6% ± 2.4% with and without the enhancer, respectively, Bonferroni test, p > 0.05, n = 6; Supplementary Fig. S1B, C). These data show that, in the absence of the CS-CRM4 element, cardiac and TA muscles express a substantial amount of microdystrophin. Furthermore, the data demonstrate that at this dose, the inclusion of the CS-CRM4 element in the AAV vector significantly increases MD1 expression in cardiac and TA muscles.

Systemic delivery of 5E+11 vg/mouse AAV9-csSpc5-12-MD1 in young mdx mice increases dystrophin expression in both skeletal and cardiac muscles compared with the delivery of AAV9-Spc5-12-MD1. Eight-week-old mdx or C57BL10 mice were intravenously injected with a dose (5E+11 vg/mouse) of AAV vectors carrying the transgene MD1 under control of Spc5-12 or csSpc5-12 promoters or with saline solution. All samples were harvested 16 weeks later.
Addition of a CS-CRM4 (CS) enhancer to the muscle-specific promoter Spc5-12 does not increase microdystrophin expression in muscles when a dose of ∼3E+13 vg/kg is used
To assess the effect of the CS enhancer on MD1 expression at higher and lower vector doses, vectors were intravenously injected in 8-week-old male mdx mice at either 1E + 12 vg (HD, corresponding to ∼3E +13 vg/kg; n = 6 mice) or 1E+11 vg (lower dose, LD, corresponding to ∼3E + 12 vg/kg; n = 6 mice). Age-matched mdx and C57BL10 mice were injected with saline as controls. Twelve weeks later, mouse forelimb strength was assessed using grip strength measurements. Subsequently, TA, DIA, and H were harvested and MD1 expression in these muscles was analyzed. Quantitative PCR performed on total DNA extracted from cardiac muscles confirmed that the same amount of vectors were initially injected in the HD-treated groups of mice (2,577 ± 688.8 vg and 2,698 ± 1,106 vg, respectively), whereas a lower amount of vector was detected in LD-injected cohorts (156.6 ± 47.72 vg) (Fig. 2A). Substantial expression of dystrophin was detected after injection of either vector at HD with most cardiomyocytes expressing dystrophin, although variability was present between representative cardiac sections from different mice (Fig. 2B and Supplementary Fig. S2). On the contrary, LD-treated mice had almost no dystrophin expression similar to saline-treated mdx mice (Fig. 2B). Dystrophin quantification by immunoblot analysis confirmed this finding and showed that MD1 is well expressed in hearts with (0.169 ± 0.060 intensity unit ratio) or without (0.189 ± 0.060 intensity unit ratio) the enhancer at the HD, whereas very low expression level of the transgene was observed in LD (0.002 ± 0.001 intensity unit ratio) treated mice (Fig. 2C). No statistically significant difference in dystrophin expression levels was observed with or without the enhancer in HD-injected mice and between either HD-treated groups and wild-type (WT) mice (0.069 ± 0.003, p = 0.672 and p = 0.517, respectively, for vectors with or without the enhancer, Fig. 2D). To assess dystrophin expression in skeletal muscles, the percentage of dystrophin-positive fibers was calculated (Fig. 3A, B). No statistically significant difference in dystrophin expression was detected between muscles injected with either AAV-Spc5-12-MD1 (12.1% ± 2.6% fibers) or AAV-csSpc5-12-MD1 (22.1% ± 7.0% fibers). Injection of LD of vector showed barely detectable presence of dystrophin-positive fibers (1.1% ± 0.3%) (Fig. 3B). These results correlated with the grip strength analysis that showed similar increase of muscle strength in the groups of mice treated with the HD of vectors, with or without an enhancer (Fig. 3C). By immunoblot MD1 was barely detectable in most muscles suggesting that very little protein was expressed at these doses (Supplementary Fig. S3A). However, a small but significant increase was measured with the enhancer element (Supplementary Fig. S3B). In DIA only few patches of dystrophin-positive fibers were detected per section in these muscles (Supplementary Fig. S4A). In those patches 31% ± 12% of fibers were positive for dystrophin in muscles treated with the AAV-Spc5-12-MD1, whereas 23.7% ± 5.0% and 0.7% ± 0.2% of dystrophin-positive fibers were detected in similar patches of fibers of mice injected with AAV-csSpc5-12-MD1 at HD and LD, respectively (Supplementary Fig. S4B). No statistical difference was detected between the two HD-treated groups. These data show that the injection of the HD of AAV-Spc5-12-MD1 (corresponding to 3E+13 vg/kg) induces relatively robust MD1 protein expression in mdx mice. In cardiac muscle, the amount of MD1 protein expressed with or without CS-CRM4 enhancer is not significantly different compared with the endogenously expressed dystrophin in WT mice. Hence, at these high vector doses, the addition of the cardiac-specific CS-CRM4 element did not further increase MD1 protein expression in either skeletal or cardiac muscles, suggesting a possible saturation effect.
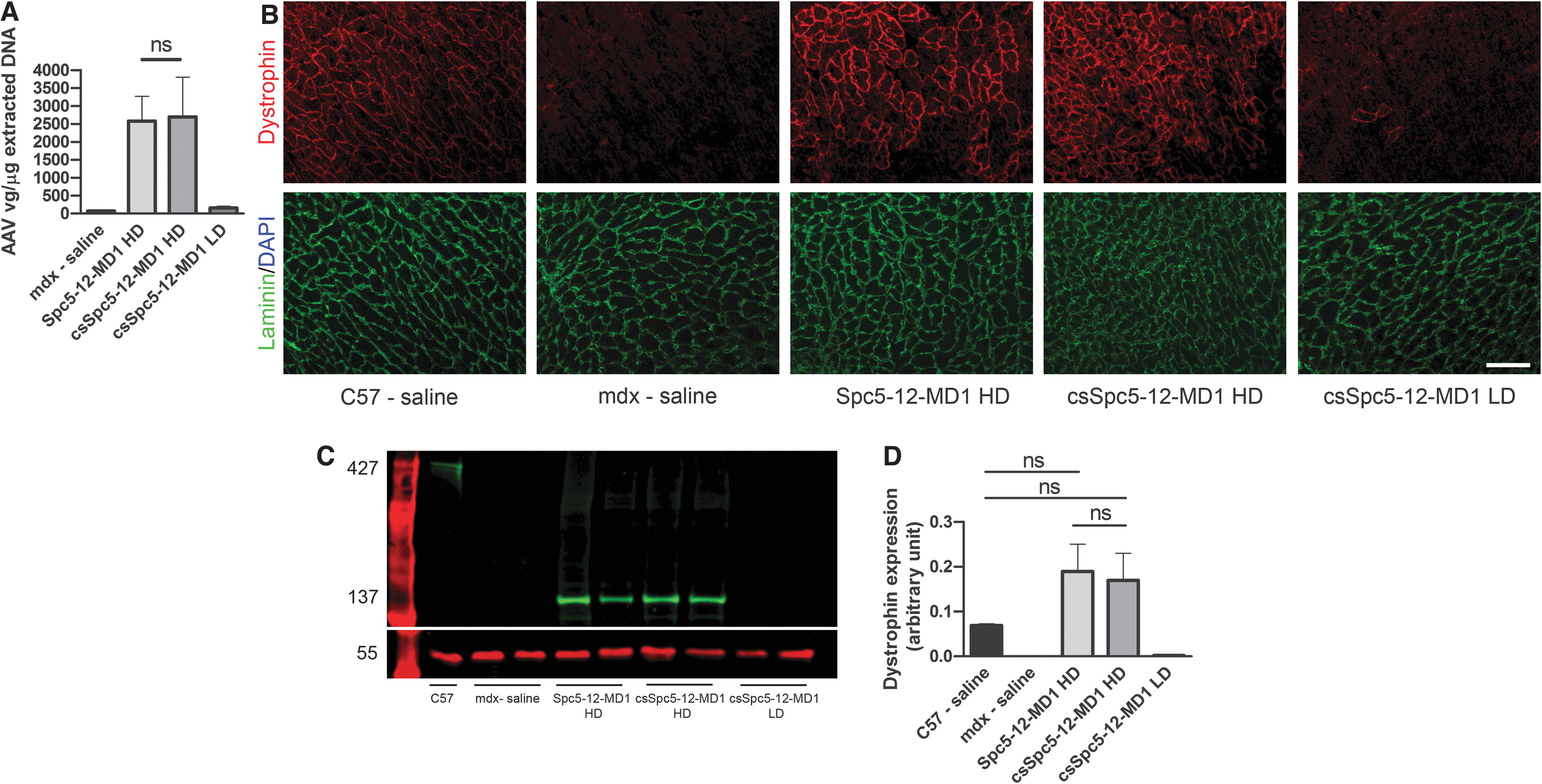
Systemic delivery of AAV9-Spc5-12-MD1 in young mdx mice restores high levels of dystrophin expression in cardiac muscles irrespective of inclusion of a CS-CRM4 enhancer. Eight-week-old mdx or C57BL10 mice were intravenously injected with high or low doses (HD and LD, respectively) of AAV vectors carrying the transgene MD1 under control of Spc5-12 or csSpc5-12 or with saline solution. Cardiac muscles were harvested 16 weeks later.
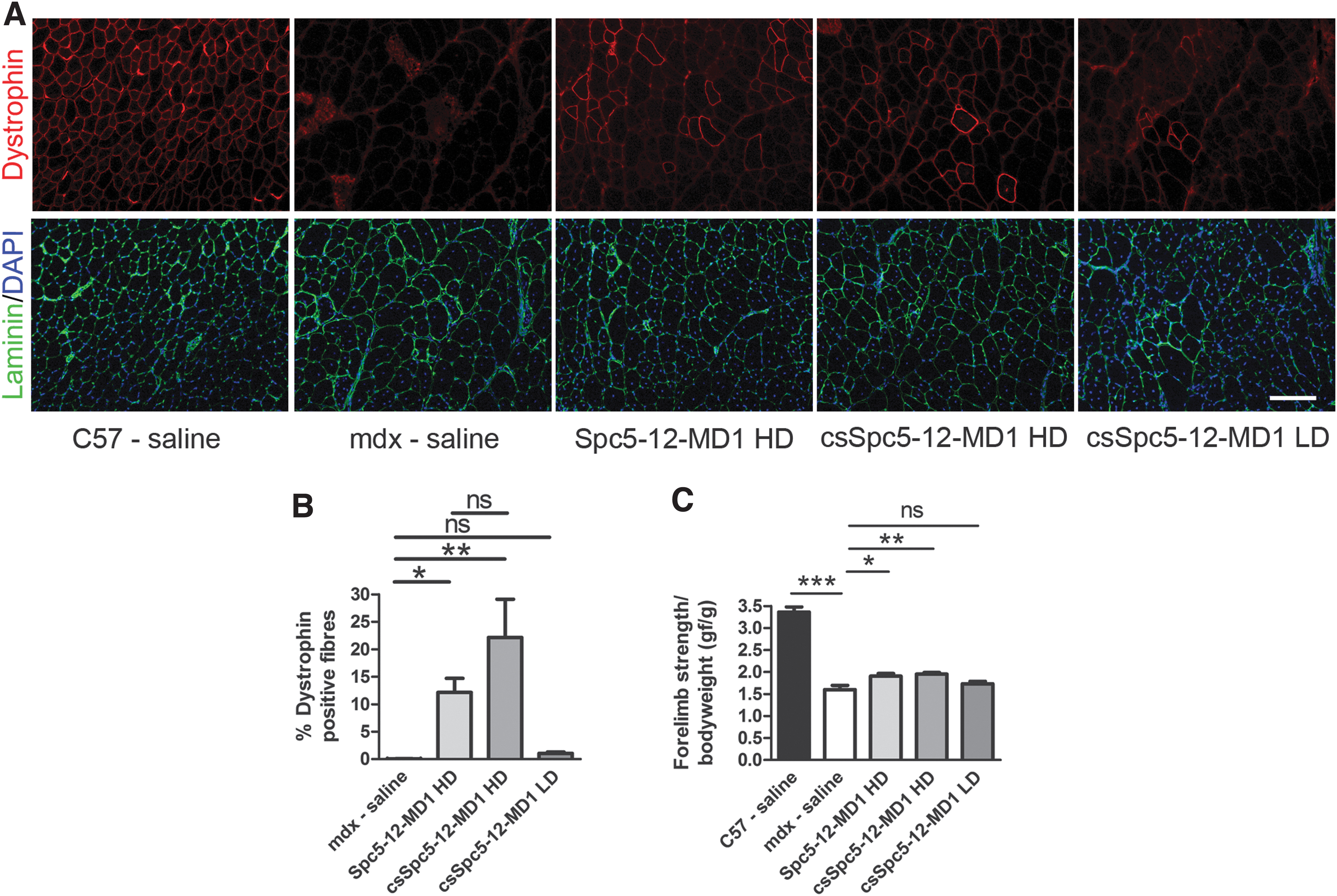
Systemic delivery of AAV9-Spc5-12-MD1 in young mdx mice restores significant percentage of dystrophin-positive fibers in TA muscles and improves muscle strength irrespective of inclusion of a CS-CRM4 enhancer. Eight-week-old mdx or C57BL10 mice were intravenously injected with high or low doses (HD and LD, respectively) of AAV vectors carrying the transgene MD1 under control of Spc5-12 or csSpc5-12 or with saline solution. TA muscles were harvested 16 weeks later.
Discussion
New methodologies have to be developed to reduce the amount of the biological product to be administered in DMD patients to ease the economic and manufacture constraints, and reduce the risk of potential immune responses to the vector and/or transgene. Recently, it was shown that using the chimeric synthetic CS-CRM4/SPc5-12 promoter to deliver luciferase resulted in a nearly threefold increase in cardiac gene expression compared with SPc5-12 alone, whereas no change in protein expression was shown in skeletal muscle. 15 Increasing dystrophin expression in cardiac muscle is particularly crucial for DMD where cardiac failure is a leading cause of death. Since the aim was seeing an improvement with enhancer inclusion, we tested a range of doses of vector considered suboptimal in dystrophic mdx mice 16,17 and in general lower than the doses used in the current DMD clinical trials (NCT03375164 and NCT03362502). The same dose tested in a canine GRMD model of DMD provided only a small functional effect in skeletal muscle, whereas the effect in the heart was not assessed as this canine model does not develop cardiomyopathy. 9 Systemic delivery of the LD of AAV-csSpc5-12-MD1 produced almost undetectable protein expression in both cardiac and skeletal muscles suggesting that this dose could be below the threshold necessary to achieve a detectable tissue transduction in mdx mice. On the other hand, the top dose, irrespective of the inclusion of the cardiac-specific element, resulted in a significant amount of dystrophin protein expression in skeletal muscle and a strong expression of dystrophin in the heart at least at the same level of the normal protein expressed by WT mice. The cardiac-specific effect of the CS-CRM4 regulatory element became evident when an intermediate vector dose was injected that was sufficient to achieve a substantial protein expression in the heart but not enough to achieve 100% dystrophin restoration. This intermediate dose of the vector, including the cardiac-specific enhancer was indeed effective in enhancing dystrophin by ∼10 times compared with the use of the Spc5-12 promoter without the CS-CRM4 element. The results indicate that the effect of CS-CRM4 on gene expression is dependent on the vector dose and suggest that there is an optimal dose window to enable its enhancing effect. Moreover, the impact of the CS-CRM4 element on gene expression levels may vary depending on the transgene, since gene-dependent post-transcriptional bottlenecks may ultimately determine the net increase in protein levels. The efficiency we observed in the heart is probably due to the combined use of a serotype with high tropism for the heart and the Spc5-12 promoter. The amount of MD1 detected in cardiac muscle using 3E+13 vg/kg of an AAV vector driven by Spc5-12 promoter confirmed previous observations obtained using either AAV8 or AAV9 with either MD1 or the Δ4173-microdystrophin. 6,18 Here the combination of AAV9 and Spc5-12 promoter partially masked the efficiency of the enhancer in the heart. Indeed, we observed a comparable dystrophin expression in cardiac muscle of HD-treated mice and WT mice suggesting that when a maximal threshold is achieved, the presence of a cardiac-specific enhancer may not be able to further enhance expression. In transgenic mice, where minidystrophin was expressed 50 or 100 times higher than the normal dystrophin amount in the heart, 19 transgene overexpression resulted in accumulation of minidystrophin vesicles in the sarcoplasm accompanied by worsening of the pathology. 19 However, this was likely due to the use of the murine α-myosin heavy chain promoter that permits supraphysiological transgene expression. 19,20 With regard to MD1 expression in skeletal muscles, at the intermediate dose, inclusion of the CS-CRM4 enhancer slightly enhanced dystrophin expression in TA muscles. This increase is in the range previously observed when a reporter gene was driven by the CS-CRM4 enhancer paired with the SPC5-12 promoter. 15 Notably, the observation that the CS-CRM4 enhancer certainly does not decrease and in fact slightly increases expression of dystrophin protein in skeletal muscles is important for the translation of such regulatory element in clinical settings. Other more efficient skeletal muscle cis-regulatory modules (SK-CRM) have been described that can be used to enhance expression of muscle-specific genes. 21 The inclusion of a 2R5S enhancer sequence, combined with either a highly truncated MCK promoter or with Spc5-12 promoter increased the expression of the minidysΔ3990 gene in skeletal muscles. 18 Our experiments represent a valuable starting point for further studies of MD1 expression in skeletal and cardiac muscles. In the future, other enhancers should be tested in conjunction with Spc5-12 promoter: examples include the use of SK-CRMs with Spc5-12 promoter and MD1 transgene. Other options include testing the efficacy of the 2R5S enhancer on expressing micro-dystrophin MD1 in cardiac and skeletal muscles and its comparison with the one offered by CS-CRM4-Spc5-12 after systemic injection in mdx mice. Similarly, it would be interesting to assess the efficiency of MD1 expression using CS-CRM4 to control activity of the CK8 promoter, a regulatory sequence currently used in a clinical trial. 17,22
Materials and Methods
AAV preparation
AAV-Spc5-12-MD1 or AAV-csSpc5-12-MD1 were prepared using the standardized double transfection protocol. 6,8 Briefly, HEK293T cells were seeded in roller bottles and cultured in DMEM at 37°C and 5% CO2. Once 50% confluent, cells were transfected using polyethylenimine with the relative pAAV plasmid and pAAV helper cap9 plasmids (pDP9rs). Cells were cultured in roller bottles for three more days. Afterward cells were lysed and recombinant pseudotyped AAV vector particles were purified by iodixanol (Sigma-Aldrich) step-gradient ultracentrifugation. The copy number of vector genomes was quantified by quantitative polymerase chain reaction (qPCR).
In vivo experiments
Groups of 8-week-old mdx male mice of similar body weights were randomly allocated to the different groups minimizing as much as possible the number of mice coming from the same litter. Mice were put in a heater and left for 10 min before being restrained and intravenously injected with the vector diluted in 200 μL sterile saline. Soon after injections, mice were put back in the cage. Two experiments were performed: In the first experiment, six 8-week-old mice were injected with5E+11 vg of either AAV-Spc5-12-MD1 or AAV-csSpc5-12-MD1. In the second experiment, six 8-week-old mdx male mice per group were injected with the same vectors at either 1E+12 vg (HD) or 1E+11 vg (LD) per mouse. In both experiments, 5-6 age-matched mdx and C57BL10 male mice were injected with sterile saline as controls. Twelve weeks postinjection, mice were sacrificed and Heart (H), DIA, and TA muscles were harvested and weighed. All samples were embedded in optimal cutting temperature medium (VWR, Leicestershire, UK) and frozen in liquid nitrogen-cooled isopentane (Sigma, Dorset, UK) for cryosectioning. They were kept at −80°C until use. In vivo experiments were conducted under statutory Home Office recommendation; regulatory, ethics, and licensing procedures and the Animals (Scientific Procedures) Act 1986. All experiments were approved by the Institutional Animal Welfare and Ethical Review Body.
Dystrophin and laminin staining
After cryosectioning on an OTF 5000 cryostat (Bright, Huntingdon, UK), sections (10-μm thickness) were kept at −80°C until use. Frozen sections of TA and DIA were immunostained for dystrophin, laminin, and 4′,6-diamidino-2-phenylindole (DAPI). Cryosections of H were immunostained for dystrophin and laminin only. The MOM Immunodetection Kit (VECTOR Laboratories) was used following the standardized protocol. Briefly slides were air dried and fixed with acetone for 5 minutes. Sections were then incubated with MOM blocking solution for 1 h at RT. Afterward, the mouse anti-dystrophin (1:50, Manex 1011c DHSB) and rabbit anti-Laminin (1:500, ab11575; Abcam) primary antibodies were incubated for 1 h at RT diluted in MOM diluent solution. After washings, the secondary antibody for laminin (1:400, goat anti-Rabbit 488 Alexa Fluor) was added for 1 h at RT. After washings, the secondary antibody from the MOM Kit was added for 10 min at RT. After further washings, slides were incubated for 5 min at RT with Streptavidin-568 (1:20, Alexa Fluor) in PBS (phosphate-buffered saline). Lastly, sections of TA and DIA were incubated with 1:1,000 DAPI in PBS and mounted. All washings were performed with PBST (PBS with 1% Tween 20) three times for 5 min each. Sections of immunostained TA muscles were imaged using a Nikon Ni-E upright microscope. Five, 20 × magnification images were randomly taken from a blinded observer and used to calculate the percentage of dystrophin-positive fibers.
Western Blot
Proteins were extracted from ∼75 tissue sections. One 3 mm tungsten carbide bead (QIAGEN, Manchester, UK) and RIPA (radio immunoprecipitation assay) buffer (150 mM sodium chloride, HEPES 5 mM, NP-40 1%, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, EDTA 0.1 Mm, Protease inhibitor tablet [Roche]) was added to each tube (30 μL for TAs and 50 μL for hearts) that was subsequently processed on a TissueLyser II (QIAGEN) at 25 Hz for 2 × 2 min. Samples were then centrifuged for 10 min at 13,000 g at 4°C and the supernatant was collected in new prechilled tubes and stored at −20°C. Proteins were quantified using DC Protein Assay (Bio-Rad, Hertfordshire, UK) using the manufacturer's instructions. Fifteen (TAs) or 5 (heart) μg of proteins were diluted in a final volume of 10 μL, denatured for 10 min at 100°C, and subsequently loaded in 3–8% Tris-Acetate Pre-Cast gel (NuPage) alongside with HiMark Prestained Protein Standard (Invitrogen). Proteins were transferred to nitrocellulose blotting membrane (GE Healthcare Life Science) for 2 h at 30 V by using transfer buffer (NuPage) with 20% methanol and 1 mL antioxidant (NuPage). Blocking was performed with 5% milk in 0.2% PBST for 1 h at RT. Overnight incubation at 4°C with the different primary antibodies (1:50 mouse anti-dystrophin Manex 1011c [DHSB] and 1:2,500 Rabbit α-tubulin [ab4074; Abcam]) in 5% milk in 0.2% PBST was performed. Alternatively, the kit, “Revert 700 Total Protein Stain for Western Blot Normalization” (Li-cor) was used. Membranes were subsequently incubated with the secondary antibodies (1:5,000, Goat anti-mouse 800 or Donkey anti-rabbit 680 (both from Li-cor) in 5% milk in 0.2% PBST for 1 h at RT. The membranes were scanned using Odyssey CLx Imager (Li-cor) and analyzed with Li-cor Image studio Lite.
AAV genome copy number quantification
The number of AAV viral genome copy number normalized by the total extracted DNA was calculated in cardiac muscles of treated mice. Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN) and quantified with the ND-1000 NanoDrop spectrophotometer (Thermo Scientific, UK). AAV.Spc512.MD1 plasmid (1,000 ng/μL) was diluted in 1:10 serial dilutions and used to generate the qPCR standard curve. qPCR assay was setup in a mix with water, SYBR Green Mastermix (Roche) and the forward (CTACAGAACCGCCATGAAGC) and the reverse (ACGTTCACCAGGTTGTTGTG) primers targeting MD1. qPCR was performed by using the Roche LigthCycler480 II machine with the following setting: 1 cycle of initializing for polymerase activation at 95°C, 45 cycles of denaturation at 95°C, 45 cycles of annealing at 60°C, and 45 cycles of extension at 72°C.
Measurement of forelimb strength
The forelimb strength was measured using a commercial grip strength monitor (Linton Instrumentation, Norfolk, UK). Five measurements per mouse were taken over a 3-day period by the same investigator, who was blind to the treatment of the mice. Mice were allowed to grasp a metal mesh attached to a force transducer using their forelimbs. The force produced during 5 sequential gentle pulls, until the mice released their grip, was recorded. Thirty seconds were waited between consecutive tests per each mouse. Data collected were expressed as gram force normalized by the initial animal body weight.
Statistical analyses
All results are reported as mean ± standard error of the mean. Differences between cohorts were determined using one-way analysis of variance with multiple comparison test or unpaired t-test. All data analyses were performed with GraphPad Prism 8 software (San Diego, CA).
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This research was supported by Muscular Dystrophy UK (grant number 16UNI-PRG48-0003, UNITE-DMD workpackage 4). TV and MC are supported by FWO and VUB IOF GEAR.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.