
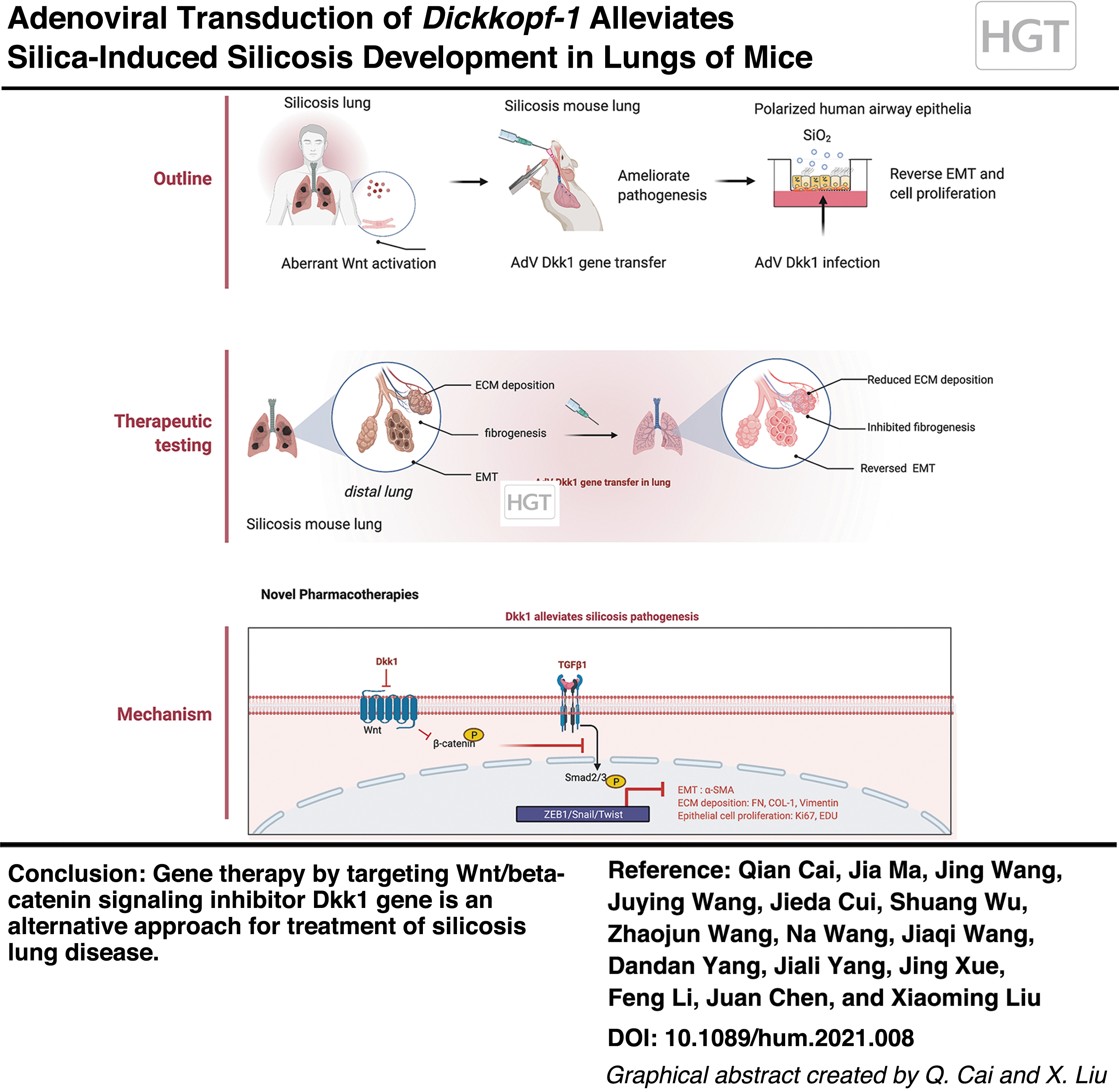
Abstract
Silicosis is an occupational disease caused by inhalation of silica dust, which is hallmarked by progressive pulmonary fibrosis associated with poor prognosis. Wnt/β-catenin signaling is implicated in the development of fibrosis and is a therapeutic target for fibrotic diseases. Previous clinical studies of patients with pneumoconiosis, including silicosis, revealed an increased concentration of circulating WNT3A and DKK1 proteins and inflammatory cells in bronchoalveolar lavage compared with healthy subjects. The present study evaluated the effects of adenovirus-mediated transduction of Dickkopf-1 (Dkk1), a Wnt/β-catenin signaling inhibitor, on the development of pulmonary silicosis in mice. Consistent with previous human clinical studies, our experimental studies in mice demonstrated an aberrant Wnt/β-catenin signaling activity coinciding with increased Wnt3a and Dkk1 proteins and inflammation in lungs of silica-induced silicosis mice compared with controls. Intratracheal delivery of adenovirus expressing murine Dkk1 (AdDkk1) inhibited Wnt/β-catenin activity in mouse lungs. The adenovirus-mediated Dkk1 gene transduction demonstrated the potential to prevent silicosis development and ameliorate silica-induced lung fibrogenesis in mice, accompanied by the reduced expression of epithelia–-mesenchymal transition markers and deposition of extracellular matrix proteins compared with mice treated with “null” adenoviral vector. Mechanistically, AdDkk1 is able to attenuate the lung silicosis by inhibiting a silica-induced spike in TGF-β/Smad signaling. In addition, the forced expression of Dkk1 suppressed silica-induced epithelial cell proliferation in polarized human bronchial epithelial cells. This study provides insight into the underlying role of Wnt/β-catenin signaling in promoting the pathogenesis of silicosis and is proof-of-concept that targeting Wnt/β-catenin signaling by Dkk1 gene transduction may be an alternative approach in the prevention and treatment of silicosis lung disease.
INTRODUCTION
Silicosis is an occupational fibrotic lung disease caused by inhalation of crystalline silica (silicon dioxide, SiO2) dust. 1 This disease has reemerged in many developing countries in recent years, particularly the increased incidence of acute silicosis, 2 due to the high inhalational risk of excessive silica exposure in modern industries by using high-powered hand tools, such as denim sandblasting, jewelry polishing, artificial stone engineering, dental trimming, building constructing, and highway repair. 3 –5 To date, silicosis remains an incurable disease with a few treatment options. Better understanding of the pathogenesis of silicosis is necessary to address this unmet medical need by developing novel therapeutic approaches that prevent and/or treat chronic lung disease.
Pathologically, airway epithelial cells and alveolar macrophages (AM) engulf inhaled silica particles and trigger an innate immune response by producing inflammatory factors and reactive oxygen species. 6 –8 Subsequent remodeling occurs through epithelial–mesenchymal transition (EMT) and pulmonary fibrogenesis characterized by massive extracellular matrix (ECM) deposition, fibroblast proliferation, and myofibroblast differentiation. 9 However, one study in mice showed that stimulation of the innate immune response alone was sufficient to drive silicosis development. 10 In addition, a reduction of functional Treg cells was found in silicosis patients, 11 and cytokines produced by AM exposed to silica dust initiate alveolitis and substantial responses in silicosis progression. 7 Therefore, cytological and biochemical assessments of cellular patterns and biomarkers in bronchoalveolar lavage fluid (BALF) and blood have demonstrated important prognostic value in diagnosing and evaluating the clinical stage of pneumoconiosis 12 such as silicosis 7,11 –13 and asbestosis. 14,15
Although the pathogenesis of silicosis remains incompletely understood, mechanistic studies have demonstrated that a number of cellular signaling pathways are involved in the development of pulmonary fibrosis and other chronic pulmonary disorders. 16 Among them, Wnt/β-catenin signaling, a key developmental signaling, is activated in idiopathic pulmonary fibrosis (IPF), 17 –20 asthma, 21 and chronic obstructive pulmonary disease (COPD). 22 Aberrant Wnt/β-catenin signaling has also been identified in the silicosis mouse lung 23 –25 and in A549 human alveolar epithelial cells in response to an exposure of silica dust. 26 Blockade of Wnt/β-catenin signaling using short hairpin RNA (shRNA), 23,24 and microRNA 25,27 by targeting the Wnt signaling mediator, β-catenin, attenuates inflammation and ameliorates fibrosis in silica-induced silicosis in mouse and rat lungs. These studies have clearly suggested that the Wnt/β-catenin pathway is a key driver in the initiation and development of pulmonary fibrosis, and inhibitors of this pathway may effectively prevent or treat fibrotic silicosis. 28
Dickkopf-1 (Dkk1), a member of the Dkk family, is one of the most investigated candidates of Wnt signaling inhibitors for therapeutic use in many diseases, particularly in cancers and bone diseases. 29 There is increasing evidence that DKK1 is a biomarker and/or therapeutic target in infectious diseases, 30 autoimmunity, 31 joint remodeling, 32 and fibrosis. 33 In terms of the inhibition of Wnt/β-catenin signaling in lungs, the overexpression of Dkk1 led to the suppression of myofibroblast differentiation in acute lung injury, 34 and amelioration of paraquat-induced pulmonary fibrosis in rats, 35 suggesting an anti-fibrotic potential of Dkk1.
Gene transfer using viral vectors has been shown to have great potential to treat many chronic lung diseases, 36 including cystic fibrosis 37 and pulmonary fibrosis. 38 –41 For instance, adeno-associated virus serotype 9 (AAV9)- and AAV1-mediated gene transduction vectors have been tested for the treatment of bleomycin-induced mouse pulmonary fibrosis 38,39,41 and aging-related IPF in telomerase-deficient mice. 40 These studies infer that therapeutic gene delivery using viral vectors may be an effective treatment for pulmonary fibrosis, including silicosis.
In light of the fact that Dkk1 is an effective inhibitor of Wnt/β-catenin signaling and reduces pulmonary fibrosis in animal models and with the knowledge that viral vectors may efficiently transduce therapeutic genes to the lung, we aimed at testing the pathogenic role of Wnt/β-catenin signaling and the effect of Dkk1 gene transduction in silicosis lung by using adenoviral vectors expressing murine Wnt3a (AdWnt3a) and Dkk1 (AdDkk1) in silicosis lungs of mice. Our results demonstrated that adenovirus-mediated Dkk1 gene transduction prevents the development of silica-induced silicosis and mitigates the fibrosis of silicosis lung in mice, partially through a mechanism of inhibiting TGF-β/Smad signaling.
MATERIALS AND METHODS
Subjects
The study and protocol were approved by the Ethics Committee for Conducting Human Research at General Hospital of Ningxia Medical University (NXMU-2020-487). All patient participants gave informed consent for the collection and analysis of their blood and bronchoalveolar lavage (BAL) samples for publication according to the protocol (NXMU-2020-487) outlined by the Ethics Committee for the Conduct of Human Research. The principal investigator of this study maintains human research records, including signed and dated consent documents for 10 years. Plasma samples of 56 clinically diagnosed patients with pneumoconiosis, silicosis, or coal worker pneumoconiosis (all males; silicosis group) were collected (Supplementary Table S1). These patients were recruited in the Department of Pulmonary and Critical Care Medicine at General Hospital of Ningxia Medical University between August 2020 and December 2020. Plasma samples of 56 sex-matched healthy individuals (control group) were also collected. These healthy control cohorts were recruited from individuals who had undergone comprehensive medical screening at the General Hospital of Ningxia Medical University. The healthy cohorts had no history of long-term exposure to insult dusts such as silica or coal and had no family history of chronic pulmonary diseases (Supplementary Table S2). Pneumoconiosis was diagnosed and classified according to the Guideline of National Occupational Health Standards of China 2015 criteria for the diagnosis of pneumoconiosis (GZB-70-2015). This guideline coincided with the requirements of International Labor Organization (ILO) International Classification of Radiographs of Pneumoconiosis (Revised edition 2011). 42 Both silicosis and control groups included a similar proportion of nonsmokers, ex-smokers, and current smokers. High-resolution computed tomography (HRCT) was performed with 1.0- or 1.5 mm-thick sections taken at 1 cm intervals throughout the entire lung during inspiration in a supine position. Each HRCT scan was reviewed by three independent radiologists.
In addition to the demographics and clinical data of individuals, the routine blood testing was also performed before antibiotic treatment and automated cytological analysis, including white blood cell (WBC) count, neutrophils (NEUT) count, and lymphocyte (LYM) count, and neutrophil–lymphocyte ratio (NLR) (Supplementary Tables S1 and S2). Patients with pulmonary fibrosis due to other causes, complications, and/or chronic pulmonary diseases, such as COPD, pulmonary infection, tuberculosis, and tumors in the lung, were excluded from this study. Patients with severe heart and renal dysfunction were also excluded. All blood was collected in tubes with heparin, and plasma was isolated and frozen in 100 μL aliquots at −80°C until subsequent analysis. No genetic relationship existed among individual subjects. All subjects were older than 30. All samples were collected with informed patient consent.
Cellular content of human BALF
Informed consent of BAL was obtained from only 6 subjects among silicosis patients, 18 patients declined to undergo bronchoscopy, and 32 patients did not undergo bronchoscopy due to health reasons such as weakness, severe respiratory or cardiac failure. In the control group, 13 participants consented to bronchoscopy. The BAL was performed via the oropharyngeal route in accordance with the American Thoracic Society guidelines. 43 Two hundred milliliters of prewarmed (37°C) saline was instilled into a segment of the right middle lobe, and the BALF was aspirated manually with a syringe and collected in a plastic bottle on ice. The BALF was centrifuged at 800 g for 10 min. The supernatant fraction was aliquoted and used to assess the presence of inflammatory factors and secretory proteins. Cell pellets were washed 3 × times before being resuspended in 10 mL of Hanks' balanced salt solution (HBSS). The cell suspension was cytospun on glass slides and stained with Wright-Giemse for cytologic analysis as per the Clinical Guidelines of the European Society of Pneumology Task Group. 44
Animal welfare statement and generation of silica (SiO2)-induced silicosis mouse model
All mouse protocols were approved by the Use of Laboratory Animal Committee of Ningxia University in accordance with guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NXULS20180123-3). 8 For the generation of a mouse model of lung silicosis, silicon dioxide (∼99% SiO2, 0.5–10 μm particle size, 14808-60-7; Sigma) particles were baked at 160°C for 2 h to inactivate endotoxin, suspended in saline at a concentration of 50 mg/mL, then further dispersed for 15 min in a water bath sonicator, and trichurated through a 25G needle before use. All mice were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China), and housed in a special pathogen-free facility with a 12/12 h light/dark cycle with food and water ad libitum in the animal facility at Ningxia Medical University (Yinchuan, China). For all experiments involving mice, 6- to 8-week-old C57BL/6 male and female mice were used in equal proportions. A statistical method was not used to predetermine the sample size. All mouse experiments were not randomized and blinded to the outcome assessment. Mice were randomly divided into different experimental groups (12 mice for each group, including 6 males and 6 females). The protocol, dose, and delivery route for the generation of silicosis in mice have been previously described and are followed here. 8 Briefly, mice were anesthetized with 3.5% isoflurane until agonal breathing was observed before a 50 μL of 50 mg/mL silica (or recombinant adenovirus) in saline or saline alone was intratracheally instilled into the lung through the oropharyngeal route. Mice were euthanized by carbon dioxide (CO2) inhalation at the end of the experiment. The blood and lung were harvested for pathological and molecular analysis at 14, 28, and 56 days post the silica challenge. 8
Cell culture
The protocol for collection of human lung biopsies and isolation of primary human bronchial epithelial cells (HBECs) was approved by the Ethics Committee for Conduction of Human Research at General Hospital of Ningxia Medical University (NXMU-2020-487). The A549 human adenocarcinoma alveolar epithelial cell line (ATCC CCL-185TM) and RAW264.7 murine macrophage cell line (ATCC TIB-71) were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured with Dulbecco's modified Eagle medium (Gibco) supplemented with 10% fetal bovine serum (Cat. No. VS500T; Ausbian) and 1% penicillin/streptomycin in a humidified atmosphere of 95% air-5% CO2 at 37°C. For polarization of primary HBEC cultures, HBECs were first seeded in collagen pre-coated dishes and expanded in Small Airway Epithelial Cell Growth Medium (SAGM™; CC-3119, Lonza, Walkersville, MD, supplemented SAGM Single Quots CC-3314 with 1% penicillin/streptomycin). The HBECs of Passage 3 (P3) were seeded in Corning 24 Transwell inserts (0.4 μm pore, 0.65 cm diameter, polyester membrane; Corning 3470, Tewksbury, MA), polarized, and maintained in PneumaCult ALI medium (STEMCELL, Vancouver, Canada) at an air
Recombinant adenoviral vectors and viral infection
Adenoviral vectors expressing mouse Wnt3a (AdWnt3a) and a “null” adenoviral backbone vector control (AdBglII) were kindly provided by the laboratory of Dr. John F. Engelhardt at the University of Iowa (Iowa City, IA). Adenoviral vectors expressing mouse Dkk1 (AdDkk1) were generated by Shanghai Genechem Co. LTD. (Shanghai, China). For adenoviral infection in A549 cells, cells were infected at ∼80% confluency with adenoviral vectors at a multiplicity of infection (MOI) of 1,000 viral particles for 24 h. Cells were cultured for an additional 48 h in the presence or absence of 100 μg/cm2 of silica dust (mesoporous, 2 nm particle size, CAS No. 7631-86-9; Sigma-Aldrich, St. Louis, MO) before being harvested for analysis. For the viral infection of ALI cultures, fully differentiated epithelial ALI cultures were infected with adenovirus at an MOI of 10,000 particles (1 × 106 cells/transwell with a diameter of 0.65 cm) basolaterally for 24 h at 37°C. 46 The cultures were then exposed to silica dust (SiO2, 2 μm in size) apically at a concentration of 100 μg/cm2 for an additional 48 h before being analyzed. For viral infection of mouse lungs, mice were intratracheally instilled with 50 μL saline containing 1 × 1011 viral particle/mouse. Mice received either AdBglII, AdWnt3a, or AdDkk1 recombinant adenovirus. Mice were challenged with silica (or saline control), as described earlier, 48 h after infection. Blood, BALF, and lung tissue were collected for analysis at 14, 28, and 56 days after silica challenge. 8
Measuring WNT3A and DKK1 protein concentration
The concentrations of WNT3A and DKK1 protein in human plasma and mouse sera were measured by an enzyme-linked immunosorbent assay (ELISA) using appropriate commercially available kits as per the manufacturer's instructions. The ELISA Kits to detect mouse Wnt3a and Dkk1 and human WNT3A and DKK1 were products of Shanghai Enzyme-linked Biotechnology Co., Ltd. (Shanghai, China). The protein concentration in each sample was determined by comparison to the standard curve and presented as ng/mL.
Lung histopathological analysis
Mouse lung tissues were fixed in 4% paraformaldehyde (PFA) solution before they were either embedded in optimal cutting temperature (OCT) compound or dehydrated and processed through paraffin. Five-micrometer-thick paraffin sections were used for histopathological analysis by hematoxylin and eosin (H&E) staining, Masson's trichrome staining, or immunohistochemical (IHC) staining as described elsewhere. 47 Images were captured under an Olympus optical microscope (Olympus TH4-200). To quantify fibrotic (silicotic nodule) area, all images were acquired by using identical settings and the affected regions of silicotic nodules were demarcated and areas were measured by using identical threshold setting in FIJI-ImageJ software. The percentage and size of silicotic nodules relative to the entire lung lobe was calculated. The average of data from five representative sections per lung of each animal was used for analysis. To assess the severity of silicosis, Ashcroft scale was used to score pulmonary fibrosis by two independent and blinded pathologists as previously described. 48 The degree of fibrosis in lungs was scored from 0 to 8 for the increasing extent of fibrosis based on histological images of lung as previously described. 49
Hydroxyproline content in lung tissues
The right lobes of the lung were homogenized for total protein isolation, and lysates from each experimental group were pooled for technically triplicate hydroxyproline measures. Hydroxyproline content was determined by using a commercially available hydroxyproline assay kit (Shanghai Jiang Lai Biology Institute) according to the instructions provided by the manufacturers. The hydroxyproline content was presented as microgram (μg) per milligrams total protein of wet lung tissues.
Immunoblotting analysis
Thirty micrograms of total protein from homogenized lung tissues as well as with A549 cells lysed in RIPA Lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Igepal, 1% sodium deoxycholate, 5 mM iodoacetamide, 0.1% sodium dodecyl sulfate [SDS], and protease inhibitor cocktail inhibitor) were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were then transferred onto a PVDF nitrocellulose membrane. The membrane was blocked with blocking buffer (4% non-fat milk containing 0.2% Tween-20 in phosphate buffered saline [PBS], or 3% bovine serum albumin [BSA] in TBST) for 1 h at room temperature (RT), then probed with the primary antibody against the protein of interest overnight at 4°C, and finally incubated with IRDye-conjugated (Li-Cor) or horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at RT. The protein of interest was then directly imaged on a Li-Cor Odyssey Image studio Ver 4.0 (LI-COR Biosciences, Lincoln, NE) or developed to enhanced chemiluminescence (ECL) reagent (Thermo Fisher Scientific, Waltham, MA) and visualized in GE ImageQuant LAS 600 (GE Healthcare, Chicago, IL). Relative expression of target protein was measured semiquantitatively by densometric analysis of blots. The intensity of blot area was calculated by optical densitometry using ImageJ Software version 2.0.0 (
IHC staining
For IHC staining, paraffin sections were deparaffinized and rehydrated. After antigen retrieval, slides were blocked with 5% BSA/TBS at RT for 1 h; samples were incubated with primary antibodies at 4°C overnight. Sections were then washed three times with PBS-tween 20 (0.1%) and then incubated with biotin- or HRP-conjugated secondary antibodies for 30 min at RT. Antibody staining was then developed with DAB substrate (Zsbio, Beijing, China) for HRP-conjugated secondary antibodies or incubated with an Avidin-Biotin Complex Staining Kit (ABC Kit; Vector Laboratories, Burlingame, CA) before DAB development as per the manufacturer's instructions. Slides were then counterstained with hematoxylin, dehydrated, mounted, and finally visualized under an upright microscope. Primary antibodies and secondary antibodies used for IHC staining are listed in Supplementary Table S3.
Immunofluorescent staining
Fixed lung tissues that were embedded in OCT compound were cryosectioned at a thickness of 10 μm for immunofluorescence (IF). Cryosections were air-dried at RT for 30 min, re-fixed in 4% PFA for 10 min, and finally permeabilized in 0.2% Triton X-100/PBS for 20 min at RT. Sections were then blocked by incubating them in 5% donkey serum in PBS for 1 h before being probed with primary antibodies in dilutant buffer (1% donkey serum, 0.03% Triton X-100, and 1 mM CaCl2 in PBS) at 4°C overnight. The sections were washed and then incubated with fluorescent dye-conjugated secondary antibodies at RT for 2 h. The slides were washed in PBS three times for 5 min, mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories), imaged under a Leica TCS SP2 A0BS Confocal System, and processed on Leica Confocal Software v.2.6.1 (Leica). For immunofluorescent staining of primary cells grown on Transwell membranes and A549 and RAW264.7 cells grown on coverslides, cells were fixed in 4% PFA for 30 min at RT, permeabilized with 0.3% Triton X-100 in PBS for 30 min at RT, and finally incubated with primary antibody for 48 h at 4°C. Samples were washed in PBS and then incubated with fluorescent dye-conjugated secondary antibody at RT for 2 h. After washing, membranes or coverslides were mounted and visualized under a Leica confocal fluorescence microscope. For imaging of single-cell resolution, cells with virous treatments at normal density were replated on coverslides at a low density for an attachment of 6 h before being processed for immunostaining. Images of each condition were captured on the same scope with the same settings and processed the same way by using FIJI-ImageJ. Image analysis was performed by using Metamorph Software's multiwavelength cell scoring application as described, 50 and the quantification of IF-stained cell markers was conducted as in previous studies. 51,52 At least five membranes or coverslides were evaluated per condition. Primary antibodies and secondary antibodies used for IF are listed in Supplementary Table S3.
Cell viability assay
Cell viability was assessed by using the Cell Counting Kit-8 (CCK-8) as per the manufacturer's instructions (Dojindo Molecular Technologies, Kumamoto, Japan). Briefly, the cells (5 × 103/well) were seeded in a 96-well plate and grown overnight before being exposed to silica dust (2 μm particle size) at a dose of 0, 50, 100, 150, or 200 μg/cm2 for 24, 48, or 72 h. Subsequently, 10 μL of CCK-8 solution was added to each well, and the plate was then incubated at 37°C for an additional 2 h. Absorbance was measured at a wavelength of 450 nm read on a microplate reader (BioTek, Winooski, VT). The relative cell viability was expressed as the percentage of (ODsilica-cells − ODsilica-medium)/(ODcells − ODmedium) × 100, where OD450 nm values represent measurements of wells containing silica-treated cells, cell-free medium containing silica, untreated control cells, and cell-free and silica-free medium alone. All experiments included biological triplicates, and data were representative of at least three independently performed experiments.
5-Ethynyl-2′-deoxyuridine incorporation assay in polarized airway epithelial cells
Click-iT EdU (5-ethynyl-2
Statistical analysis
All experiments were performed with biological triplicates, and data represent at least three independently performed experiments. Data are presented as mean ± standard deviation. All analyses were assessed by using GraphPad Prism version 8 software (GraphPad Software, Inc., La Jolla, CA). Two-tailed, unpaired Student's t-tests or one-way analysis of variance were used for pair-wise comparisons. All p-values of <0.05 were considered statistically significant.
RESULTS
Demographic data
All 56 pneumoconiosis patients enrolled in this study were male with a mean age of 67.34 ± 13.19 years. The control group included 56 male individuals with a mean age of 61.7 ± 6.15 years. Parameters assessed in the differential blood counts between silicosis and control patients included the following: WBCs, 6.71 ± 1.72 × 109/L and 6.79 ± 2.42 × 109/L; NEUT, 3.87 ± 1.38 × 109/L and 4.68 ± 2.07 × 109/L; and NLR, 2.01 ± 0. 85 × 109/L and 3.54 ± 1.86 × 109/L. Further, the LYM count was significantly decreased in silicosis patients, at 1.47 ± 0.57, compared with control individuals at 2.1 ± 0.67 (p = 0.034, Table 1 and Supplementary Tables S1 and S2). These data led us to hypothesize that patients develop silicosis due to a diminished local capacity to elicit an immune response in lungs.
Demographics of pneumoconiosis patients and healthy cohorts
p < 0.05.
NLR, neutrophil–lymphocyte ratio; WBC, white blood cell.
Elevated circulating WNT3A and DKK1 proteins in pneumoconiosis patients
To determine whether Wnt/β-catenin signaling is clinically implicated in pathogenesis of human silicosis, concentrations of Wnt signaling ligands, WNT3A and DKK1 proteins were evaluated in plasma of 56 patients with pneumoconiosis from silicosis and coal worker pneumoconiosis. 53 The HRCT image showed normal lung images in both nonsmoking and smoking healthy subjects (Fig. 1A), but fibrotic nodules in the lungs of silicosis patients, regardless of smoking or nonsmoking (Fig. 1B). Cytologic examination revealed an increased numbers and frequency of dust-laden macrophages (dust cells, DCs) in BALF collected from silicosis patients compared with healthy subjects (Fig. 1A, B and Table 2). The distribution of cell types in BAL of the silicosis and control cohorts was also different (Table 2). The number and frequency of both NEUT and DCs were more abundant in the BAL of silicosis patients relative to that of healthy controls. They were 40.71 ± 34.45 × 103/mL and 18.50% ± 9.22% versus 16.08 ± 8.55 × 103/mL and 10.67% ± 4.99% for NEUT; 31.94 ± 24.43 × 103/mL and 16.33% ± 9.03% versus 10.85 ± 10.29 × 103/mL and 7.13 ± 6.40 for DCs, respectively (Table 2). Biochemical analysis revealed more abundant plasma WNT3A protein in pneumoconiosis patients (2.577 ± 1.42 ng/mL, N = 56) relative to that of healthy cohorts (0.8355 ± 0.7816 ng/mL, N = 56; p < 0.0001) (Fig. 1C). Unexpectedly, a higher level of DKK1 protein was also observed in patients with pneumoconiosis (52.04 ± 31.11 ng/mL, N = 56) compared with healthy cohorts (33.01 ± 4.756 ng/mL, N = 34; p = 0.0003) (Fig. 1D). Smoking statuses had no impact on WNT3A and DKK1 levels in plasma of both healthy individuals and pneumoconiosis patients (Supplementary Fig. S1A–D). Although the circulating WNT3A and DKK1 proteins were highly correlated in humans (Supplementary Fig. S1E), neither the plasma WNT3A nor the DKK1 level had a correlation with the WBC count, NEUT count, and NLR (Supplementary Fig. S2A–C, E–G). Of note, both WNT3A (r = −0.2127, 95% confidence interval, CI [−0.3831 to −0.0283], p = 0.0243) and DKK1 (r = −0.2481, 95% CI [−0.4146 to −0.0656], p = 0.0243) were inversely correlated with LYM count (r = −0.2127, 95% CI [−0.3831 to −0.0283], p = 0.0083) (Supplementary Fig. S2D, H). These results suggested that aberrant Wnt/β-catenin signaling was clinically relevant to the pathogenesis of silicosis, which is consistent with findings in IPF pathogenesis. 17
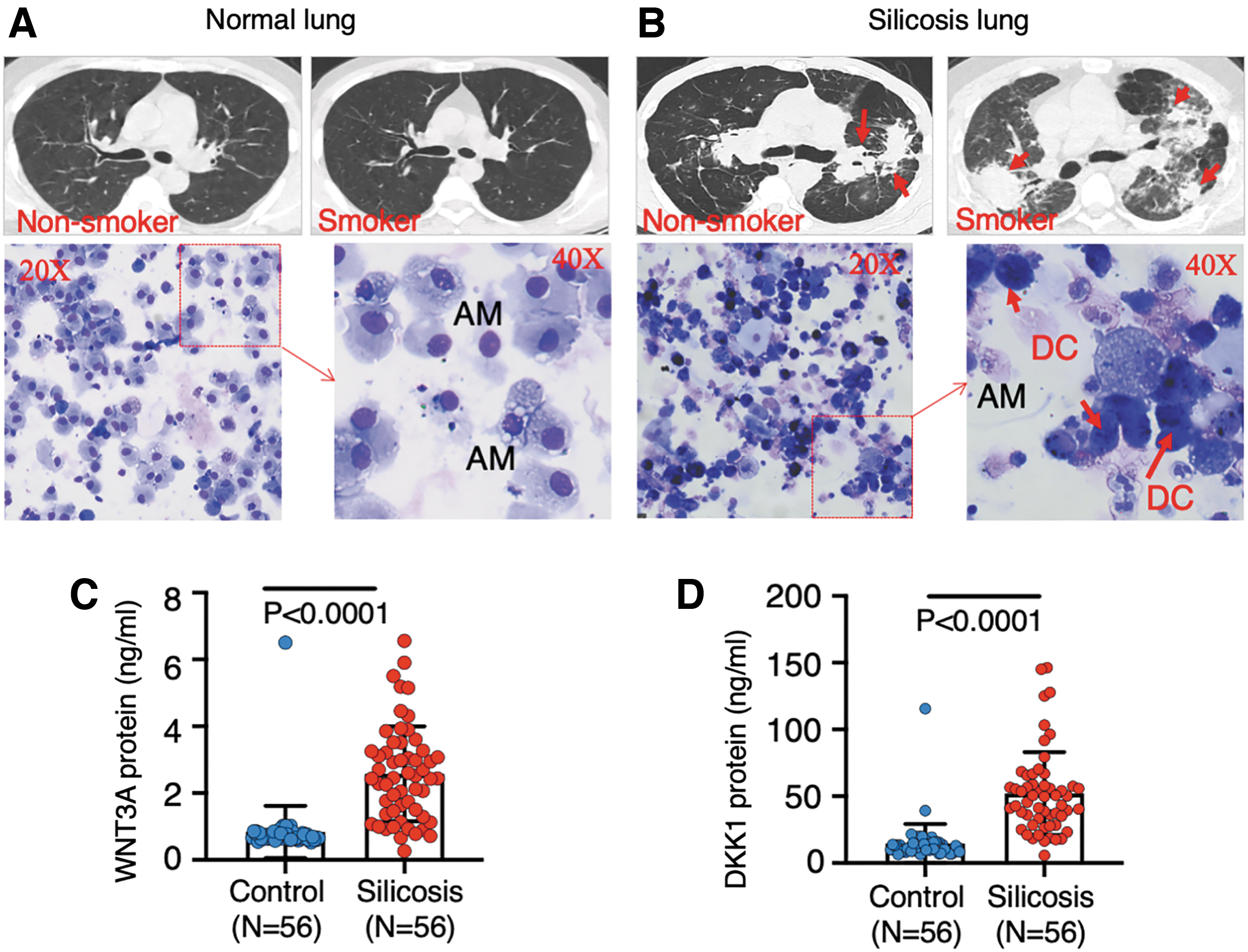
An increased concentration of WNT3A and DKK1 proteins in plasma of patients with silicosis and pneumoconiosis.
Bronchoalveolar lavage differential counts in silicosis patients and healthy cohorts
p < 0.05.
BAL, bronchoalveolar lavage.
An elevated TGF-β signaling and ECM deposition in lungs of silicosis mice
To further interrogate the potential of Wnt/β-catenin signaling as targets for treatment of silicosis lung disease, a silicosis mouse model was generated by instillation of crystalline silica (SiO2) in the lung (Fig. 2A). 54 Pathohistological evaluation demonstrated a progressive development of silicosis with time as determined with an increased area of silicotic nodules, a hallmark of silicosis (Supplementary Fig. S3A, B). In addition, mice challenged with silica had significantly poor performance of growth compared with mice receiving saline (p = 0.0072) (Supplementary Fig. S3C). On days 14, 28, and 56 after silica challenge fibrotic nodules covered 18.03%, 32.08%, and 45.51% area of the entire lobe on average, respectively (Supplementary Fig. S3B), and severity Ascroft scores were 5.167 ± 0.291, 6.166 ± 0.105, and 7.000 ± 0.333, respectively (Fig. 2B). Consistently, progressive pulmonary fibrosis ascertained by a more abundant hydroxyproline content in lung tissues was observed in silica-challenged mice compared with control mice at d14 (p = 0.045), d28 (p = 0.020), and d56 (p < 0.0001) post the first challenge (N = 9) (Fig. 2C). The histological changes in lungs of silica-challenged mice were corroborated by activated TGF-β signaling and increased ECM deposition as assessed by key TGF-β signaling factors and target genes (Fig. 2D, E). Immunoblotting showed an increased abundance of TGF-β1, Phos-Smad2/3, Snail, ZEB1, and Twist, as well as the profibrogenic factor alpha smooth muscle actin (α-SMA) and ECM proteins Collagen I (COLI), but the epithelial marker E-cad decreased in lungs of silicosis mice, in comparison to control animals (Fig. 2D, E). The IHC staining also showed a tonic expression of COLI, fibronectin (FN), and TGF-β1 in silicosis nodules of silicosis lungs (Supplementary Fig. S4). Of note, no difference in the pathogenesis of silicosis was observed between male and female mice.
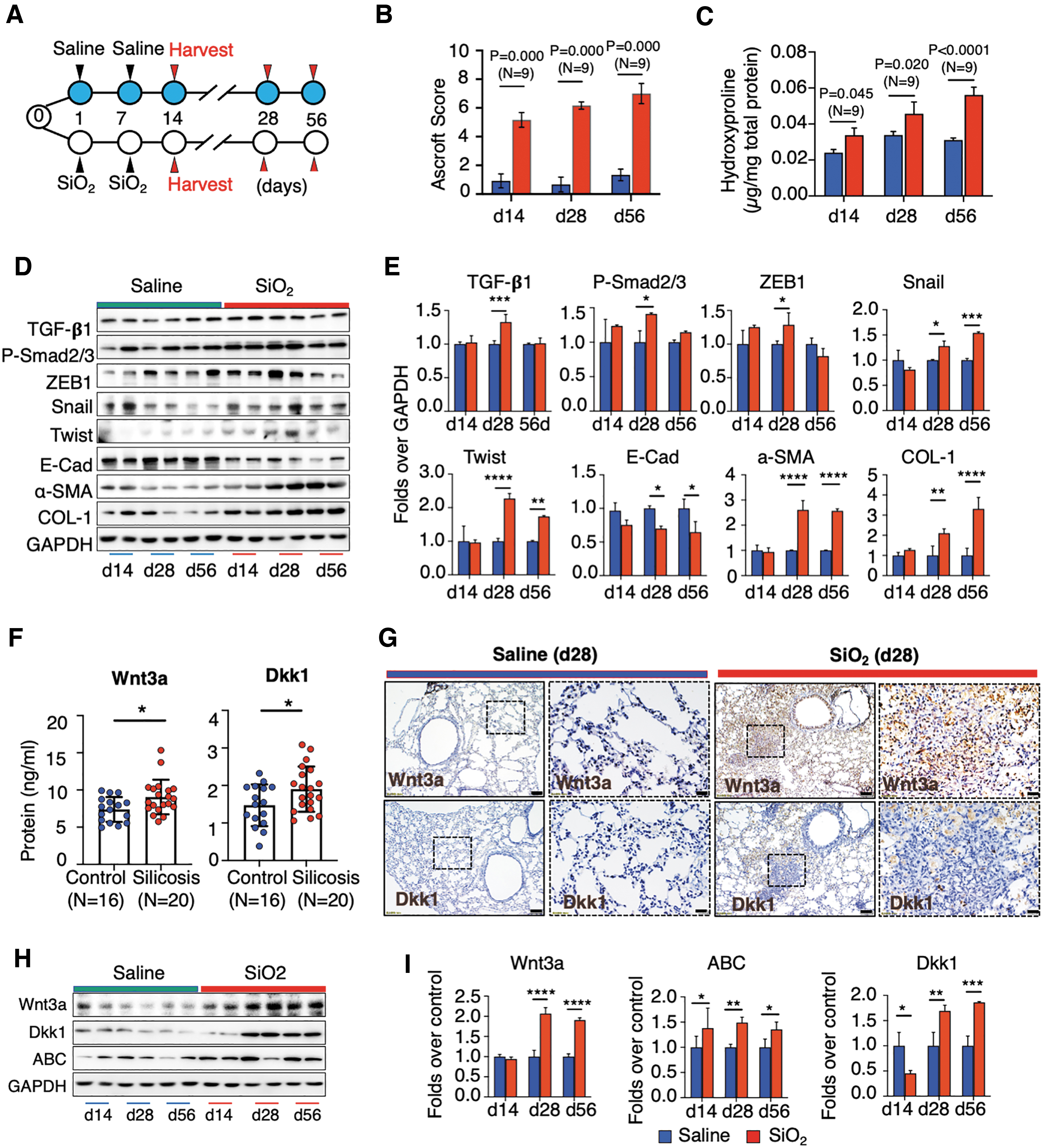
Activation of TGF-β and Wnt/β-catenin signaling in silicosis mouse lung.
Reactivated Wnt/β-catenin signaling in lungs of silicosis mice
To investigate whether Wnt/β-catenin signaling was activated in silicosis mouse lungs as seen in human IPF lungs, 17 the abundance of Wnt3a and Dkk1 proteins in sera of mice was first evaluated by ELISA. Similar to what is seen in humans, circulating Wnt3a and Dkk1 was elevated in silicosis compared with controls (Fig. 2F). By IHC staining, Wnt3a and Dkk1 were increased within silicosis lungs 28 days after the silica challenge compared with saline-instilled lungs (Fig. 2G). Immunoblotting further corroborated an increased abundance of Wnt3a and active β-catenin (ABC) proteins, in lungs of silicosis mice at d14, d28, and d56 post-silica challenge, compared with controls (Fig. 2H, I). Of note, Dkk1 was decreased in lungs of mice at day 14, but increased at d28 and d56 post-silica challenge, relative to controls (Fig. 2H, I), implying that dynamic changes in Dkk1 levels might contribute to the development of silicosis in mouse lungs. A reduction in Dkk1 could help explain aberrant Wnt/β-catenin activity, and insufficient Dkk1 levels might initiate silicosis. These data clearly indicate that Wnt/β-catenin signaling was altered and activated in silicosis mouse lungs with the progression of pathogenesis and led us to hypothesize that targeting Dkk1 could correct Wnt/β-catenin signaling activity to treat silicosis. 23 –25 Of interest, similar to what is seen in human patients, increased NF-κB p65 and F4/80 double-positive macrophages were measured in BALF of mice 14 days after silica dust challenge, which was indicative of an evoked inflammatory response (Supplementary Fig. S5A, B). In agreement with this observation, a robust increase in Toll-like receptor 4 (TLR4) and NF-κB p65 was induced in murine macrophage RAW264.7 cells on exposure to silica as determined by immunoblotting (Supplementary Fig. S5C, D) and IF analysis (Supplementary Fig. S5E). Nonetheless, the expression of TLR4 and NF-κB p65 waned at 48 h post-stimulation (Supplementary Fig. S5C, E). This funding is concurrent with the notion that silicosis is an inflammatory pulmonary disease. 55,56
Adenovirus-mediated Dkk1 gene transduction mitigates the development of silicosis in mouse lungs
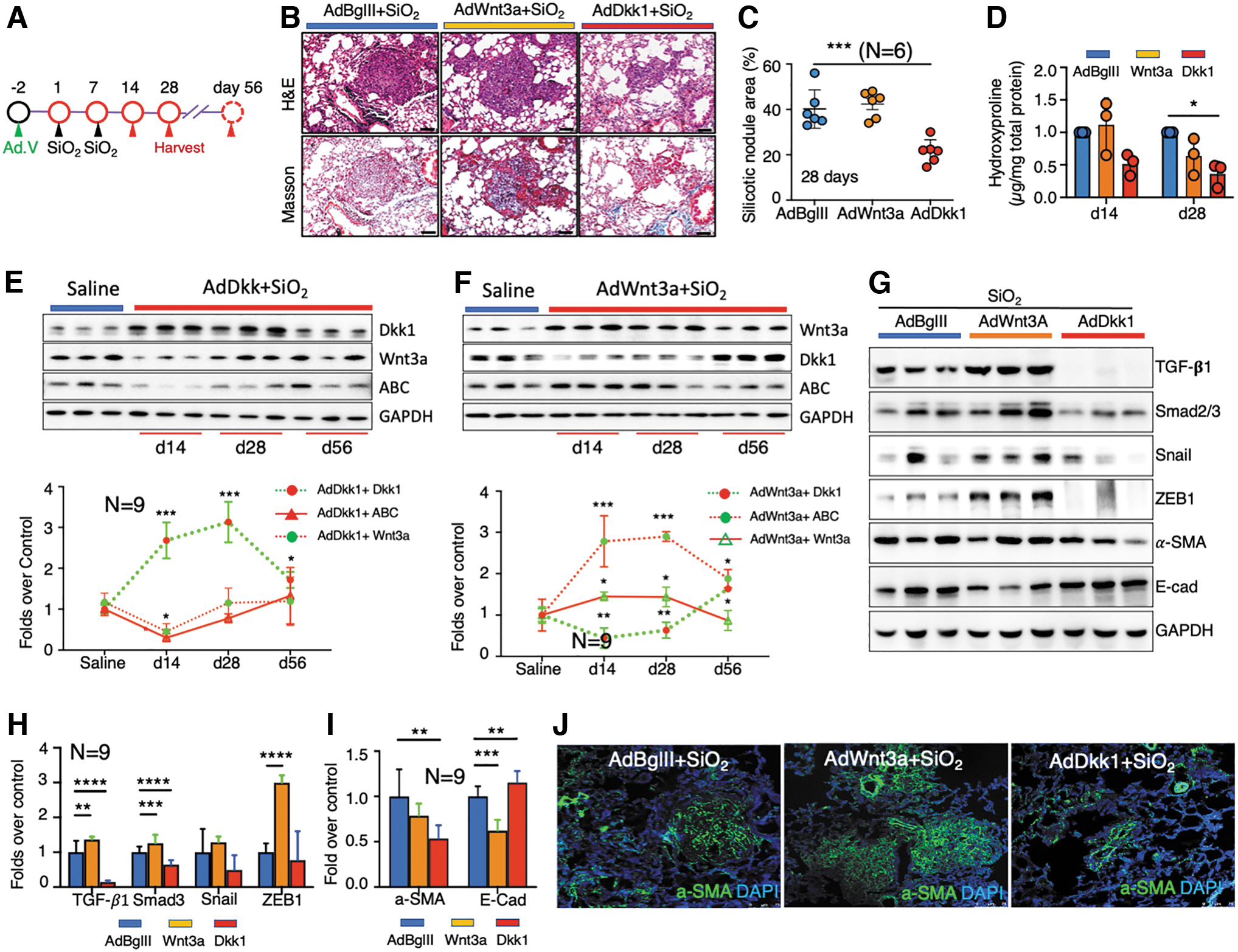
A compelling body of research has demonstrated that Wnt/β-catenin signaling may be a therapeutic target to treat pulmonary fibrosis 17,19,35,57 and silicosis. 23 –25 In addition, Dkk1 is a well-characterized Wnt inhibitor and has been extensively evaluated as a potential therapeutic target for several disorders, including cancers, 58 Alzheimer's disease, 59 and osteoarthritis (OA). 32 We, therefore, aimed at evaluating the potential of driving Dkk1 expression to prevent/alleviate silicosis development in a mouse model by using an adenoviral vector that is able to efficiently mediate gene transfer to lungs (Fig. 3A). 60 Surprisingly, a significant improvement in body growth was observed in silica-challenged mice that received adenovirus expressing Dkk1 (AdDkk1) in comparison with that of Wnt3a (AdWnt3a) and control “null” vector (AdBglII) (Supplementary Fig. S6A). The adenovirus-mediated Wnt3a and Dkk1 gene transduction in silicosis mouse lungs was confirmed by IHC (Supplementary Fig. S6B). Notably, histopathologic evaluation demonstrated partial rescue of silicosis pathology, as indicated by the size and abundance of silicotic nodules and ECM deposition (Fig. 3B). Silica-challenged mice infected with AdDkk1 possessed fewer silicotic nodules of smaller size compared with that of mice infected with AdWnt3a or AdBglII (Fig. 3C). Quantitative fibrotic score analysis revealed a reduction of nodular area (21.4%) of lung in silica-challenged mice treated with AdDkk1 compared with mice infected with AdWnt3a (42.33%) and AdBglII (40.17%) at 14 days after the first challenge (p < 0.001) (Fig. 3C). Consistently, a reduction of hydroxyproline content was detected in AdDkk1-infected silicosis mice as compared with AdWnt3a and AdBglII mice (p < 0.05) (Fig. 3D).
Adenoviral vector-mediated Dkk1 gene transduction inhibits Wnt/β-catenin and TGF-β signaling in the lungs of silica-challenged mice.
Of interest, AdWnt3a did not worsen silicosis outcomes (Fig. 3B–D). As expected, immunoblotting revealed that lung tissues from mice infected with AdDkk1 or AdWnt3a showed increased Dkk1 or Wnt3a expression and also decreased or increased ABC staining, respectively, compared with tissues from the AdBglII group (Fig. 3E, F). Given the fact that adenoviral vector transgene expression is transient, we next sought to examine the persistence of Dkk1 and Wnt3a gene transduction during the pathogenetic time course of silicosis. To this end, the abundance of Wnt3a, Dkk1, and ABC proteins in silicosis lungs of mice transduced by AdDkk1 or AdWnt3a from d14 to d56 was evaluated by immunoblotting. As expected, the abundance of Dkk1 (Fig. 3E) and Wnt3a (Fig. 3F) was increased in lung tissues of mice at d14 and d28, but it dropped to normal levels by d56. Moreover, increased Dkk1 accompanied a reduction in Wnt3a and ABC in lungs infected with AdDkk1 (Fig. 3E). Likewise, increased Wnt3a coincided with a reduction of Dkk1 and increased ABC in lungs infected by AdWnt3a (Fig. 3F). These results suggested that the adenovirus-mediated gene transduction persisted in silica-challenged lungs for at least 28 days. These data also indicate that inhibition of Wnt/β-catenin signaling by Dkk1 early after silica challenge can reduce the progression of silicosis development.
Dkk1 suppresses TGF-β signaling in silicosis mouse lung
Next, we sought to understand the mechanism of AdDkk1 in mitigating silicosis development in silica-challenged mice. The profibrotic role of TGF-β signaling in pulmonary fibrosis has been well established 61 ; we, therefore, first examined alterations of key factors in TGF-β signaling in silicosis lungs of mice infected with AdWnt3a or AdDkk1. TGF-β signaling was elevated in silica-induced silicosis mouse lung (Fig. 2D, E and Supplementary Fig. S4). However, silica-induced activation of TGF-β signaling was suppressed by AdDkk1 at d28 post-challenge, as indicated by a reduction of TGF-β1, P-Smad2/3, ZEB1, Snail, and Twist and α-SMA abundance and an increase of E-Cad abundance compared with mice infected with AdBglII (Fig. 3G–I). Moreover, by IF α-SMA decreased predominantly within silicosis nodules (Fig. 3J). Of note, AdWnt3a treatment exhibited the opposite effect and enhanced TGF-β signaling in challenged mice compared with AdBglII-treated mice (Fig. 3G–J). To investigate whether the activation of TGF-β1/Smad signaling impacts Wnt/β-catenin signaling, we exposed A549 cells, a human lung carcinoma cell line, to TGF-β1 protein and measured Wnt signaling activity by quantifying levels of ABC, WNT3A, and DKK1 (Supplementary Fig. S7). We observed a dose-dependent induction of ABC and WNT3A along with increased expression of the EMT marker SMA and decreased E-Cad by immunoblotting (Supplementary Fig. S7A, B) and IF (Supplementary Fig. S7C). Intriguingly, TGF-β1 had a limited impact on DKK1 protein expression levels in A549 cells (Supplementary Fig. S7). However, these results suggest that there is an interaction between Wnt/β-catenin and TGF-β signaling in the pathogenesis of silicosis.
Adenovirus-mediated Dkk1 gene transfer alleviates the existing silicosis in mouse lungs
To examine whether the adenoviral vector-mediated Dkk1 gene transduction had the potential to ameliorate existing silicosis lung disease, AdDkk1 or AdBglII virus was administrated into silica-induced silicosis lungs via the laryngotrachea route at d14 post the first silica challenge, after the silicosis lung was developed (Fig. 4A). Histological observations revealed reduced silicotic nodules with ECM deposition in the silicosis lungs of mice infected with AdDkk1, compared with the AdBglII-infected lungs at both d28 and d56 post the silica challenge (Fig. 4B). Pathohistological analysis by fibrotic score further demonstrated a significant reduction of nodular area (20.55%) of lung in silicosis lungs infected with AdDkk1, compared with that of AdBglII (33.17%) at d56 (p = 0.009), despite no statistically significant reduction being detected between the AdDkk1 (22.38%) and Ad.BglII (32.83%) infected silicosis lungs at d28 post the challenge (p = 0.009) (Fig. 4C). A significant reduction of hydroxyproline content was also observed in lung tissues of silicosis mice infected with AdDkk1, as compared with that of AdBglII at d56 post the challenge (p < 0.001) (Fig. 4D). Molecular analysis of the persistent inhibition of Wnt/β-catenin signaling with immunoblotting assay showed an increased Dkk1 at d28 and d56 silicosis lungs infected with AdDkk1, along with the reduction of Wnt3a and ABC at d28 but not d56 post the challenge, compared with the AdBglII group (Fig. 4E, F). The AdDkk1-mediated inhibition of Wnt/β-catenin signaling resulted in a reduced α-SMA abundance (Fig. 4G–I) and key molecules of TGF-β/Smad signaling cascade in silicosis mouse lungs, as accessed by IHC and/or immunoblotting assays (Fig. 4G–I). These data suggest that the inhibition of Wnt/β-catenin signaling by targeting Dkk1 using approaches of pulmonary gene transfer has potential for the treatment of existing silicosis lung disease.
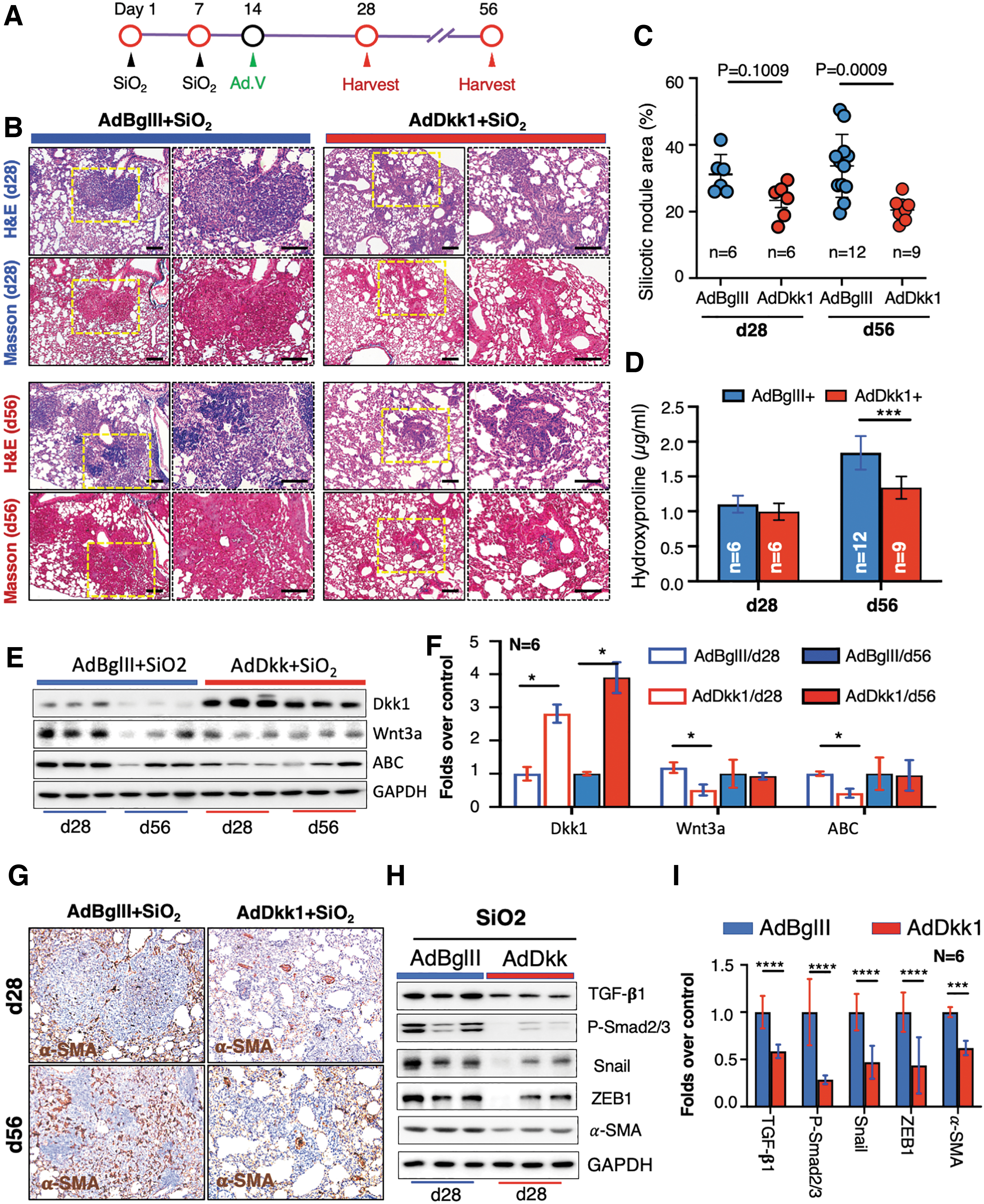
Adenovirus-transduced Dkk1 ameliorates existing silicosis in the lungs of silica-challenged mice.
Dkk1 inhibits silica-induced EMT in lung epithelial cells
The EMT is a key feature of the pathogenesis of pulmonary fibrosis, including silicosis. 8,62 To further delineate whether Dkk1 alters silica-induced EMT in epithelial cells, we assessed the viability of A549 human cells in response to silica by using a CCK-8 assay. We observed a time-dependent reduction in cell viability in cells exposed to silica at a concentration of between 50 and 200 μg/cm2 (p < 0.05). However, at 50 μg/cm2 or less, silica had only a moderate impact on cell viability by 24 and 48 h post-exposure (Fig. 5A). Therefore, further in vitro studies utilized a concentration of 100 μg/cm2. As seen in silicosis mouse lungs, IF staining showed that exposure to silica activated Wnt/β-catenin signaling and induced EMT in A549 cells, as assessed by increased abundance of ABC and α-SMA (Fig. 5B). In addition, proteins of Wnt/β-catenin signaling cascade, including Wnt3a, ABC, and Cyclin D1, and EMT markers, including vimentin and α-SMA, were also increased in response to exposure as determined by immunoblotting assay (Fig. 5C, D). The IF staining indicated that adenoviral vectors efficiently mediated Wnt3a and Dkk1 gene transduction into A549 cells, and AdWnt3a enhanced Wnt/β-catenin signaling as assessed by the increased nuclear translocation of ABC regardless of exposure to silica (Fig. 5E). The AdWnt3a augmented the silica-induced TGF-β signaling transcriptional factor, ZEB and reduced the EMT marker α-SMA in A549 cells, but the infection of AdDkk1 produced the opposite results (Fig. 5F). Immunoblotting analysis further demonstrated that the adenoviral-mediated Dkk1 gene transduction suppressed the expression of TGF-β signaling factors, including TGF-β1, Phos-Smad2/3, and ZEB1, and reduced EMT markers α-SMA and Vimentin in A549 cells and offset silica-activated TGF-β signaling (Fig. 5G, H).

Wnt/β-catenin enhances silica-induced TGF-β signaling and EMT in A549 epithelial cells.
Dkk1 inhibited silica-induced HBEC proliferation and EMT
The results cited earlier indicated that exposure to silica leads to the activation of Wnt/β-catenin signaling and the induction of EMT in mouse lung and A549 cells. Given the fact that Wnt/β-catenin signaling is critical for cell proliferation and maintenance of tissue homeostasis, we next sought to test the impact of Dkk1 on silica-induced EMT and cell proliferation in polarized epithelial cells generated with primary HBECs (Fig. 6A). The silica exposure caused the polarized HBECs to significantly upregulate α-SMA but downregulate E-cad expression (Fig. 6B). Subsequential analysis of fluorescent integrated intensity revealed a significant downregulation of α-SMA (p < 0.05) but upregulation of E-cad (p > 0.01) in polarized HBECs infected with AdDkk1 compared with the AdBglII regardless of SiO2 exposure (Fig. 6C). The adenovirus-mediated ectopic expression of Dkk1 resulted in a partial rescue of silica-induced α-SMA upregulation and E-cad suppression, compared with the AdBglII control (Fig. 6D). However, AdWnt3a had the opposite effect (Fig. 6D, E). Effects on proliferation were assessed by staining for the proliferation marker, Ki67 (Fig. 6D, E) and assessing the incorporation of the nucleotide analogue, EdU (Supplementary Fig. S8). Results showed an enhanced cell proliferation in cells exposed to silica dust, as determined by the number of cells expressing Ki67 (Fig. 6D, E). The silica-induced cell proliferation was increased by infection of AdWnt3a but inhibited by AdDkk1, as compared with AdBglII (Fig. 6D, E). Dkk1 over-expression also repressed EdU incorporation and EMT markers in silica-induced cells in vitro (Supplementary Fig. S8). These data suggest that Dkk1-mediated inhibition of Wnt/β-catenin signaling reduces EMT and cell proliferation in human airway epithelial cells exposed to silica dust.
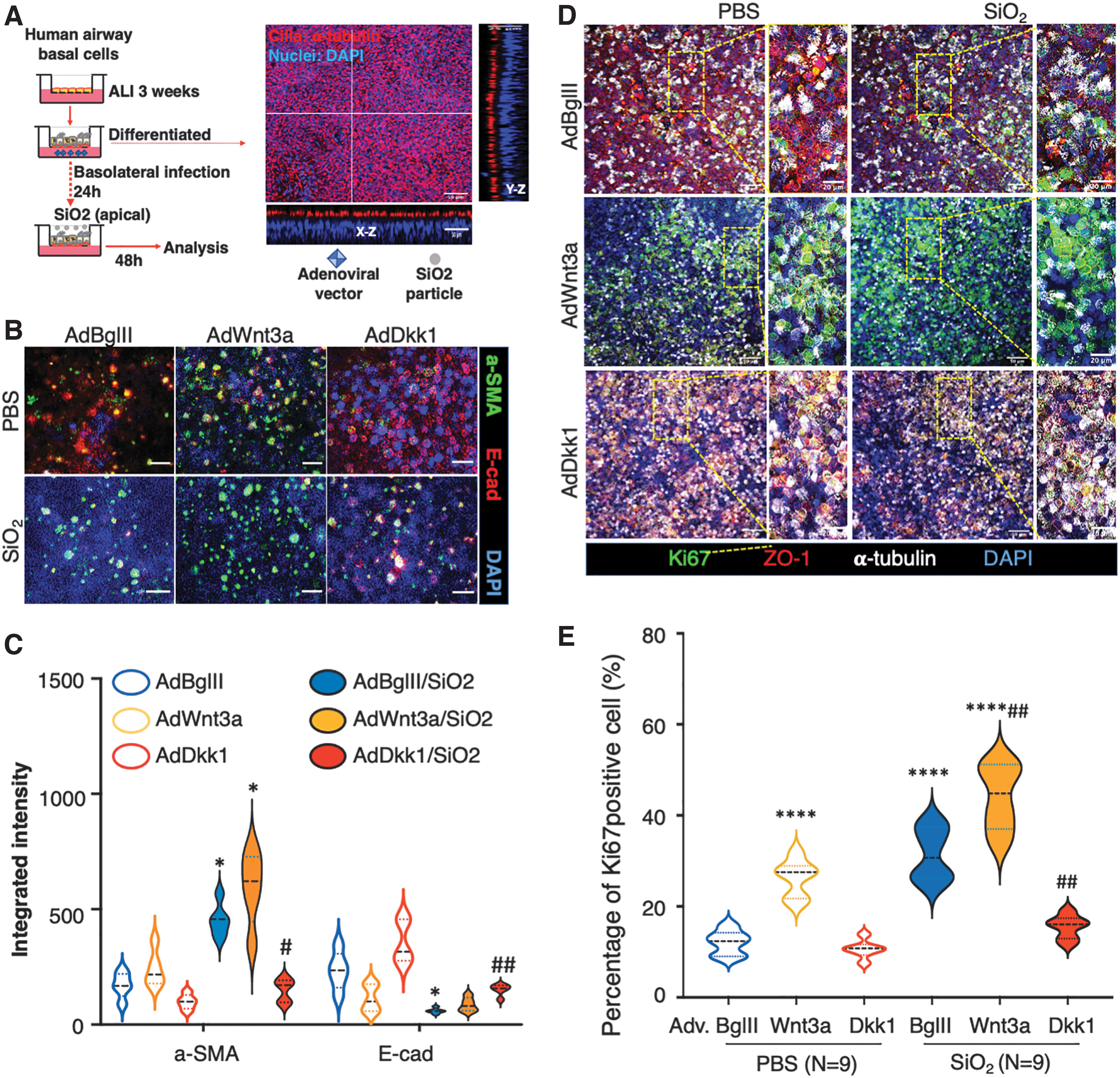
Dkk1 inhibited silica-induced cell proliferation in polarized human airway epithelial cells.
DISCUSSION
Pneumoconioses such as silicosis is an irreversible pulmonary disease mainly characterized by nodular pulmonary fibrosis. Environmental exposures that activate Wnt/β-catenin signaling have been demonstrated to play a pathogenic role in silicosis, and repression of Wnt/β-catenin signaling is a potential treatment to prevent chronic disease. In the present, proof-of-concept study, we demonstrated that intratracheal delivery of adenoviral vector expressing Wnt inhibitor Dkk1 (AdDkk1) could prevent the development of silica-induced lung silicosis and alleviate the existing silicosis in mice lungs. The AdDkk1-mediated amelioration of silicosis was, in part, orchestrated through inhibiting silica-induced activation of Wnt/β-catenin and TGF-β/Smad signaling and suppressed silica-induced cell proliferation, EMT, ECM deposition, and myofibrotic differentiation. Thus, this study provides evidence that viral vector-mediated Dkk1 gene transfer may be an effective strategy in reducing silicosis development.
Accumulating evidence has demonstrated that the dysregulation of multiple developmental signaling pathways can drive pulmonary fibrosis. 16 Among them, the Wnt/β-catenin signaling pathway is well known to drive pathogenesis. 17 –20 Dysregulated Wnt/β-catenin signaling induces TGF-β/Smad-mediated EMT and myofibroblast differentiation, resulting in excessive production and deposition of ECM in the lung. 63 It has been well documented that aberrant Wnt/β-catenin activity and TGF-β signaling has been observed in the lungs of patients with IPF, 17 and experimental silicosis animal models. 8,23 –25 Therefore, Wnt/β-catenin signaling has been considered a potential therapeutic target for the treatment of pulmonary fibrosis, including silicosis. Indeed, the therapeutic potential of targeting Wnt/β-catenin signaling was tested by using experimental silicosis models and provides encouragement for being effective against disease. 23 –25
In this article, we observed that forced activation of Wnt/β-catenin was accompanied by increased TGF-β/Smad signaling, increased EMT, and increased ECM deposition in silica (SiO2)-exposed silicosis mouse lungs, which was in line with findings from previous studies. 23 –25 Intriguingly, increased levels of circulating WNT3A and the Wnt inhibitor, DKK1, were also detected in the plasma of patients with pneumoconiosis, including silicosis and the sera of silicosis mice. Increased WNT3A protein was consistent with findings from IPF patients and bleomycin-induced mouse pulmonary fibrosis. 17 In addition, an elevated level of DKK1 expression is consistent with findings in airway epithelia 64 and BALF of airway lumen 65 of IPF patients. Previous studies have demonstrated that circulating DKK1 is linked to pathological inflammation and autoimmune diseases. 30,31 These findings provide rationality for targeting Wnt/β-catenin signaling as a silicosis treatment.
Dkk1 is a member of the Dkk family and is a well-studied potent inhibitor of Wnt/β-catenin signaling. 66 Expression of DKK1 increased in HBECs on exposure to silica dusts. 26 In line with an increased DKK1 in plasma samples from pneumoconiosis patients and BALF from IPF patients, an elevated Dkk1 level was also determined in sera and total protein of lungs of mice exposed to silica in this study. It is worth noting that induced Dkk1 expression was seen in silica-exposed mice older than 4 weeks but was not seen in developing mice exposed to silica in the first 2 weeks after the challenge. The mechanism controlling dynamic Dkk1 expression during the pathogenesis of silicosis is currently unknown. However, it may imply that the silica-induced activation of Wnt signaling ultimately triggers an autoinhibitory pathway in epithelial cells that act to suppress Wnt signaling and restore epithelial homeostasis. Nevertheless, the reduced Dkk1 in the first 2 weeks after a silica challenge may imply the key role of Dkk1 in the initiation of the pathogenesis of silicosis. Therefore, in adult animals, over-expression of Dkk1 at an early stage of silicosis development may alleviate disease progression after the exposure of silica dust.
Dkk1 is secreted by a variety of cell types, including platelets and epithelial cells, on stimuli to regulate immunity. 30 Increased circulating DKK1 was involved in facilitating T helper 2 (Th2) cell polarization and cytokine production, working to ultimately protect tissues from injury caused by environmental insults (such as silica dust) or inflammations. 67 The Wnt inhibition-mediated regulation of Th cells in inflammation was also reported in silica-induced mice, 23 and silica-activated TLR and NF-κB signaling was observed in mice in vivo and macrophages in vitro. Of interest, however, in human IPF lungs, epithelial-secreted DKK1 was insufficient to effectively inhibit Wnt induction and aberrant alveolar epithelial cell proliferation. 65 In addition, an adeno-associated virus (AAV)-transduced Dkk1 has been tested to treat OA and improve bone architectural parameters in a rat OA model. 32 Together with our findings, these studies suggest that ectopic expression of DKK1 by gene addition may be a safe and effective approach for suppressing Wnt/β-catenin signaling in lungs. Therefore, Dkk1 gene transfer may be an alternative approach for the treatment of inflammatory diseases, such as pulmonary fibrosis and silicosis.
The inhibitory role of DKK1 in Wnt/β-catenin signaling has been shown to be involved in the fibrogenesis of several organs, including the livers, lungs, and kidneys, 33 and forced expression of Dkk1 suppresses fibrosis in animal models. 68 With respect to silicosis, several lines of evidence demonstrate that blocking Wnt/β-catenin signaling attenuates silicosis lung fibrosis in experimental animals. 23 –25 For instance, shRNA to β-catenin-mediated inhibition of Wnt/β-catenin signaling could alleviate silica-induced fibrosis in mice. 23,24 Such an attenuation of silica-induced pulmonary fibrosis was also reported by targeting Wnt/β-catenin signaling using microRNA-29c expressed in bone marrow mesenchymal stromal cells in rats. 57 As a key regulator of Wnt signaling, epithelial-derived DKK1 could alter the pathogenic effects of Wnt in IPF. 64 Together with the findings from IPF lungs, 65 this led us to test the potential of recombinant adenovirus-mediated gene transfer of Dkk1 to treat silicosis in a mouse model. Our results demonstrated an ameliorated degree of silica-induced fibrosis, coinciding with the reduction of EMT, ECM deposition, and hydroxyproline accumulation in silicosis mouse lungs administered AdDkk1 2 days before and 14 days after they were challenged with silica.
It is worth noting that although the gross histology showed an ameliorated pathogenesis of silicosis in the lungs of mice infected with AdDkk1 compared with AdBglII null virus at both d28 and d56 post the silica challenge (Fig. 4B), a statistically significant amelioration was only observed in AdDkk1-infected silicosis lungs at d56 (d42 post the viral infection) but not at d28 post the silica challenge, as ascertained by silicotic nodular area and hydroxyproline content (Fig. 4C, D). We reasoned that the Dkk1 gene transduction at an early stage of silicosis pathogenesis could alleviate the disease progression and ultimately yield a therapeutic effect in the silica-induced silicosis mouse lungs at late time points. Despite the pathogenesis being ameliorated in the lungs of mice infected with AdDkk1 relative to the AdBglII null virus at d28 (14 days post the gene transfer), it reached a statistically significant difference between these two viral infections. Therefore, it is worthwhile to examine the therapeutic effect of Dkk1 gene transfer in the late stages of silicosis mouse lungs (such as d28 and/or d56 post the silica challenge) in future studies. Nonetheless, this proof-of-concept study, thus, provides evidence that the viral vector-mediated Dkk1 gene transduction in lungs has the potential to alleviate silicosis progression, although the specific mechanism underpinning the anti-fibrotic effect of Dkk1 in the silicosis mouse lung needs further investigation.
It has been well recognized that Wnt signaling is crucial for tissue or organ homeostasis by regulating cell proliferation and differentiation and tissue regeneration on injury. Activation of Wnt/β-catenin signaling usually promotes epithelial proliferation, and inhibition of the signaling by Dkk1 leads to suppression of Wnt-mediated cell proliferation and regeneration in the pathogenesis of IPF. 30 This suggests that the balance between Wnt/Dkk1 signaling may be critical for a proper function of stem/progenitor cells, such as basal cells during airway epithelial injury repair. 69 In addition, in vitro studies demonstrated that primary bronchial epithelial cells are potentially responsive to Wnt signaling, as chronic Wnt activation led to epithelial cell senescence. 70 By contrast, the Wnt inhibitor DKK1 was able to regulate the human bronchial and alveolar epithelial cell line proliferation in a dose-dependent manner. 65 These results mirrored our finding in A549 cells on a silica stimulation. Further, exposure to silica activated Wnt/β-catenin and TGF-β/Smad signaling in mouse lungs. Moreover, the silica-induced Wnt activity was enhanced by AdWnt3a-mediated expression of Wnt3a, which coincided with the increased expression of EMT markers and ECM proteins in both A549 cells and polarized primary HBECs. However, forced expression of Dkk1 inhibited the silica-activated Wnt and TGF-β signaling, and it reduced the expression of EMT markers. The exposure of silica and AdWnt3a infection alone was able to induce the proliferation of primary HBECs in ALI cultures. The infection of AdDkk1 reduced cell proliferation to pre-silica exposure levels in HBEC cultures. These findings suggest that Wnt/β-catenin signaling positively regulates TGF-β signaling and EMT pathogenesis in silicosis. 71
Despite us showing that adenovirus-mediated Dkk1 gene transfer could suppress the pathogenesis of experimental silicosis lung in mice, there are limitations to this proof-of-concept study. First, although DKK1 and WNT3A proteins were elevated in patients diagnosed with pneumoconiosis, the lack of pneumoconiosis lung biopsy, particularly the silicosis lung, limited us to assess the Wnt/β-catenin signaling in silicosis lungs in clinical settings. Second, this study focused on adenovirus-mediated gene transfer because adenoviral vectors are among the most efficient vectors and are able to infect a broad range of tissues and cell types. However, there are several drawbacks to adenoviral vectors, such as their ability to provide only transient gene expression, their potential to evoke strong inflammatory responses due to pre-existing immunity in humans. 72 The helper-dependent AAV is much less likely to evoke an immune response in humans. 73 In terms of AAV-mediated gene transfer, great efforts have been placed in developing novel vectors that are able to penetrate the airway mucus barrier 74 and delivery approaches of AAV delivery in the respiratory tract. 75 For instance, experimental studies of AAV-mediated telomerase transduction demonstrated a therapeutic potential for treatments of pulmonary fibrosis and aging-related IPF. 40,41 Interestingly, telomere shortening and gene variants were also found in patients with silicosis. 76 Third, in this study, we tested the potential of AdDkk1-mediated gene transfer as a prophylactic treatment to prevent silicosis and reduce fibrogenesis of an early stage of silicosis in mice. As such, the vector was delivered 2 days before the mice were challenged with silica or 14 days post the challenge when the silicosis was developed. Prophylactic treatment may be appropriate in scenarios where silica exposure is highly eminent and help us to better understand the mechanism of pathogenesis of silicosis, but such a real-world scenario is unlikely. Thus, it remains necessary to investigate whether AdDkk1 treatment mitigates silicosis if administered at various timepoints after silica exposure. Fourth, although our results demonstrated the potential of gene transfer of Dkk1 in the treatment of silicosis at some extent in mice, it remains unclear as to whether the therapeutic approach with gene transfer is an effective means for the treatment of established silicosis in clinic settings. Reducing occupational exposure to respirable crystalline silica obviously is the best way to prevent future cases of silicosis. Finally, to more precisely identify how the dysregulation of Wnt signaling contributes to the pathogenesis of pneumoconiosis in human patients, continued collection and analysis of clinically obtained specimens are necessary to achieve a larger aggregate sample size. These studies will also enhance our understanding of the mechanism of pathogenesis of this currently uncurable disease.
CONCLUSION
Circulating levels of WNT3A and DKK1 were elevated in patients with pneumoconiosis and specifically silicosis. Elevated Wnt/β-catenin signaling activity was observed in the lungs of silica-induced silicosis mice. The adenoviral vector-mediated Dkk1 gene transduction displayed potencies in both prophylactic treatment and therapy of early onset of silicosis lung disease in mice, by alleviating silicosis pathogenesis, including reduced EMT and ECM deposition in lungs. AdDkk1 treatment ameliorated silicosis development through a mechanism by partially suppressing the TGF-β/Smad signaling response to silica. In addition, Dkk1 inhibited silica-induced epithelial cell proliferation in A549 cells and polarized primary HBECs. Thus, this article provides evidence that viral vector-mediated gene delivery of DKK1 in lungs can ameliorate silicosis. This proof-of-concept provides an entry point to develop gene therapies for the treatment of exposure-induced pneumoconioses such as silicosis.
ETHICS STATEMENT
All human studies were performed according to protocols approved by the Institutional Review Boards (ethics committee for conduction of human research) of General Hospital of Ningxia Medical University (NXMU-2020-487), and all animal experimentation was performed according to protocols approved by the Institutional Animal Care and Use Committee of Ningxia University (NXULS20180123-3).
Footnotes
AUTHORs' CONTRIBUTIONS
X.L. conceived and designed the experiments; Q.C., J.M., J.W., D.Y., J.Y., and J.X. conducted the experiments, acquired and analyzed data; J.Y.W., Z.W., N.W., J.Q.W., S.W., and F.L. collected and analyzed clinic samples; Q.C. drafted the article; and J.C. and X.L. interpreted data and critically revised the article. All authors read and approved the final version of the article.
ACKNOWLEDGMENTS
The authors thank Dr. Engelhardt at the University of Iowa for providing adenoviral vectors used in this study and for scientific discussions. They especially thank Dr. Thomas J. Lynch at the University of Iowa for English editing and scientific discussions.
AUTHOR DISCLOSURE
The authors declare no competing financial interests.
FUNDING INFORMATION
This work was financially supported by grants from the National Natural Science Foundation of China (Nos. 31860318, 32070863, 81760004, 81860566) and a grant from the Natural Science Foundation of Ningxia (No. 2018AAC03171). Funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
SUPPLEMENTARY MATERIAL
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.