
Abstract
Severe acute respiratory syndrome (SARS) is a newly emerging infectious disease (COVID-19) caused by the novel coronavirus SARS-coronavirus 2 (CoV-2). To combat the devastating spread of SARS-CoV-2, extraordinary efforts from numerous laboratories have focused on the development of effective and safe vaccines. Traditional live-attenuated or inactivated viral vaccines are not recommended for immunocompromised patients as the attenuated virus can still cause disease via phenotypic or genotypic reversion. Subunit vaccines require repeated dosing and adjuvant use to be effective, and DNA vaccines exhibit lower immune responses. mRNA vaccines can be highly unstable under physiological conditions. On the contrary, naturally antigenic viral vectors with well-characterized structure and safety profile serve as among the most effective gene carriers to provoke immune response via heterologous gene transfer. Viral vector-based vaccines induce both an effective cellular immune response and a humoral immune response owing to their natural adjuvant properties via transduction of immune cells. Consequently, viral vectored vaccines carrying the SARS-CoV-2 spike protein have recently been generated and successfully used to activate cytotoxic T cells and develop a neutralizing antibody response. Recent progress in SARS-CoV-2 vaccines, with an emphasis on gene therapy viral vector-based vaccine development, is discussed in this review.
INTRODUCTION
The novel coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) first emerged in December 2019 in China as an outbreak of pneumonia, but soon spread to other countries leading to a serious worldwide pandemic. 1,2 The outbreak was declared a public health emergency of international concern on January 30, 2020, and a pandemic on March 11, 2020, by The World Health Organization. Because of its genetic similarity to bat coronaviruses, the virus is believed to have emerged from a bat-borne virus with a zoonotic origin. 3 Based on epidemiological studies conducted with the Wuhan strain, each infection is estimated to result in 5.7 new ones in the absence of immunity or vaccination. 4 COVID-19 disease symptoms include (but are not limited to) fever, dry cough, breathing difficulties (dyspnoea), headache, and pneumonia leading to eventual death (Fig. 1). 5 Acute lung injury, systemic inflammatory response syndrome, and acute respiratory distress syndrome (ARDS) have been observed in patients with COVID-19 similar to what has been described in SARS-CoV and Middle East Respiratory Syndrome (MERS)-CoV-infected patients. 6 Patients with severe COVID-19 exhibited a cytokine storm characterized by the overproduction of inflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-12, interferon (IFN)-γ, and tumor necrosis factor-α. 5 The cytokine storm is responsible for causing ARDS, a systemic inflammatory response, and multiple organ failure. 7 Contrary to an anticipated inflammatory response as a result of a viral infection, a cytokine storm can cause serious damages to patients' health leading to high mortality rates if not handled properly. 8 As of April 2021, more than 133 million coronavirus cases in 220 countries were reported with 2.9 million causalities around the world.
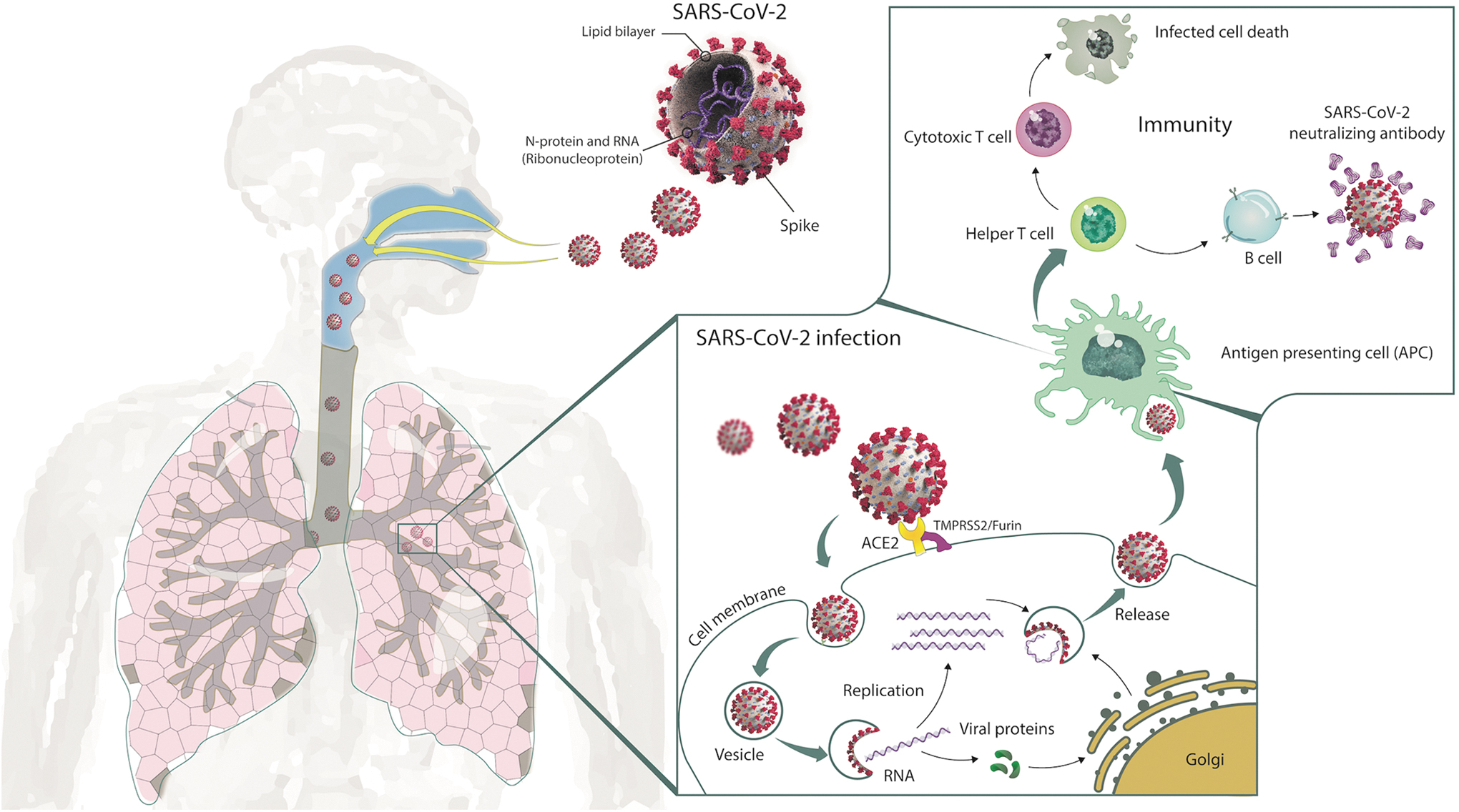
The mechanisms of host cell entry and immune system activation by SARS-CoV-2. 113 The life cycle and main route of transmission of SARS-CoV-2 are also depicted. Direct, indirect, or close contact with infected people may result in the transmission of SARS-CoV-2 via respiratory droplets expelled from an infected person through coughing, sneezing, or talking. Angiotensin I-converting enzyme-2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) act as receptors involved in SARS-CoV-2 (or 2019-nCoV) entry into host target cells. Spike is cleaved by TMPRSS2 protease on the cell surface to ensure virus and host-cell membrane fusion. Then SARS-CoV-2 virus replicates within the cell and passes to the lower airways potentially leading to severe pneumonia. Antigen-presenting cells deliver viral antigens to immune cells for the activation of humoral and cellular immune responses. Helper T cells mediate cytotoxic T cell generation and activation leading to the destruction of SARS-CoV-2 infected cells. Neutralizing antibodies produced from the activated B cells bind and interfere with the SARS-CoV-2 binding to the targeted respiratory cells. The duration of protection depends on the presence and longevity of memory T and B cells in circulation. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Vaccination is the most effective way for protection against infectious diseases, and the development of a vaccine against SARS-CoV-2 has been a priority. 9 A guidance document on COVID-19 vaccines has recently been issued by The United States Food and Drug Administration. 10 An ideal vaccine is expected to have an excellent safety profile across multiple population groups (e.g., immunocompromised individuals, children, older adults, pregnant women, and so on). It should ideally induce protective immunity within 2 weeks, and it should demonstrate at least 70% efficacy with minimal adverse events. Furthermore, long-lasting protection that engages both humoral and cellular immune responses lasting for at least 1 year is desired. The vaccine should also be stable at room temperature to avoid cold chain and transportation issues, and it should be amenable to mass production. Even though more than 90 SARS-CoV-2 vaccines have been under development by scientists in companies and universities around the globe, the process of vaccine development can not be rushed and may take even longer than expected. It is noteworthy to mention that more than 70% of the scientists leading vaccine research are from industrial or private firms. Furthermore, vaccine development might be hampered due to differences in severity, transmission rate, and the geographic distribution of different viral strains. To date, 149 mutations have been detected in 103 SARS-CoV2 strains sequenced, revealing L and S subtypes and these numbers are increasing each day. 11 For example, SARS-CoV-2 with a D614G substitution in the spike protein appeared in early 2020. 12 Soon after, the D614G mutation successfully substituted the SARS-CoV-2 strain originally discovered in China, becoming the dominant form of the virus around the globe. 13 Despite the increased infectivity and transmission rate, the strain with the D614G substitution neither caused severe illness nor modified the effectiveness of vaccines or therapeutics. 14 Another SARS-CoV-2 variant namely SARS-CoV-2 VOC 202012/01 recently emerged in the United Kingdom on December 14, 2020. This variant possessed 23 nucleotide substitutions leading to increased transmissibility without affecting disease severity. By the end of 2020, VOC-202012/01 variant (lineage B.1.1.7) had been reported in 31 other countries. 15 The other observed variants such as B.1.351 (501Y.V2, South African variant) 16 and P.1 (501Y.V3, Brazilian variant) 17 were also claimed to be more transmissible. Despite the variants, B.1.1.7, B.1.351, and P.1 did not manifest enhanced host cell entry, variants B.1.351 and P.1 but not B.1.1.7 could escape from therapeutic antibodies (monoclonal antibodies with emergency use authorization [EUA] for COVID-19 treatment) or antibodies induced by natural infection or vaccination (convalescent plasma and sera). 18 These results suggest that the B.1.351 and P.1 variants might be able to successfully spread even in vaccinated individuals or convalescent patients elevating the risk for human health.
The development of an effective SARS-CoV-2 vaccine also requires the understanding of the molecular structure of the virus, since proper antigen and the vector type are essential to inducing optimum immune responses following vaccination. 19 SARS-CoV-2 is a member of Betacoronaviruses sharing 79.5% sequence identity to SARS-CoV and 50% homology to the MERS coronavirus. SARS-CoV-2 carries a positive-strand RNA genome of nearly 30 kbp in length (Fig. 2A, B). The 5′ end of the genome contains a long ORF1ab polyprotein consisting of 15–16 nonstructural proteins, 20 while the 3′ end of the genome encodes four major structural proteins, including the spike (S), nucleocapsid (N), membrane (M), and the envelope (E). 21 Importantly, all of these viral structural proteins may serve as antigen candidates for vaccine development as described below.

Virion structure and genome composition of SARS-CoV-2 virus.
187
Assessment of viral structural proteins for vaccination studies
Even though SARS-CoV-2 has an affinity for angiotensin-converting enzyme 2 (ACE2)-expressing epithelial cells of the respiratory tract, systemic hyperinflammation coordinated by the adaptive immune system involving T cell-mediated cellular and B cell-mediated humoral immunity has been observed in patients with severe COVID-19. 22 Following infection with the virus, viral peptides are loaded and presented on the major histocompatibility complex (MHC) I of epithelial cells, monocytes, and dendritic cells (DCs). Cytotoxic T lymphocytes recognize the viral peptides and kill virus-infected cells by inducing apoptosis. Viral peptides presented on MHC II activate CD4+ helper T cells leading to the release of cytokines such as IL-2 and IL-6. Activation and proliferation of virus-specific B cells generate plasma cells secreting immunoglobulin M, immunoglobulin G (IgG), and immunoglobulin A antibodies that mediate virus neutralization. During SARS-CoV-2 infection, an antigen-specific immune response generated by activated T and B cells is essential for clearing the virus and protecting the patient from a deadly infection. As a result of uncontrolled virus replication, SARS-CoV-2 dampens antiviral IFN responses to evade the innate immune response. The infiltration of monocytes/macrophages, neutrophils, and adaptive immune cells results in the overproduction of proinflammatory cytokines. 23 Systemic inflammation eventually leads to vasodilation resulting in inflammatory lymphocytic and monocytic infiltration of the lung and heart. Pathogenic T cells secreting granulocyte-macrophage colony-stimulating factor have been implicated in the recruitment of inflammatory IL-6-secreting monocytes and severe lung pathology in patients with COVID-19. 24 The severity of the inflammation has been attributed to the magnitude of the cytokine storm—especially IL-1β, IFN-γ, CXCL10, and CCL2. 25 Thus, the analysis of SARS-CoV-2 structural protein-derived antigenic peptides is essential to produce a vaccine to achieve optimum stimulation of innate and adaptive immune responses to restrain the spread of SARS-CoV-2. 26
Whole-cell antigens (WCA), such as proteins, lipids, polysaccharides, and nucleic acids, of the virus are mainly used in the development of either dead or live-attenuated vaccines. 27,28 Although WCA is a quick and easy way of producing a vaccine and routinely used in many laboratories worldwide, quality assurance and efficacy studies are unreliable due to their complex nature. Conversely, the spike protein appears to be the most promising antigen candidate in SARS-CoV-2 vaccine development for the following reasons. First, spike proteins of SARS-CoV and MERS-CoV viruses have already been shown to be effective for vaccination. 29 –33 Second, the spike protein is present on the virus surface, which can easily be recognized by the host defense system resulting in the activation of a robust immune response against the virus itself. 34 Finally, the spike protein mediates viral transduction via interaction with the ACE2 receptors followed by endocytosis. 35 These results suggested that vaccines based on the spike protein could induce antibodies that prevent the virus from both binding to the host cell and fusing with the host cell membrane. Compared to all structural proteins of SARS-CoV, the spike protein appeared to be the main immunogenic protein capable of inducing both cellular and humoral immunity against virus infection. Intriguingly, the structure of spike protein carries some clues for the development of an effective vaccine against SARS-CoV-2 as explained below.
SARS-CoV-2 spike protein, in monomeric form, consists of 1,273 amino acids and has a molecular weight of about 140 kDa (Fig. 2B). Spike protein contains two subunits (S1 and S2) and can naturally form a trimer. Moreover, the spike protein plays essential roles in viral transduction and pathogenesis; specifically, the S1 subunit recognizes and binds to host ACE2 receptors, and then the S2 subunit undergoes conformational changes resulting in the fusion of the viral envelope with the host cell membrane. 30 While the receptor-binding domain (RBD) of the spike protein is located at the C-terminal domain of the S1 subunit, membrane fusion is mediated by the fusion peptide (FP) located in the S2 subunit. 36 The discovery of prefusion conformation of spike protein in trimeric form and the interaction of RBD with ACE2 receptors drastically influenced vaccine design against SARS-CoV-2. 34,35
Various fragments of the spike protein (the full-length spike protein, the RBD domain, the S1 subunit, N-terminal domain [NTD], and FP) have been all individually tested as antigens in vaccine development with varying degrees of success. Full-length spike proteins in their natural forms are expected to serve as better antigens as compared to truncated versions due to the presentation of more immunogenic epitopes. As expected, numerous vaccine candidates based on the full-length spike protein of SARS-CoV have already been reported. For example, the full-length spike protein SARS-CoV Urbani strain encoded by a DNA vaccine generated both protective neutralizing antibody and T cell responses in mice. 37 Mice immunized with baculovirus-produced spike protein complexed with nanoparticles in an alum adjuvant formulation yielded a high titer of neutralizing antibodies. 38 Vaccination of mice or monkeys with attenuated modified vaccinia Ankara (MVA) virus encoding the full-length spike protein of the SARS-CoV Urbani strain or HKU39849 strain-induced neutralizing antibodies and decreased viral titers in the respiratory tracts of animals after homologous SARS-CoV challenge. 39,40 Mice, camels, and rhesus macaques immunized with a DNA vaccine encoding MERS-CoV spike protein had reduced typical clinical symptoms of the disease during the infection. 41 Finally, vaccination of mice and hamsters with a full-length S protein trimer protected the animals from the homologous SARS-CoV (HKU39849 strain) challenge. 42 These data collectively highlight the potential for the highly immunogenic full-length S protein to elicit neutralizing antibodies capable of suppressing virus proliferation and protection against SARS-CoV challenge. However, it is important to keep in mind that SARS vaccines encoding the full-length S protein may also activate destructive immune responses yielding liver and lung damage of the vaccinated animals by antibody-dependent enhancement (ADE). 30,43 –45 The region of the SARS-CoV-2 spike protein responsible for the generation of harmful immune responses remains unknown, and ADE has not been detected in cases where effective neutralizing antibody formation was achieved.
Since the RBD of the spike protein in the S1 subunit interacts with ACE2 receptors on the host cell surface, antibodies generated by RBD-based vaccines are expected to interfere with virus binding and entry into the cells. 46 Most of the SARS-CoV 47 and MERS-CoV 48 subunit vaccines were developed using the RBD domain carrying multiple conformational neutralizing epitopes as an antigen. 49 Consequently, intramuscular and mucosal administration of an adeno-associated virus (AAV)-based vaccine encoding RBD (RBD-rAAV) stimulated satisfactory neutralizing antibody production that inhibited homologous SARS-CoV (GZ50) challenge in mice. 50,51 However, this approach has been disputed since coronaviruses can display antibody escape mutations in the RBD domain. 52 Because of this reason, the use of different epitopes of SARS-CoV-2 to induce an effective immune response has been advised to avoid the immune escape of the virus. Other virus surface proteins, except the E Protein (E Protein), are suitable antigens with varying degrees of effectiveness for vaccine development, such as the NTDs, S1 Subunit, the FP domain of the S2 subunit, N Protein, and M Protein. 19
SARS-Co V-2 VACCINES UNDER DEVELOPMENT
Live-attenuated or inactivated vaccines
Live-attenuated vaccines prepared from weakened viruses under laboratory conditions have been used since the 1950s. 53 These vaccines can replicate in a vaccinated individual and produce an immune response with mild or no disease. Compared to an infection with the wild-type virus, live-attenuated viral vaccines can generate an exceptional immune response providing recurrent antigenic stimulation needed for the generation of memory cells. However, there are some safety concerns regarding the use of these vaccines. Live-attenuated pathogens have the potential (although rare) to revert to a pathogenic form and cause disease in vaccinated individuals. Furthermore, individuals with compromised immune systems may not be able to respond effectively. For instance, the formalin-inactivated poliovirus vaccine (IPV) developed by Jonas Salk was licensed for use in 1955. 54 While polio was finally brought under control after implementing the use of IPV, making Jonas Salk a national hero in the United States, failure to properly inactivate IPV in the early batches resulted in vaccine-associated outbreaks of polio—known as Cutter incident. 55 In the 1960s, Albert Sabin developed attenuated versions of the oral poliovirus vaccine (OPV) by passaging virulent viruses through different animals and cells. 56 However, the Sabin infectious/attenuated poliovirus vaccines also caused vaccine-associated paralytic poliomyelitis in a small number of recipients. 57 Since properly inactivated Salk vaccine did not accidentally cause poliomyelitis, OPV was eventually replaced with Salk's vaccine as recommended by federal authorities in 1999. 58 Nevertheless, Codagenix, Inc., in collaboration with the Serum Institute of India, Ltd. agreed to develop a live-attenuated vaccine against SARS-CoV-2. COVI-VAC, which is a single-dose intranasal, live-attenuated vaccine against SARS-CoV-2, was developed with Codagenix's Synthetic Attenuated Virus Engineering (SAVE) platform using synthetic biology to recode the genes of viruses into a safe and stable vaccine. 59 This vaccine is currently being tested in a randomized, double-blind, placebo-controlled, dose-escalation trial involving 48 volunteers. 60
Whole-cell killed/inactivated vaccines are made from viruses that have been killed through physical (heat) or chemical (formaldehyde) processes. Individuals vaccinated with whole-cell killed or live-attenuated vaccines may display diverse immune responses against the pathogen since these vaccines present various immunogenic components to the host. 61 Because whole-cell killed/inactivated vaccines are considered to be traditional vaccines, easy to make and produce, they can quickly enter into clinical trials. Several institutions around the world have successfully isolated SARS-CoV-2 strains and immediately initiated traditional vaccine development to save time. China approved a clinical trial for a candidate COVID-19 vaccine named CoronaVac developed by Sinovac in mid-April 2020. 62 CoronaVac, which is a chemically inactivated whole virus vaccine for COVID-19, has since entered Phase 3 clinical trials in Brazil, Chile, Indonesia, Philippines, and Turkey. 9 The Butantan Institute has revealed the vaccine efficacy to be 50.4% in Brazil. 63 Another inactivated virus-based COVID-19 vaccine known as Covaxin (BBV152) has been developed by Bharat Biotech in collaboration with the Indian Council of Medical Research. 64 This whole-virion inactivated SARS-CoV-2 vaccine produced in Vero cells was prepared with a Toll-like receptor7/8 agonist molecule adsorbed to alum. The safety and tolerability of Covaxin were revealed in the phase 1 vaccine trial, which evaluated 375 participants in four groups. 65 Tolerable safety outcomes and enhanced humoral- and cell-mediated immune responses were observed in phase 2 clinical trial involving 380 participants. 66 A randomized, double-blinded, placebo-controlled Phase 3 study involving 25,800 participants between 18 and 98 years of age covering a total of 22 sites across India was initiated in November 2020. On January 3, 2021, the Drugs Controller General of India granted the EUA for the vaccine, which is still in clinical trials. As of March 2021, the interim efficacy rate for the phase 3 trial was reported to be ∼81%. 67
mRNA vaccines
Nucleic acid therapeutics, particularly those involving mRNA constructs, have appeared as promising alternatives to traditional vaccine approaches in recent years. Major technological innovation and research in mRNA synthesis, modification, and delivery technology have allowed mRNA to become a promising therapeutic approach in vaccine development. Compared to live-attenuated, whole-cell killed/inactivated vaccines, including DNA-based approaches, mRNA-based vaccines offer several benefits. Because mRNA is noninfectious and does not integrate into the genome, there is no potential risk of infection or insertional mutagenesis. The half-life and immunogenicity of the mRNA in cells can easily be modulated through genetic modification and delivery methods. 68 Covalent conjugates, protamine complexes, nanoparticles based on lipids or polymers, and hybrid formulations can be formulated into carrier molecules for efficient in vivo delivery of mRNA. 69 Furthermore, mRNA vaccines can be administered repeatedly without any concern over antivector immunity. Rapid, inexpensive, and scalable manufacturing of mRNA are the other benefits of using mRNA-based vaccines as nucleic acid therapeutics. 70 mRNA-based vaccine development involves the selection of antigen, sequence optimization, and modification, assessment of immune response, and safety following administration. 71 Storage and handling requirements related to the need for freezing temperatures due to rapid degradation and increased reactogenicity in a subset of vaccinated individuals regarding side effects such as fever, muscle aches, and fatigue appeared to be the main limitations to mRNA-based vaccine platforms. 72
BioNTech in cooperation with Pfizer has developed an mRNA-based COVID-19 vaccine sold under the brand name Comirnaty (Tozinameran code-named BNT162b2). The Pfizer-BioNTech COVID-19 vaccine is composed of nucleoside-modified mRNA (modRNA) encoding a mutated form of the spike protein of SARS-CoV-2 (D614G), which is encapsulated in lipid nanoparticles. Modification of select nucleosides was necessary to suppress intrinsic immunogenic properties of the mRNA of interest. Structural engineering of the modRNA and nucleoside modification converted a virus mRNA encoding spike protein into a surrogate cellular mRNA with higher protein expression efficacy. BioNTech has demonstrated that the modification of select nucleosides in the manufactured mRNA suppresses its intrinsic immunogenic properties while resulting in superior protein production. Clinical trials of the Pfizer-BioNTech COVID-19 vaccine began in April 2020. 73 After testing a total of 43,548 participants at 152 sites worldwide by December 2020, a two-dose regimen of the Pfizer-BioNTech COVID-19 vaccine conferred 95% protection against Covid-19 in persons 16 years of age or older. 74 This vaccine became the first COVID-19 vaccine to be authorized by a stringent regulatory authority for emergency use in individuals 16 years of age and older on December 11, 2020 and was the first to be cleared for regular use. 75
The Moderna COVID-19 vaccine, known as mRNA-1273, is an mRNA-based COVID-19 vaccine developed by the National Institute of Allergy and Infectious Diseases, Moderna, and the Biomedical Advanced Research and Development Authority. This vaccine uses a technology involving a modRNA compound to induce immunity to SARS-CoV-2 by encoding a prefusion-stabilized spike protein naturally present on the surface of SARS-CoV-2 particles. 76 On December 30, 2020, the Phase 3 randomized, observer-blinded, placebo-controlled clinical trial enrolling 30,420 volunteers was conducted at 99 centers across the United States demonstrating 94.1% efficacy in preventing COVID-19 infection, including severe disease. 77 Apart from transient local and systemic reactions, no safety concerns were revealed. The Moderna COVID-19 vaccine was issued an EUA by the United States Food and Drug Administration (FDA) on December 18, 2020. Despite the Moderna and the Pfizer-BioNTech vaccines having similar efficacy, the Moderna vaccine requires storage at the temperature of 2–8°C for up to 30 days or −20°C for up to 4 months, whereas the Pfizer-BioNTech vaccine requires ultracold freezer storage between −80°C and −60°C. Although differences in lipid nanoparticle formulations or mRNA secondary structures were reasoned for the thermostability differences between the two vaccines, variations in storage requirements and shelf lives do not negatively impact their efficacy. 78
DNA vaccines
DNA vaccines offer several benefits compared with conventional live-attenuated vaccines; for example, they induce both cellular and humoral immunity without replication, enable the creation of a vector encoding different antigens in a single preparation, and large-scale but low-cost manufacture. 79 Preserving the quality of the live vaccines requires cold storage, but DNA vaccines are highly stable without the need for refrigeration at low temperatures. DNA-based vaccines comprised plasmids encoding one or more antigens. Compared to mRNA-based vaccines, they are more stable, but carry insertional mutagenesis risks due to vector integration. 80 Intriguingly, all current clinical trials of DNA vaccines utilize the delivery of the spike protein as the antigen. 81 Inovio Pharmaceuticals (Plymouth Meeting, PA) developed a DNA vaccine using a plasmid pGX9501 encoding the SARS-CoV-2 spike protein as the antigen (INO-4800). The vaccine administered intradermally through electroporation is now under a phase 1 open-label study to evaluate the safety, tolerability, and immunogenicity of INO-4800.
Subunit vaccines
Similar to inactivated whole-cell vaccines, subunit vaccines do not contain live virus particles and are considered very safe. However, they only contain the antigenic parts of the virus to elicit a protective immune response different from the inactivated whole-cell vaccines. To achieve this, antigenic properties of the various subunits of the virus must first be studied in detail to determine which particular mixtures will yield an effective immune response. Nevertheless, the generation of a strong protective immune response requires the addition of adjuvants. Currently, almost all SARS-CoV-2 subunit vaccines under testing use the spike protein as the antigen. Clover Biopharmaceuticals, Inc., developed a subunit vaccine candidate against SARS-CoV-2 using the Trimer-Tag technology for the expression of trimeric spike protein in mammalian cells. 82 After completing primate studies, a Phase 1 randomized, observer-blind, placebo-controlled clinical trial involving 150 adult and elderly participants was started in June 2020 to assess the safety and immunogenicity of the adjuvanted COVID-19 S-Trimer vaccine at multiple-dose levels. A similar subunit vaccine based on the “molecular clamp” technology was also developed by the University of Queensland in collaboration with a global biotech company CSL. 83 A molecular clamp is a polypeptide-based technology designed to preserve the prefusion configuration of peptides to generate a better immune response in experimental vaccines. However, this vaccine failed to proceed to further clinical trials due to “false positive” results to human immunodeficiency virus (HIV) tests. 84
Peptide-based vaccines are chemically synthesized small fragments of intact antigens. They are very easy to make and produce in large quantities. Unfortunately, due to their small size, they often exhibit weak immunogenicity requiring detailed structural modification and the addition of adjuvants. 85 Currently, many peptide-based SARS-CoV-2 vaccine candidates are being used in clinical trials. 86
Viral vector-based vaccines
One of the effective tools for controlling infectious diseases is genetic immunization by way of induction of strong protective immunity. DCs acting as professional antigen-presenting cells (APCs) trigger strong T and B cell-mediated immunity. Delivery of antigens into DCs ex vivo, and then transfer into patients was the first method used in DC-based vaccines developed against infectious diseases. The discovery of viral vectors paved the way to the virus-mediated direct transfer of antigens in vivo leading to prolonged immunity. 87 Viral vector-based vaccine development against infectious agents involves the introduction of antigenic full-length or truncated viral surface protein-encoding genes (e.g., SARS-COV2 spike) into viral expression vectors. After verification of viral vector-driven antigen expression in cell lines, immunization in preclinical animal models is carried out to assess the vaccine efficacy before conducting clinical trials. 88 In this scenario, viral vectors serve as gene transfer vehicles generating immune responses by way of heterologous gene transfer. Because they are naturally immunogenic and well-characterized, they soon became the preferred vector of choice in vaccine development. 89 The most used viral vectors for vaccination studies are adenovirus (Ad), AAV, poxvirus, and lentiviruses. Viral vector-based vaccines generate a very strong cellular immune response, as infection of immune cells leads to the stimulation of humoral immune response due to their inherent adjuvant properties. Compared to conventional vaccines, they can be modified for specific targeting and provide prolonged antigen presentation. The potential application of viral vectors in humans ranges from infectious diseases to cancer treatments. High levels of recombinant protein expression is achievable by viral vectors, which provided the basis for modern vaccine development. One benefit of using viral vectors to transport vaccine antigens is that the live viral vectors may generate robust mucosal humoral immune response 90 much better than what is accomplished by other vaccine candidates, such as protein and DNA vaccines. 91,92
Ad structure and vector design strategy
Ad is a nonenveloped, double-stranded DNA virus with a 90–100 nm icosahedral capsid composed of penton and hexon subunits. Attachment to host cells is mediated by fiber and knob domains, while the penton base is involved in the secondary interactions required for virus entry into the cell (Fig. 3A). Based on the virus serotype, the affinity of the knob domain varies primarily depending on the use of coxsackievirus Ad receptor, CD46, and various integrins for cellular entry. Ads possess a linear dsDNA genome of 36 kb (26–48 kb), categorized into early (E1, E2A, E2B, E3, and E4), intermediate (IVa2 and IX), and late (L1, L2, L3, L4, and L5) genes flanked by inverted terminal repeats (ITRs) sequences (Fig. 3B). Since E1 is involved in virus replication and E3 interferes with the host immune response, E1 deletion in association with E3 results in the generation of replication-deficient Ad called first-generation vectors creating a transgene capacity of 6.5 kb for cloning, just as the E1A/E1B deletion frees up space for a transgene that is 4.5 kb in length. 93 The E2 and E4 regions of Ad play essential roles in viral DNA replication and viral mRNA export, respectively. Deletion of these regions in addition to E1 and E3 reduces immunogenicity and prevents leaky viral gene expression, which led to the second-generation Ad vectors. Unfortunately, apart from increasing transgene capacity to 10.5 kb, second-generation Ad vectors have had limited use since these vectors do not provide much benefit over their first-generation counterparts. Despite prolonged transgene expression, second-generation Ad vectors can still trigger host immunogenicity and cellular toxicity. 94 The removal of the entire protein-coding region of the Ad backbone, except for the ψ packaging signal and flanking ITR sequences, results in the generation of gutless viruses called third-generation vectors. These changes are essential to decrease the immunogenicity of the vector while prolonging transgene expression and improving the safety of Ad vectors. Gutless vectors have a cargo-carrying capacity of 36 kb with a minimized chance of producing replication-competent virus. Current adenoviral vectors specific for COVID-19 primarily use the first-generation Ad, which only supplies short-term transgene expression in vivo. However, if prolonged transgene expression is desired without sacrificing the natural adjuvant property of the virus, then gutless adenoviral vectors might be preferred so the vector-transduced cells only express the vaccine antigen (spike) and not Ad antigens (Fig. 3C).
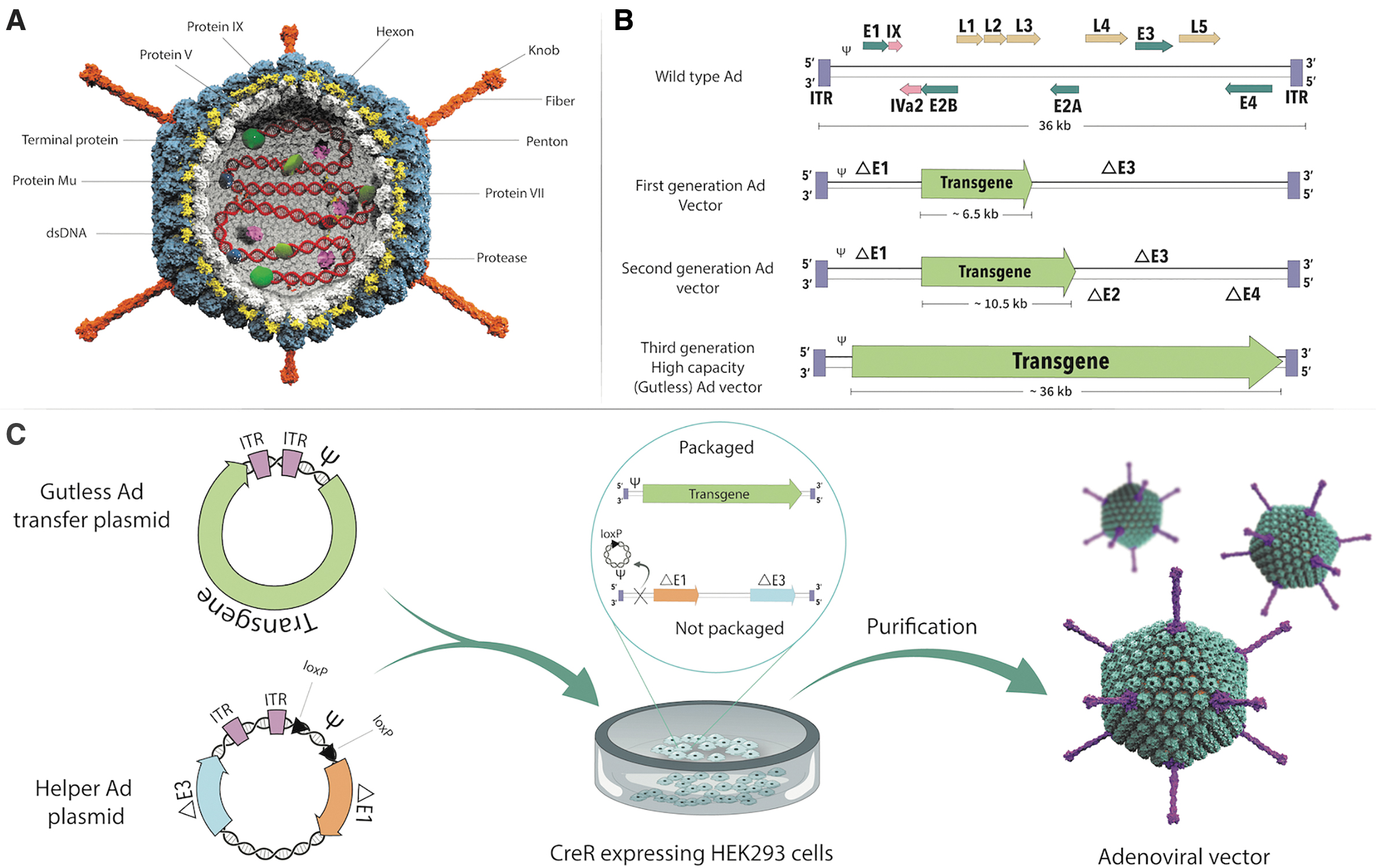
Ad structure and vector design strategy.
189
Among the gene therapy vectors tested, Ad vectors are the most effective gene delivery vehicles in carrying foreign antigens to host cells. 95 Compared to other virus-based gene delivery systems such as lentivirus, retrovirus, and AAVs, they are more immunogenic and activate both the innate and adaptive immune response. 96 Moreover, they are the preferred vector of choice in cancer gene therapy applications. 97 –105 Contrary to stable and high levels of transgene expression required in target cells for classical gene therapy approaches, transient antigen expression might be clinically more beneficial for vaccination purposes. In comparison to short-term overexpression, prolonged low levels of antigen expression minimize antigen-induced apoptosis of T cells and increase the persistence of memory T cells. 106 By this token, Ad vectors provide only transient gene expression due to their antigenicity. Ad-based vectors can function as a “self-adjuvant,” leading to the activation of multiple innate immunostimulatory pathways following infection, which may boost the immunogenicity of the encoded antigen. Conversely, the triggering of undesired innate immunity pathways also has the potential to be damaging to their existence resulting in the clearance of Ad-transduced cells. 107 Vigilant engineering of adenoviral vectors is needed to use Ad vectors for both gene therapy and vaccination purposes. Besides, their natural affinity for upper airway epithelial cells makes them useful in intranasal vaccination against mucosal surface-infecting viruses such as influenza and SARS-CoV-2. Ad-based vaccines can be used as nasal sprays making them practical to use in mass. 108
Ad-based vaccines
Compared to other viral vectors, Ad vector-based vaccines are easy to design and produce on a mass scale, which is of paramount significance for clinical use. In addition to inherent adjuvant properties, and the ability to induce robust transgene-specific T and B cell responses, Ad vectors are easy to purify to high titer, are genetically stable, and can be delivered via oral, intramuscular, intradermal, and aerosol routes. 109 Ad triggers several innate immune signaling pathways resulting in the secretion of several proinflammatory cytokines. These proinflammatory cytokines pave the way to effective immune cell stimulation leading to the induction of robust adaptive humoral and cellular immune responses. Recent studies have demonstrated that Ad-vector vaccines induced better humoral and cellular immune responses than recombinant protein vaccines, plasmid-based DNA vaccines, and other recombinant vector systems currently available (Table 1). Since the nasal mucosa is the first line of defense against SARS-CoV-2 before viral spread to the lung, Ad-mediated intranasal vaccination might be an attractive strategy to prevent COVID-19. An intranasal Ad type 5 (Ad5)-vectored vaccine encoding the RBD of the SARS-CoV-2 spike protein called AdCOVID has activated both systemic and local immune responses successfully inducing mucosal immunity against COVID-19 in mice. 110
Adenovirus-based severe acute respiratory syndrome coronavirus 2 vaccines under development
Ad, adenovirus; EUA, emergency use authorization; FDA, Food and Drug Administration; NA, not available.
Building on CanSinoBIO's Ad-based viral vector vaccine technology platform, Convidicea (Ad5-nCoV) has become the first novel coronavirus vaccine for COVID-19 in a clinical trial in China. 111 This vaccine is a recombinant Ad5-vectored COVID-19 vaccine expressing the spike glycoprotein of SARS-CoV-2. Immunogenicity and safety of a recombinant Ad5-vectored COVID-19 vaccine in healthy adults aged 18 years or older were assessed in a randomized, double-blind, placebo-controlled phase 2 trial involving 508 eligible participants. 112 The Ad5-vectored COVID-19 vaccine (consisting of 5 × 1010 viral particles) was safe and induced significant immune responses in the majority of recipients after a single immunization. In February 2021, data obtained from an interim analysis of Phase 3 trials involving 30,000 participants and 101 COVID cases revealed that the vaccine had an efficacy of 65.7% at preventing moderate cases of COVID-19 and 90.98% efficacy at preventing severe cases. This vaccine is currently licensed for use in the Chinese military. In February 2021, China approved the vaccine for general use.
Johnson and Johnson (J&J) and Janssen Pharmaceutical research teams in collaboration with Beth Israel Deaconess Medical Center, constructed a vaccine candidate
The University of Oxford in collaboration with AstraZeneca has developed a replication-deficient simian Ad vector (ChAdOx1) carrying the full-length codon-optimized coding sequence of the structural spike protein of SARS-CoV-2 with a tissue plasminogen activator (tPA) leader sequence as an nCoV-19 vaccine (AZD1222). The safety, reactogenicity, and immunogenicity of a chimpanzee Ad-vectored vaccine (ChAdOx1 nCoV-19) expressing the spike protein of SARS-CoV-2 were assessed in a phase 1/2, single-blind, randomized controlled trial in healthy adults aged 18–55 years in five trial sites across the United Kingdom. 114 Results indicated the ChAdOx1 nCoV-19 vaccine given at a dose of 5 × 1010 viral particles was safe, well-tolerated, and immunogenic. Even though a single dose was sufficient to provoke both humoral and cellular responses against SARS-CoV-2 without serious adverse reactions, booster immunization was needed to enhance neutralizing antibody titers paving the way for phase 3 trials. Based on this encouraging data, phase 3 trials were launched in the United Kingdom (2020-001228-32), Brazil (ISRCTN89951424), United States (NCT04516746), Russia (NCT04540393), and India (CTRI/2020/08/027170). 115 The interim analysis of the clinical trial in the United Kingdom (2020-001228-32) and Brazil (ISRCTN89951424) demonstrated the nCoV-19 vaccine ChAdOx1 to have an average efficacy of 70%, based on analyzing a total of 131 COVID-19 cases from 11,636 volunteers as revealed on November 23, 2020. However, 90% efficacy was achieved using the dosing regimen where the vaccine was initially given as a half dose followed by a second full dose (n = 2,741). This finding is very intriguing considering that the regimen consisting of two full doses had only 62% efficacy (n = 8,895). The most suitable regimen of AZD1222 awaited further clinical trials with the hope of resolving the dose-response discrepancy between different subgroups. Due to a severe adverse reaction (transverse myelitis) observed in a previously healthy 37-year-old woman, AstraZeneca gave a voluntary pause to its phase 3 COVID-19 vaccine trial in the United Kingdom on September 9, 2020. Accordingly, scientists advised caution and pointed out the necessity for addressing the safety concerns. 116 After a brief pause, AstraZeneca resumed the trial in the United Kingdom on September 12, 2020. The vaccine has been approved by the United Kingdom, Argentina, El Salvador, India, and the Dominican Republic regulatory authorities for emergency usage. However, there are still some concerns regarding the possibility of dominant immunogenicity toward the Ad vector genes rather than the transgenes. On January 29, 2021, Oxford/AstraZeneca vaccine was recommended for the European Medicines Agency approval for granting conditional marketing authorization for people 18 years of age and older. Even though both the AZD1222 and Janssen COVID-19 vaccines use spike protein as an antigen, differences in transgene design (tPA attached spike vs the stabilized prefusion spike) and vector choice (chimpanzee vs. human) might account for differences in efficacy and dose requirements (one vs. two-dose regiments) to achieve protection from COVID-19.
Gam-COVID-Vac, trade-named Sputnik V and developed by Gamaleya Research Institute Epidemiology and Microbiology, Health Ministry of the Russian Federation, is a viral two-vector vaccine (rAd26-S + rAd5-S) based on two human Ad vectors, which encode spike protein of SARS-CoV-2 to stimulate an antiviral immune response. The Ad26-based vaccine is used for the initial immunization, and the Ad5 vaccine is given 21 days later to boost the immune response. The safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine were studied in Phase 1–2 trials involving 76 participants from Russia. The results suggested that the heterologous rAd26 and rAd5 vector-based COVID-19 vaccine has a good safety profile and induced strong humoral and cellular immune responses in participants. On August 11, 2020, Russia was the first country to approve a vaccine against COVID-19 (Sputnik V). However, scientists around the world have raised concerns about the clinical trials conducted to evaluate its safety and efficacy. 117 As of December 2020, only Belarus and Argentina have received EUAs for Gam-COVID-Vac outside of the Russian Federation. The federal authorities announced the start of a large-scale free-of-charge vaccination program with Gam-COVID-Vac for Russian citizens in early December 2020. Currently, Gam-COVID-Vac is in Phase 3 trials involving more than 40,000 participants in Russia and Belarus with the intent of expanding to various other countries such as UAE, India, Venezuela, Egypt, and Brazil. An interim analysis of the safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine was recently revealed in a randomized controlled phase 3 trial (NCT04530396) involving 25 hospitals and polyclinics in Moscow, Russia. 118 91.6% efficacy against COVID-19 was reported in this interim analysis of Gam-COVID-Vac study involving 21,977 enrolled adults between September 7 and November 24, 2020.
Lentivirus structure and vector design
Lentiviruses comprising nine species (seven animal and two human lentiviruses) are enveloped single-stranded RNA viruses belonging to the Retroviridae family (Fig. 4A). HIV-1 is the most extensively characterized human lentivirus used as the backbone for lentiviral-vectored gene therapies. 119,120 Compared to retroviral vectors, lentiviral vectors have superior properties, such as the ability to transduce diving and nondividing cells, high infection efficiency, and low probability of insertional mutagenesis. 121 Contrary to AAV (weakly immunogenic) and Ad-vectored (highly immunogenic) therapies, lentiviral vectors are fairly nonimmunogenic and provide long-term gene expression due to permanent integration into the host genome. Two copies of the viral RNA genome and some other viral enzymes such as reverse transcriptase and integrase are present within the icosahedral viral capsid. The viral RNA genome is ∼9.7 kb in size and encodes a total of nine genes flanked by 5′ and 3′ long terminal repeat (LTR) sequences. Structural proteins such as the viral nucleocapsid and matrix are encoded by GAG, while reverse transcriptase, integrase, and protease functions are encoded by POL. Surface glycoproteins necessary for host cell receptor recognition are encoded by ENV (Fig. 4B). The regulatory genes tat and rev play essential roles during viral replication, whereas the remaining accessory genes nef, vif, vpr, and vpu are needed to increase infectivity, but they are dispensable for viral vector preparation.
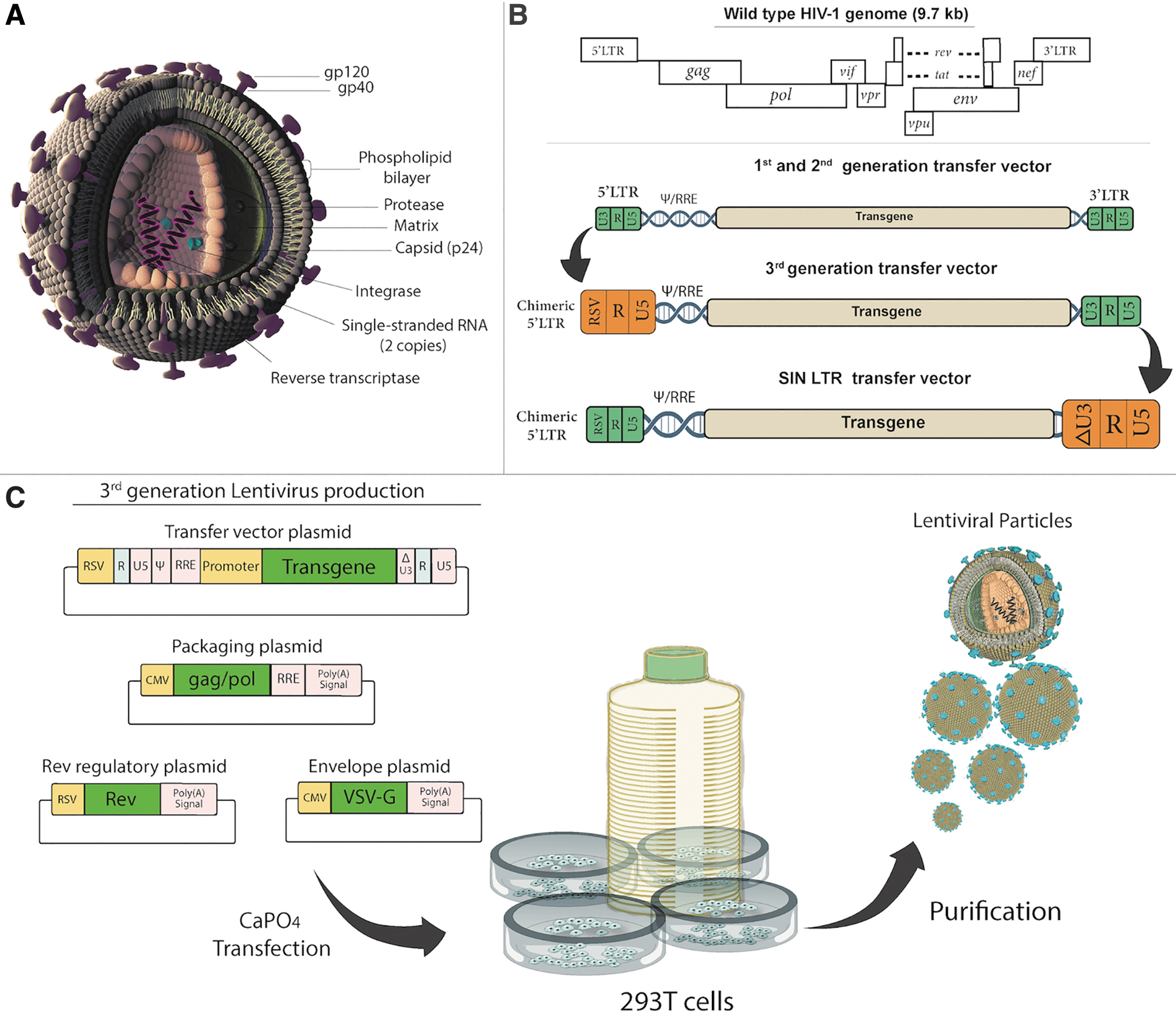
Lentivirus structure and vector design strategy
First-generation lentiviral vector production involves the use of three plasmids, including a transfer plasmid, a packaging plasmid, and an envelope plasmid. Transfer plasmid contains the lentiviral vector genome composed of the wild-type 5′ and′ LTRs, the ψ sequence, a part of the env gene containing the rev response element, an internal promoter, and the transgene. 122 The packaging plasmid harbors the HIV-1 genome with all viral genes except the env gene. The envelope plasmid contains envelope proteins from another virus such as the vesicular stomatitis virus G (VSV-G) protein that improves the stability and widens the cellular tropism of the viral particles produced. Unfortunately, the first-generation vector system is considered to have an unacceptably high risk of generating replication-competent viral particles (RCLs) via recombination. 123 Second-generation vectors were developed by the removal of virulence factors (nef, vif, vpr, and vpu) after the discovery that these accessory proteins were not needed for efficient viral replication and production. 124 Removal of the accessory genes does not interfere with the transfer of genetic material to the host cell. Second-generation lentiviral vectors are safer than first-generation vectors and can be used routinely in a scientific research laboratory. To rule out even the theoretical possibility of creating RCL, third-generation vectors were developed to use lentiviral vectors not only in a laboratory but also in a clinical setting (Fig. 4C). 125 In this scenario, the promoter of the 5′LTR has been deleted to reduce its activity and Rev is expressed from a separate plasmid in addition to the removal of the HIV Tat gene used to drive expression from the LTRs. Furthermore, an RSV or CMV promoter is commonly inserted in this LTR to transcribe the viral genome in producing cells. Deletion of the promoter/enhancer elements in the 3′ LTR resulted in self-inactivating (SIN) third-generation vectors unable to transcribe full-length RNA after integration, further improving safety. Since third-generation vectors require only three HIV-1 genes (gag, pol, and rev) for production, 126,127 this system offers the best safety profile. 128 Consequently, VSV-G pseudotyped third-generation SIN lentiviral vectors became the preferred vector of choice to achieve long-term gene expression in vivo. 129,130 Integration-deficient lentiviral vectors were developed to rule out the risk of insertional mutagenesis completely. 131 These integrase-deficient lentiviral vectors have mutations in the catalytic domain of the viral integrase, preventing the integration of vector cDNA into the host genome. Instead, they produce circular forms, which are lost eventually due to cell division, since they do not replicate inside the cell.
Lentivirus-based vaccines
DCs are the most effective immune-stimulatory cells that induce antigen-specific T cell responses against infectious pathogens in vivo. Contrary to nonviral gene delivery approaches, viral vectors are the most effective gene delivery vehicles for stimulating T cell response. 132 However, the dominant immunity against the viral antigens might represent a clear handicap to establish optimal transgene-specific immune response. Even though higher levels of transgene expression are desired for optimal immunogenicity, the efficacy of Ad-vectored vaccines in clinics is limited due to preexisting and/or acquired immunity to Ad, that is, following natural infection or previous vaccination. In contrast, lentiviruses demonstrated superior properties at inducing T cell-mediated immune response compared to other viral vectors (e.g., Ad), such that 2- to 10-fold higher transduction levels were achieved in human and murine DCs using lentiviral vectors 133,134 without interfering with the antigen presentation machinery of DCs. 135 Based on these studies, 10- to 100-fold more Ad vector was needed to achieve the same levels of transduction attained with lentivirus vectors. In conclusion, lentivirus-mediated gene transfer to DCs results in effective antigen presentation and activation of transgene-specific cytotoxic T cells. 133,136 The immune responses elicited via direct injection of lentivirus vectors appear to be less dependent on CD4+ T cells in terms of primary and memory CTL response. 137 Compared to vaccinia vectors, lentivirus-mediated CD8+ T cell responses have been reported to last longer. 138 The fact that lentivirus vectored immunization yielded more CD127+ antigen-specific CD8+ T cells, compared to peptide-based applications, suggests lentiviral vectors generate an effective memory T cell response. 139
Since antigen-specific T cells not only eliminate cancer cells but also virally infected cells, the expansion of a large population of T cells with anti-SARS-CoV-2 viral antigen specificity might be helpful to stop an ongoing COVID-19 infection. 140 Similarly, genetically modified APCs expressing the conserved domains of the viral structural proteins carried by the lentivirus are expected to stimulate the differentiation of naive T cells into effector cytotoxic T cells. By this token, clinical trials are underway evaluating the safety and efficacy of genetically modified APCs alone and in combination with antigen-specific cytotoxic T cells (NCT04299724, NCT04276896). These trials involve the testing of multiple Covid-19 minigenes using a lentiviral vector system to express viral proteins and immune-modulatory genes to modify DCs leading to the activation of T cells.
Preclinical studies of lentivirus-mediated spike gene delivery against COVID-19 have recently begun. A lentiviral vector encoding the full-length, membrane-anchored form of SARS-CoV-2 spike glycoprotein was generated to test the vaccine efficacy of lentivirus-mediated spike gene delivery in a preclinical animal model of COVID-19. 141 The animal model was generated by transduction of respiratory tract cells using Ad expressing the SARS-CoV-2 receptor hACE2. Systemic injection of lentivirus carrying spike gene in mice resulted only in partial protection despite high levels of serum neutralizing activity and T cell responses. Intriguingly, an intranasal boost strategy generated a stronger localized immune response in the upper respiratory tract yielding complete protection against SARS-CoV-2 in rodents.
AAV structure and vector design
AAVs are naturally nonpathogenic, nonenveloped, and small DNA viruses (25 nm in size) of the Parvoviridae family (Fig. 5A). 142 AAV is a replication-defective virus requiring a helper virus such as Ad to initiate productive infection. AAV2 genome size is nearly 4.7 kbp and the genome encodes four open reading frames (ORFs), Rep (Replication), Cap (Capsid), aap (Assembly), and maap (membrane-associated accessory protein) flanked by two 145 base ITRs (Fig. 5B). 143,144 The complementary DNA strand is synthesized from these ITRs. 145 At least 13 serotypes of AAV have been identified, 146 but most vector systems utilize AAV2 ITR as the packaging signal. Type 2 ITRs can be packaged with a variety of capsid types leading to further refinement of the tissue tropism of AAV vectors. 147 –149 Translation of Rep and Cap using different promoters and alternative splicing generates multiple distinct gene products (Rep78, Rep68, Rep52, and Rep40—required for the AAV life cycle; VP1, VP2, and VP3-capsid proteins). 150 Four replication proteins are encoded from the first ORF, while the second ORF encodes three capsid proteins. The assembly-activating protein (AAP) and the recently discovered membrane-associated accessory protein (MAAP) with unknown function are encoded from the third and fourth ORFs, respectively. 93 AAP is derived from the aap gene in an alternate reading frame overlapping the cap gene. The expression of the AAP together with capsid proteins VP1, VP2, and VP3 is needed for AAV capsid assembly. 151 AAV transfer plasmid is constructed by placing the transgene between the two ITRs and cotransfection of the transfer plasmid with Rep and Cap supplied in trans in addition to an adenoviral helper plasmid is needed to produce AAV particles in 293 cells (Fig. 5C).
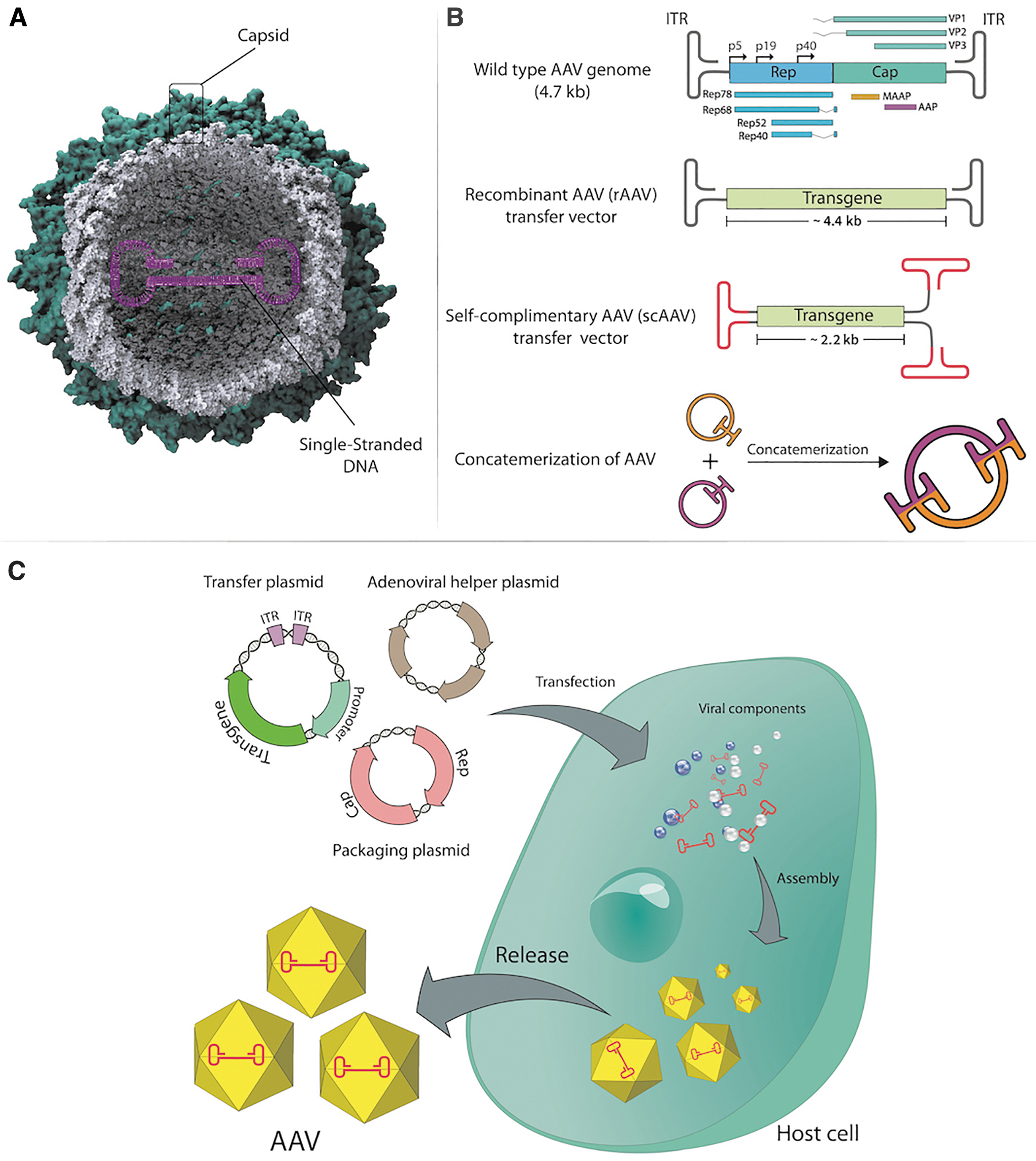
AAV structure and vector design strategy.
The conversion of ssDNA genomes to dsDNA is one of the rate-limiting steps in AAV transduction. 152 This causes delays in transgene expression because the vector depends on the DNA replication machinery of the host cell to synthesize the complementary strand. Consequently, second-generation self-complimentary (sc) AAV vectors were developed to resolve the inefficient process of second-strand DNA synthesis causing low transduction efficiency with AAV vectors. 153 scAAV vectors possess complementary sequences capable of spontaneously annealing following transduction, which abolishes the requirement for host cell DNA synthesis. However, this approach reduces the packaging capacity of AAV to 2.2 kb (Fig. 5B). Trans-splicing of AAV vectors allowed for the cloning of larger transgenes into AAV vectors. 154 In this strategy, the transgene is cloned into two AAV transfer plasmids, the first with a 3′ splice donor and the second with a 5′ splice acceptor. Following cotransduction of target cells by these two AAV vectors, viral genomes form concatemers through trans-splicing allowing for the full-length transgene expression. Homologous recombination relying on substantial sequence overlap between two transfer plasmids is another method used to increase the packaging capacity of AAV vectors.
AAV-based vaccines
AAV-based gene therapy vectors have a significant potential for treating viral infectious diseases. The rAAV-based vaccine (RBD-rAAV) encoding the RBD of SARS-CoV S protein, which is a major target of neutralizing antibodies, was generated. 155 A single dose of RBD-rAAV in BALB/c mice resulted in the production of sufficient neutralizing antibodies to protect against SARS-CoV infection. Compared to intramuscular injection, a single intranasal prime dose vaccination with RBD-rAAV could induce a detectable systemic humoral immune response, but the addition of a booster immunization resulted in a much stronger and prolonged mucosal immune response with neutralizing activity. 51 Furthermore, RBD-rAAV priming and boosting with RBD-specific peptides for T cell epitopes increased both humoral and cellular immune responses against SARS-CoV infection in female BALB/c mice. 50
AAV-based COVID-19 vaccines are currently in early-stage development. On May 5, 2020, Massachusetts Eye and Ear and Massachusetts General Hospital delivered a press release on the testing of an experimental vaccine called AAVCOVID, which is being developed in the laboratory of Luk H. Vandenberghe, PhD, director of the Grousbeck Gene Therapy Center at Massachusetts Eye and Ear at Harvard Medical School. The AAVCOVID vaccine candidate uses AAV vector technology to deliver the SARS-CoV-2 spike antigen-encoding gene to induce a protective immune response against COVID-19. The preclinical safety and immunogenicity of two novel AAV vector-based COVID-19 vaccines were recently reported. 156 In this particular study, AAVrh32.33, a novel vector developed from rhesus macaques isolates with highly reduced antibody prevalence in humans, 157 was utilized to express a full-length SARS-CoV-2 S protein locked in a prefusion conformation and a truncated version of SARS-CoV-2 S protein designed to be secreted. Both vaccine candidates manifested robust long-lived humoral and cellular immunogenic responses following a single intramuscular injection in rodents and nonhuman primates. Furthermore, AAVCOVID vaccine candidates could be stored at room temperature for up to 1 month without losing their potency and effectiveness. These vaccine candidates are currently in preclinical trials and testing in humans is expected following the completion of animal experiments.
In November, a cocktail of two of Regeneron's laboratory-made antibodies, casirivimab and imdevimab, received an EUA from the FDA to treat COVID-19 via intravenous infusion. 158 This is a recombinant monoclonal antibody cocktail intended to yield resistance to the SARS-CoV-2 coronavirus. The University of Pennsylvania in collaboration with Regeneron Pharmaceuticals, Inc., agreed to test whether Regeneron's casirivimab and imdevimab investigational antibody cocktail could stop COVID-19 transmission when delivered intranasally using AAV vectors. 159 In cases where vaccines are not effective due to mutations, delivering the AAV vector through a nasal spray was proposed to express well-characterized anti-SARS-CoV-2 antibodies in cells that line the nose and throat to neutralize COVID-19.
Current hurdles regarding immune responses to gene therapy viral vectors used in vaccination
The suitability of a viral vector for gene therapy relies on many factors, including intrinsic immunotoxicity of the vector, in vivo versus ex vivo gene transfer, target organs, tissue tropism, cargo capacity, and the potential for genome integration concerning insertional mutagenesis. Viral vectors used in gene therapy might be indistinguishable from their wild-type counterparts to the immune system exposing them to similar innate and adaptive immune responses. Thus, the immune response to viral vectors represents one of the most important hurdles to clinical gene therapy. 160 Upon recognition of viral structures such as nucleic acids and capsids, innate immune cells stimulate the secretion of IFN-α/β resulting in reducing viral transduction and activation of the adaptive immune response. Activation of DCs and antigen presentation lead to further expansion and differentiation of T cells. While MHC I-restricted CD8+ T cells recognize and lyse virus-infected cells, MHC II-restricted CD4+ T cells (helper T cells) participate in the generation of memory CD8+ T cells and B cells leading to antibody production.
Ad is one of the first viruses to be explored as a potential gene therapy vector due to its high transduction efficiency and packaging capacity. Despite high levels of transgene expression, severe immunotoxicity associated with a high degree of inflammatory response resulted in transient gene expression and limited use against genetic diseases. 161 On the contrary, since they strongly activate CD8+ T cells, adenoviral vectors are favored in vaccination and cancer gene therapy applications. Ease of manipulation, their capacity for high titer growth, and intrinsic antigenic properties have made nonreplicating Ad vectors a preferred vector for vaccination studies. 95 However, preexisting immunity to Ad-based vectors also represents a handicap for repeated use in vivo. To overcome this, a variety of Ads isolated from humans (Ad types-26 and 5) and chimpanzees (simian adenovirus ChAd serotype Y25), which have low seroprevalence in humans, have been cloned, modified as a gene transfer vector, and tested as vaccines. 107 Compared to other viral vectors, AAV manifested superior qualities for in vivo gene transfer for vaccination due to the availability of a variety of viral capsids with diverse tropism, low immunogenicity, and satisfactory safety profile. 162 Despite these features, the seroprevalence issue is still a great concern in AAV-mediated gene therapy or vaccination approaches. This is why patients are screened for neutralizing Ab titers against the AAV vector capsid before enrollment in clinical trials. In addition to alternative serotypes or capsid modifications, the use of decoy capsids or plasmapheresis has been advised to overcome preexisting neutralizing Ab. 163,164 On the contrary, Ad type-26 (Janssen COVID-19 vaccine) and simian adenovirus ChAd serotype Y25 (AstraZeneca-AZD1222) vaccinated individuals do not require screening for preexisting neutralizing antibody due to low seroprevalence of vaccine vectors in the human population.
In vivo gene transfer to DCs is a new strategy in vaccine development, and HIV-based lentiviral vectors are also becoming very popular owing to their effectiveness in DC transduction. 165 Generation of integrase deficient lentiviral vectors for vaccination was essential to relieve safety concerns regarding integration. 166 Pseudotyping of lentiviral vectors appeared to be the main method of targeting and induction of immune response to vaccine antigens. 167 Finally, preexisting immunity to lentiviral vectors is low in humans, which further increases their appeal for use in vaccination.
Other viral vectors under development against the COVID-19
Live-attenuated viral vectors have the potential to induce long-lasting protective immunity after a single dose and are low-cost to manufacture at a large scale. 168 Owing to safe history, well-established manufacturing processes, and induction of strong, long-lasting immunity, the measles virus-vectored (MeV) vaccine is an attractive candidate for the development of an effective vaccine against COVID-19. Thus, MeV vaccine candidates expressing the full-length SARS-CoV-2 spike glycoprotein have been generated. 169 The SARS-CoV-2 S protein-encoding gene sequence was inserted into two different positions of the MeV genome to control antigen gene expression. Both protective neutralizing IgG antibody responses and cytotoxic T cell responses with S protein-specific killing activity were detected in vaccinated hamsters and mice. In another study, the MeV-based SARS-CoV-2 vaccine using the prefusion stabilized, full-length spike antigen, but not the S2 subunit, generated Th1-dominant T cell and neutralizing antibody responses leading to long-term immunity and protection from SARS-CoV-2 challenge in mice. 170 A Randomized, Placebo-controlled Phase 1 trial is underway to evaluate the safety and immunogenicity of the Measles Vector-based Vaccine Candidate Against COVID-19 in Healthy Volunteers (NCT04497298).
MVA is a vastly weakened poxvirus vector broadly used to develop vaccines for infectious diseases and cancer. Poxviruses are easy to modify since they require a little manipulation to yield robust protein expression. A novel vaccine platform based on a unique three-plasmid system to effectively produce recombinant MVA vectors from chemically synthesized DNA (sMVA) was generated in response to the ongoing global pandemic caused by SARS-CoV-2. Strong SARS-CoV-2 antigen-specific humoral and cellular immune responses in addition to potent neutralizing antibodies were obtained from mice immunized with these sMVA vectors. 171 The safety, tolerability, and immunogenicity of the Candidate Vaccine MVA-SARS-2-S is underway in an open, single-center Phase 1 trial (NCT04569383).
VSV, belonging to the Rhabdoviridae family, is a nonsegmented single-stranded negative-sense RNA virus. Due to their replicating nature, VSV-based vaccines can be deployed as a single-dose regimen to efficiently stimulate both humoral and cellular immunity. Recombinant VSV-based vaccines encoding viral glycoproteins manifest several advantages as a vaccine platform such as efficient, simple, and large-scale production in mammalian cell culture in addition to the absence of preexisting immunity to the vector. The immunogenicity and in vivo efficacy of a replication-competent VSV-eGFP-SARS-CoV-2 was tested in a mouse model of COVID-19. 172 A single dose of VSV-eGFP-SARS-CoV-2 injection was sufficient to produce a vigorous neutralizing antibody response in BALB/c mice that neutralized SARS-CoV-2 infection and decreased viral load in lung and peripheral organs. In another study, a recombinant replication-competent VSV-based vaccine candidate expressing the SARS-CoV-2 S protein (rVSV-ΔG-spike), in which the glycoprotein of VSV is replaced by the spike protein of SARS-CoV-2, was generated. 173 A single-dose vaccination of the golden Syrian hamster, which is used as a model for SARS-CoV-2 pathogenesis and transmission, with rVSV-ΔG-spike revealed a safe, effective, and sufficient neutralizing antibody response against SARS-CoV-2 challenge paving the way for clinical trials (NCT04608305). The International AIDS Vaccine Initiative, a nonprofit scientific research organization, and Merck developed SARS-CoV-2 vaccine candidate V590 using the rVSV vector-based platform. Due to inferior immune responses observed in Phase 1 clinical trial (NCT04569786, unpublished results) compared to those seen following natural infection and to those reported for other SARS-CoV-2 vaccines, the development of the SARS-CoV-2 vaccine candidate V590 was discontinued.
REMAINING CONCERNS RELEVANT TO COVID-19 VACCINE DEVELOPMENT AND CONCLUDING REMARKS
Due to the COVID-19 pandemic, we are experiencing an accelerated track for vaccine development. We are not even sure if the SARS-CoV-2 infection will protect us from future infection and how long protection will last. Currently, it is unclear if vaccine-induced immune responses are long- or short-lived than immune responses induced by natural infection. What is clear is that the best vaccine candidates are expected to induce durable, long-term protective cellular and humoral immune responses to prevent the need for boost injections over time. Vaccine candidates demonstrating short-term immunogenicity might undermine the feasibility of vaccine application in humans. A single dose vaccine regiment is preferred compared to a multiple-dose regimen to reduce the cost, increase compliance and minimize the manufacturing requirements. Most of the currently developed vaccines against COVID-19 utilize spike protein of the SARS-CoV-2 virus. However, RNA viruses accumulate mutations that can sometimes interfere with vaccine-induced immunity, as previously seen with influenza viruses. The recent unexpected high mutation rate of the SARS-CoV-2 RNA virus may eventually overshadow or diminish the efficacy of the first-generation vaccines. 174 Almost all of the vaccine candidates currently in clinical trials are injected intramuscularly inducing strong IgG responses. Luckily, viral vectors have the benefit of intranasal application with the potential of inducing strong mucosal immune responses in addition to IgG responses.
Long-term data concerning vaccination studies are also imperative to evaluate if there is any risk of vaccine-associated enhanced disease (VAED). 175 VAED is a disorder that would arise when an immunized individual subsequently infected with a virus develops a more severe illness than they would have had if they were not immunized. 176 Intriguingly, instead of providing protection, adenoviral vectors used as HIV vaccines have enhanced the susceptibility to HIV infection due to previous exposure to Ad infection in vaccinated subjects. 177 ADE is another concern that remains to be explored as was observed in previous respiratory syncytial virus and dengue virus vaccine studies, which revealed human clinical safety risks. 178 In addition to ongoing research into SARS-CoV-2-induced immunity (innate, humoral, cellular), the efficacy and longevity of vaccine-induced protection need to be established. In particular, we are not sure how to protect the elderly from lethal variants and heterologous SARS-CoV-2 strains. Since it is more challenging to achieve neutralizing antibody titers against infection in older individuals for protection, different vaccine formulations involving a virus-vectored prime-boost regimen, in particular, might be needed to augment immune responses in individuals from this age group. There is also preexisting cross-reactive immunity issue between CD4+ T cells specific for SARS-CoV-2 and CD4+ T cells specific for human common cold coronaviruses. Adults who had been exposed to common cold coronaviruses may manifest preexisting cross-reactive immunity to SARS-CoV-2 antigens resulting in differing susceptibility to SARS-CoV-2 infection. 179
Currently, vaccine supply is not sufficient to meet the worldwide demand and encounter the first wave of the pandemic for most countries. Yet, future waves are also anticipated since SARS-CoV-2 is expected to circulate as a seasonal virus around the globe. With the current limitations on vaccine supply, many nations are asking to what degree the recommended dosing regimen of COVID-19 vaccines could be altered (dose-sparing strategies) without impacting effectiveness. 180 For instance, the trials of the Oxford-AstraZeneca vaccine concluded that a longer gap between doses (2–3 months) led to greater immune responses. 115,181 Andrew Pollard, the head of the Oxford Vaccine Group and chief investigator into the trial of this vaccine, claimed: “a longer gap between vaccine doses leads to a better immune response, with the second dose causing a better boost.” The Oxford vaccine trials involving dose comparison of the second dose after 4 weeks versus the second dose after 2–4 months, revealed 70% protection after the first dose up to the second dose, and the immune response being three times greater after the second dose when the second dose was delayed. 182 Accordingly, on December 30, 2020, the four United Kingdom chief medical officers recommended the second doses of the covid vaccines should be given toward the end of 12 weeks rather than in the previously recommended 3–4 weeks. 183 Concerning both the Pfizer-BioNTech and Oxford-AstraZeneca vaccines, the Joint Committee on Vaccines and Immunization, along with the United Kingdom Chief Medical Officers have approved a prolonged second dosing interval up to 12 weeks to spread the initial phases of vaccination to a greater number of people. 184,185 Furthermore, pooled analysis of four randomized trials of the ChAdOx1 nCoV-19 (AZD1222) vaccine revealed that, contrary to the expectation, a prolonged dosing interval is linked to superior efficacy and better postdose-2 antibody titers. 186 Last but not least, SARS-CoV-2 may not be the last coronavirus to cause a pandemic requiring constant virus surveillance, as well as ongoing vaccine development.
Footnotes
Authors' Contributions
A.B. and Y.E.E. drafted the review article. S.S. designed and Y.E.E. composed the figures. A.D.S. and T.S.G. edited and improved the review content. S.S. supervised the study. All authors commented on the article.
ACKNOWLEDGMENTS
This work is supported by the Akdeniz University Scientific Research Administration Division and the Small and Medium Enterprises Development Organization of Turkey (KOSGEB-42MUD).
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
No funding was received for this study.