
Abstract
Adeno-associated viruses (AAVs) are advantageous as gene-transfer vectors due to their favorable biological and safety characteristics, with discovering novel AAV variants being key to improving this treatment platform. To date, researchers have isolated over 200 AAVs from natural sources using PCR-based methods. We compared two modern DNA polymerases and their utility for isolating and amplifying the AAV genome. Compared to the HotStar polymerase, the higher-fidelity Q5 Hot Start High-Fidelity DNA Polymerase provided more precise and accurate amplification of the input AAV sequences. The lower-fidelity HotStar DNA polymerase introduced mutations during the isolation and amplification processes, thus generating multiple mutant capsids with variable bioactivity compared to the input AAV gene. The Q5 polymerase enabled the successful discovery of novel AAV capsid sequences from human and nonhuman primate tissue sources. Novel AAV sequences from these sources showed evidence of positive evolutionary selection. This study highlights the importance of using the highest fidelity DNA polymerases available to accurately isolate and characterize AAV genomes from natural sources to ultimately develop more effective gene therapy vectors.
INTRODUCTION
Adeno-associated viruses (AAVs) are safe and effective vehicles used in gene transfer for several clinical indications. 1 The U.S. Food and Drug Administration has approved AAV-mediated gene therapy drugs for treating spinal muscular atrophy and Leber congenital amaurosis. 2,3 These approved gene therapy products, as well as many others currently under development, utilize AAV capsids isolated from natural sources as the delivery vehicle. 4 The AAV genome consists of two major open reading frames (ORFs)—Rep and Cap—which encode sequences for translating multiple protein products. The Cap ORF translation occurs from multiple start sites to produce three AAV structural proteins: VP1, VP2, and VP3. These structural protein subunits are assembled into icosahedral virions, 5 which carry a genetic payload to their target. The sequence and structural diversity of AAV capsid genes contribute to the variability in viral tropism, antigenicity, and packaging efficiency observed among viral clades. 6 Thus, discovering novel capsids with an array of tissue tropisms is necessary to advance the efficacy and utility of gene therapy.
Researchers have isolated AAV Cap sequences from natural sources using a variety of techniques that have emerged and evolved over time. The most common approach involves PCR amplification. In some cases, extracted viral DNA can be directly sequenced; this method resulted in the identification of AAV2, 7 which propagates with helper adenovirus in cell culture. In addition, extracted viral DNA can be cloned into a plasmid backbone, and sequenced (AAV1, 8 AAV3, 9 AAV3B and AAV6, 10 and AAV511). Viral genomes can also be amplified through PCR, and the amplicons can be cloned into plasmids before Sanger sequencing. Researchers have isolated many AAVs from primate, 12,13 bovine, 14 porcine, 15 rodent, 16 and other species using this method. Next-generation sequencing (NGS) analyses of mammalian genomic DNA have detected fragments of endogenous AAV genomic elements. 17 More recently, metagenomic virome sequencing studies—which use shotgun NGS to simultaneously sequence thousands of DNA molecules in complex samples—have identified many novel AAV sequences. 18,19
Using PCR to amplify AAV is a straightforward and effective means for discovering novel AAV capsid sequences. However, it is important to use PCR enzymes with high-fidelity replication capabilities to amplify the viral sequences as accurately as possible. Enzymes with high misincorporation and template-switching rates can significantly confound sequencing data and interfere with novel AAV capsid discovery. 20 Indeed, the artificial variability introduced by low-fidelity polymerases, while amplifying capsid sequences, can impair the study of AAV biology and diversity due to amplification errors that skew the “true” genetic variation in a sample.
In this study, we aimed to compare multiple AAV PCR methods to screen tissue samples for AAV natural isolate genomes to expand the breadth of capsid sequences available for characterization as potential gene delivery vectors. Discovering more capsids increases the chance of successfully identifying those that can transfer therapeutic transgenes to a range of tissues at high efficiency, have reduced immunogenicity at high doses, and have less prevalent neutralizing antibody profiles in the human population than existing AAV capsids. Given that DNA polymerase technology has undergone significant development since the last wave of AAV discovery almost 20 years ago, we compared two modern DNA polymerases and amplification methods to isolate AAV sequences. We found that the Q5 Hot Start High-Fidelity DNA Polymerase produced PCR products from the input templates at higher accuracy compared to the lower-fidelity HotStar DNA polymerase. Using the Q5 DNA polymerase, we also studied the genetic diversity of the newly isolated AAV capsid sequences by performing phylogenetic analyses. Furthermore, we found that the novel AAV natural isolates showed evidence of evolution by positive selection.
MATERIALS AND METHODS
DNA extraction from nonhuman primate and human tissue
Nonhuman primate (Macaca mulatta) tissue samples were collected postmortem from the Gene Therapy Program at the University of Pennsylvania's Perelman School of Medicine. Human tissue samples (including aortic valve 1, bone marrow 9, brain 5, breast 2, cervix 1, colon 53, heart 23, intestine 20, kidney 20, liver 54, lung 33, lymph node 1, ovary 4, pancreas 7, pericardium 1, skeleton muscle 3, and spleen 34) were obtained from the Cooperative Human Tissue Network, National Disease Research Interchange, and BioIVT tissue sources. Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN Inc., Germantown, MD).
Conventional AAV isolation
To amplify 3.1 kb AAV genome sequences from host genomic DNA, we utilized the Q5 Hot Start High-Fidelity DNA polymerase, using working conditions determined by the manufacturer (New England Biolabs, Ipswich, MA). We used the previously described ‘AV1NS’ forward primer and the ‘AV2CAS’ reverse primer to isolate AAV genomes; we replaced the degenerate base Y in AV1NS with a T (AV1NS' 5′-GCTGCGTCAACTGGACCAATGAGAAC-3′ and AV2CAS 5′-CGCAGAGACCAAAGTTCAACTGAAACGA-3′) 21 because T is the primary nucleotide that is represented in the AAV sequence phylogeny across many clades of AAV. Each primer was used at a 0.5 μM final concentration, as described in the Q5 protocol (New England Biolabs). The following thermal cycling conditions were applied: 98°C for 30 s; 98°C for 10 s, 59°C for 10 s, and 72°C for 93 s, 50 cycles; and a 72°C extension for 120 s. PCR products were TOPO cloned (Thermo Fisher Scientific, Waltham, MA) and Sanger sequenced (GENEWIZ, South Plainfield, NJ). For most PCR products, we sequenced at least three clones.
AAV isolation by AAV-Single Genome Amplification
Genomic DNA from a human heart tissue sample that was previously found to be AAV positive by conventional AAV isolation PCR was subjected to AAV-Single Genome Amplification (SGA). AAV-containing genomic DNA was endpoint diluted in 20 ng/μL sheared salmon sperm DNA (Ambion, Inc., Austin, TX) by serial dilutions. Material from each serial dilution was used as the template for 96 PCR reactions using the AV1NS and AV2CAS primers.
22
We utilized Q5 Hot Start High-Fidelity DNA polymerase (New England Biolabs) to amplify AAV DNA using the following cycling conditions: 98°C for 30 s; 98°C for 10 s, 59°C for 10 s, and 72°C for 93 s, 50 cycles; and a 72°C extension for 120 s. For a Poisson distribution, the DNA dilution that yields PCR products in no more than 30% of wells contains one amplifiable AAV DNA template per positive PCR in more than 80% of cases.
23
AAV DNA amplicons from positive PCR reactions were purified using Agencourt Ampure XP Beads (Beckman Coulter, Brea, CA), libraries were constructed using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (NEB, Ipswich, MA) and sequenced using the Illumina MiSeq 2x250 (Illumina, San Diego, CA) paired-end sequencing platform, and the resulting reads were assembled de novo using the SPAdes assembler (
Sequence analysis
We aligned AAV sequences using the AlignX component of Vector NTI Advance® 11.5.4 (Thermo Fisher Scientific) or Geneious Prime version 2019.2 (
Polymerase fidelity comparison
The pAAV2/9 trans plasmid was used as the template. To make sure the template was pure, we first re-transformed the plasmid into Stable Competent Escherichia coli cells (Thermo Fisher Scientific), and sequenced two, single-colony clones through NGS (Illumina) as described previously. 24 To ensure complete sequence identity to the input pAAV2/9 trans plasmid, we used one of the two sequenced plasmids as the template for subsequent experiments. In this comparative study, the Hot Star HiFidelity polymerase (“HiFi”) (QIAGEN Inc.) was the lower-fidelity polymerase, whereas the Q5 Hot Start High-Fidelity DNA polymerase (Q5) (New England Biolabs) was the higher-fidelity polymerase. For “HiFi circular” and “Q5 circular,” the pAAV2/9 trans plasmid was diluted and used as the PCR template. For “HiFi linear” and “Q5 linear,” the pAAV2/9 trans plasmid was linearized with the restriction enzyme PvuII (New England Biolabs) and then diluted for use as the template. For all first-round PCRs, we utilized five copies of the template in a 25-μL reaction. In the second round, we used 1 μL of the first-round PCR product as the template in a 50-μL reaction. PCR conditions were based on the manufacturer's guidelines.
For all “HiFi” experiments, we employed the HotStar HiFidelity polymerase (QIAGEN Inc.). AV1NS' and AV2CAS primers were used in accordance with the manufacturer's protocol. We applied the following thermal cycling conditions for the first-round PCR: 95°C for 300 s; 94°C for 15 s, 63°C for 60 s, and 68°C for 371 s, 40 cycles; and a 72°C extension for 600 s. For the second round of PCR, we used the primers McapF3SpeI (5′-ATCGATACTAGTCCATCGACGTCAGACGCGGAAG-3′) and McapR1NotI' (5′-ATCGATGCGGCCGCAGTTCAACTGAAACGAATTAAACGGT-3′) to perform a nested reaction. McapF3SpeI and McapR1NotI' were described in a previous publication on an AAV PCR technique. 25 McapR1NotI' is a modified version of the primer McapR1NotI from the aforementioned publication; we modified McapR1NotI to correct for two base pairs near its 3′ end, which do not align with any reported AAV sequences, including the isolates reported in the previous publication. One microliter of the first-round PCR product was used as the template in the second, nested, round of PCR. The following thermal cycling conditions were used for the second round of PCR: 95°C for 300 s; 94°C for 15 s, 63°C for 60 s, and 68°C for 315 s, 40 cycles; and a 72°C extension for 600 s.
For the first round of the “Q5” reaction, we used the Q5 Hot Start High-Fidelity DNA polymerase master mix (New England Biolabs). We used AV1NS' and AV2CAS primers in each reaction in accordance with the manufacturer's protocol. The thermal cycling conditions were as follows: 98°C for 30 s; 98°C for 10 s, 59°C for 30 s, and 72°C for 186 s, 40 cycles; and a 72°C extension for 120 s. For the second round of “Q5” reactions, we utilized the primers McapF3SpeI and McapR1NotI'. One microliter of the first-round “Q5” PCR product was used as the template in the second, nested, round of PCR in each 50-μL reaction. The thermal cycling conditions were as follows: 98°C for 30 s; 98°C for 10 s, 66°C for 30 s, and 72°C for 164 s, 40 cycles; and a 72°C extension for 120 s. The PCR products were then TOPO cloned and sequenced.
Vector production, quantitative PCR titration, and Huh7 transduction assay
For AAV vector production in six-well plates, we adapted a previously described 1-cell-stack-scale HEK293 triple-transfection protocol based on the reduced culture areas, with a few modifications: (1) the plasmid ratio used was 2:1:0.1 (helper plasmid containing the required Adenovirus helper genes: trans plasmid containing AAV2 Rep and AAV capsid genes: cis plasmid containing the CB7 promoter, Firefly luciferase gene, and the rabbit beta globin polyadenylation sequence transgene (i.e., CB7.ffluciferase.rBG), by weight, and (2) at harvest, no other treatment was performed beyond freezing/thawing. 26 We measured the vector production titer by quantitative PCR using primers and probe against the vector poly A sequence.
Infectious titer was determined in vitro according to previously described methods. 27,28 After triple transfection and vector harvesting, equal volumes of each vector lysate were serially diluted with fresh complete medium and then used to transduce Huh7 cells (University of Pennsylvania Gene Therapy Program Immunology Core, Philadelphia, PA), which were seeded at 1 × 105 cells/well 1 day prior. We detected luciferase activity with a luminometer (Biotek, Winooski, VT).
AAV VP1 sequence evolution analysis
Geneious version 2019.2 (
We constructed all phylogenetic trees using the MAFFT version 7 server (
Statistics
For Figure 1A, we performed pairwise comparison between each group using the Wilcoxon rank-sum test using the “wilcox.test” function within the R Program (version 3.5.0;

Variable fidelity of DNA polymerases and bioactivity of PCR mutants.
Data availability statement
Supplementary data deposited at NCBI GenBank: Supplementary Dataset 1 accession numbers MZ668383-MZ668416 and Supplementary Dataset 2 accession numbers MZ708646-MZ708705.
RESULTS
A lower-fidelity DNA polymerase produces more random mismatch errors
We first evaluated the impact of polymerase fidelity on AAV isolation to test the assertion that lower-fidelity DNA polymerases would produce amplicons with a higher frequency of PCR error. We used a pure, NGS-verified, AAV9 trans plasmid (i.e., pAAV2/9) containing the AAV2 Rep gene and the AAV9 Cap gene as the PCR template in reactions containing DNA polymerases with varying levels of replication fidelity. We applied a high-fidelity polymerase, the Q5 Hot Start High-Fidelity DNA polymerase (Q5), and a relatively lower-fidelity polymerase, the HotStar HiFidelity (HiFi) polymerase, due to their varying levels of known polymerase fidelity. 20,29 Employing the same protocol used to isolate AAV natural isolates AAVHSC1-1725 with the HiFi polymerase, we found that plasmids cloned and sequenced from the HiFi polymerase PCR products contained 30–60% more occurrences of random errors across the VP1 region compared to those generated using the high-fidelity Q5 DNA polymerase: eleven out of nineteen and six out of twenty total sequenced PCR product clones from the HiFi Circular and Linear groups, respectively, contained at least one mismatch. In contrast, only one out of 20 and 24 sequenced PCR product clones had a mismatch in the Q5 linear and circular groups, respectively (Fig. 1A, D; Supplementary Table S1).
We next aimed to determine whether the AAV9 PCR isolate capsid sequences generated from the HiFi polymerase experiments were functional. We cloned the isolates into pAAV2/9 trans plasmids containing the AAV2 Rep gene such that each plasmid contained a mutant AAV9 VP1 Cap gene, with these mutant trans plasmids then producing AAV vectors containing the firefly luciferase transgene (i.e., CB7.ffluciferase.rBG). Two of the mutant capsids produced vector titers at levels similar to those of wild-type AAV9 (D87G and G174D). The remainder of the mutants showed reduced vector production capacity compared to AAV9 (Fig. 1B). P32S had a titer that was 17% lower than AAV9, while G177S, Q299H, and Q678R showed an 80–90% reduction in production titer. S632F, K33T L648I, and S348P M436T showed a 60–65% reduction compared to AAV9. The mutants' Huh7 infectious titers (Fig. 1C) show a pattern similar to their vector production titers, with a few exceptions—for example, the mutant P32S has a production titer of ∼83% of AAV9, but its Huh7 infectious titer is only ∼6% of AAV9, implying the mutation P32S may impair the capsid's Huh7 transduction, which warrants further investigation. Together, these results indicate that the lower-fidelity HiFi DNA polymerase produces mutants with variable functional properties in an unpredictable manner that can impair the discovery and characterization of novel isolates.
Novel AAV sequences from multiple clades were isolated from nonhuman primate and human tissues using a high-fidelity DNA polymerase
The advancement of gene therapy requires the identification of novel AAV capsids. The majority of currently used AAV natural variants have been derived from primate tissue. 11,13,21,30 Using our validated high-fidelity Q5 PCR-based technique, we investigated whether new capsid sequences can be isolated from a panel of primate tissue samples. We used primers that bind to conserved regions of the capsid sequence to amplify a 3.1-kb AAV amplicon, to detect and amplify the AAV genomes present in 50 nonhuman primate intestinal tissue samples. In this manner, we discovered 12 AAV natural isolate sequences. Most of these isolates belonged to clades D or E or the primate outgroup clade containing AAVrh32.33 (Table 1; Supplementary Dataset S1).
Novel adeno-associated virus (AAV) natural isolates recovered from nonhuman primate intestinal tissue samples and sequence similarity to closest known AAVs
The DNA sequence of AAVrh81 was substantially different from that of all AAVs in the GenBank database; hence, the DNA difference value is not included in this table.
AAVs, adeno-associated viruses; NHP, nonhuman primate.
We also screened genomic DNA from 271 human tissue samples using the Q5 polymerase and obtained 22 new AAV natural isolate capsid sequences, including clade F member AAVhu68. Those new AAV sequences were isolated from heart, intestine, kidney, liver, lung, and spleen. Overall, 8% of the human samples were positive for AAV. Most of the novel human isolates could be classified as clade B and C viruses or were similar to AAV2 and AAV2-AAV3 hybrids (Table 2; Supplementary Dataset S1). Three human-derived natural isolates exhibited novel DNA sequences, despite having the same protein sequences as previously reported GenBank entries (i.e., AAVhu.32, AAV9, and CHC367_AAV).
Novel adeno-associated virus (AAV) natural isolates recovered from human tissue samples and sequence similarity to closest known AAVs
Recovered clones have the same amino acid sequence as previously reported AAVs, but exhibit variation in their DNA sequences.
The protein sequences of AAVhu71/AAVhu74 and AAVhu78/AAVhu88 are identical (AAVhu71 = AAVhu74 and AAVhu78 = AAVhu88), while their DNA sequences are different.
AAV-SGA identifies natural isolate AAVhu68 capsid sequences with high precision and accuracy
SGA can accurately amplify individual virus sequences from a mixed sample. Based on previous reports by Salazar-Gonzalez et al. 23 and Simmonds et al. 31 and others for the amplification and study of HIV genome dynamics in infected patients, we adapted SGA to accurately isolate AAV sequences from mammalian tissue samples using the aforementioned high-fidelity Q5 polymerase (data not shown). In this technique, endpoint-diluted genomic DNA acts as the PCR template and contains only one amplifiable AAV genome in each amplicon-positive PCR. This method prevents sequence ambiguity caused by DNA polymerase-induced mutations due to the method's replicative nature. This technique also mitigates possible DNA polymerase template-switching issues that can occur in DNA mixtures (thus leading to the recovery of artificially recombined amplicons) because only one AAV genome is amplified in each reaction.
We sought to verify the sequence of previously isolated AAVhu68 by performing AAV-SGA on the same tissue sample from which it originated, as described in Table 2. This technique, combined with the use of the high-fidelity Q5 polymerase, allowed us to confirm the identity of this sequence with high precision and accuracy. Our results show that all of the single AAV genomes recovered from this sample had 99.94–100% capsid-sequence identity to the previous, conventional Q5 PCR-isolated AAVhu68 sequence. Of the 61 single AAV genome-derived amplicons recovered from this sample, only seven amplicons had 1 to 2 nucleotide mismatches from the original sequence. The vast majority (54/61) of amplicons had 100% DNA-sequence identity to the previously isolated AAVhu68 capsid sequence (Supplementary Table S2; Supplementary Dataset S2), indicating that sequence data generated using the Q5 polymerase can be interpreted with a high degree of confidence.
Naturally isolated AAV sequences show evidence of positive evolutionary selection
Using the Q5 polymerase AAV isolation strategy, we were able to investigate the evolutionary properties of AAV genomes with minimal influence from PCR-mediated errors. We observed that several recovered AAV natural isolate capsid sequences had greater numbers of DNA differences than corresponding protein sequence changes when compared with their closest, previously reported AAV sequence according to the GenBank sequence database. For example, the protein sequence of AAVrh75 differs by only two amino acids from AAVrh.8; however, their DNA sequences exhibited differences for 170 nucleotides (Table 1). In this case, the majority of differences in DNA sequence were transitions.
If the virus experiences selective pressure in favor of a particular genetic mutation, we would expect the nonsynonymous mutation rate (dN) to be higher than the synonymous mutation rate (dS) in that region. The contrary is true for deleterious mutations within a sequence. To evaluate the evolutionary stability of the AAV sequences isolated from primate tissues, we performed statistical analyses to determine whether there was evidence of positive, diversifying selection across the entire VP1 genes of our novel AAV when compared to their closest natural isolate sequence. We used the BUSTED due to its ease of use for evolutionary analyses on small sets of similar sequences. 32 BUSTED determines whether the dN/dS rates over the entire gene of interest—across different groups of branches within a phylogenetic tree—are suggestive of positive selection. We detected statistical significance (p < 0.05) at several branch points, indicating that at least one site in the VP1 gene experienced diversifying selection between test branches in the phylogeny (Fig. 2; Supplementary Fig. S1; Table 3).
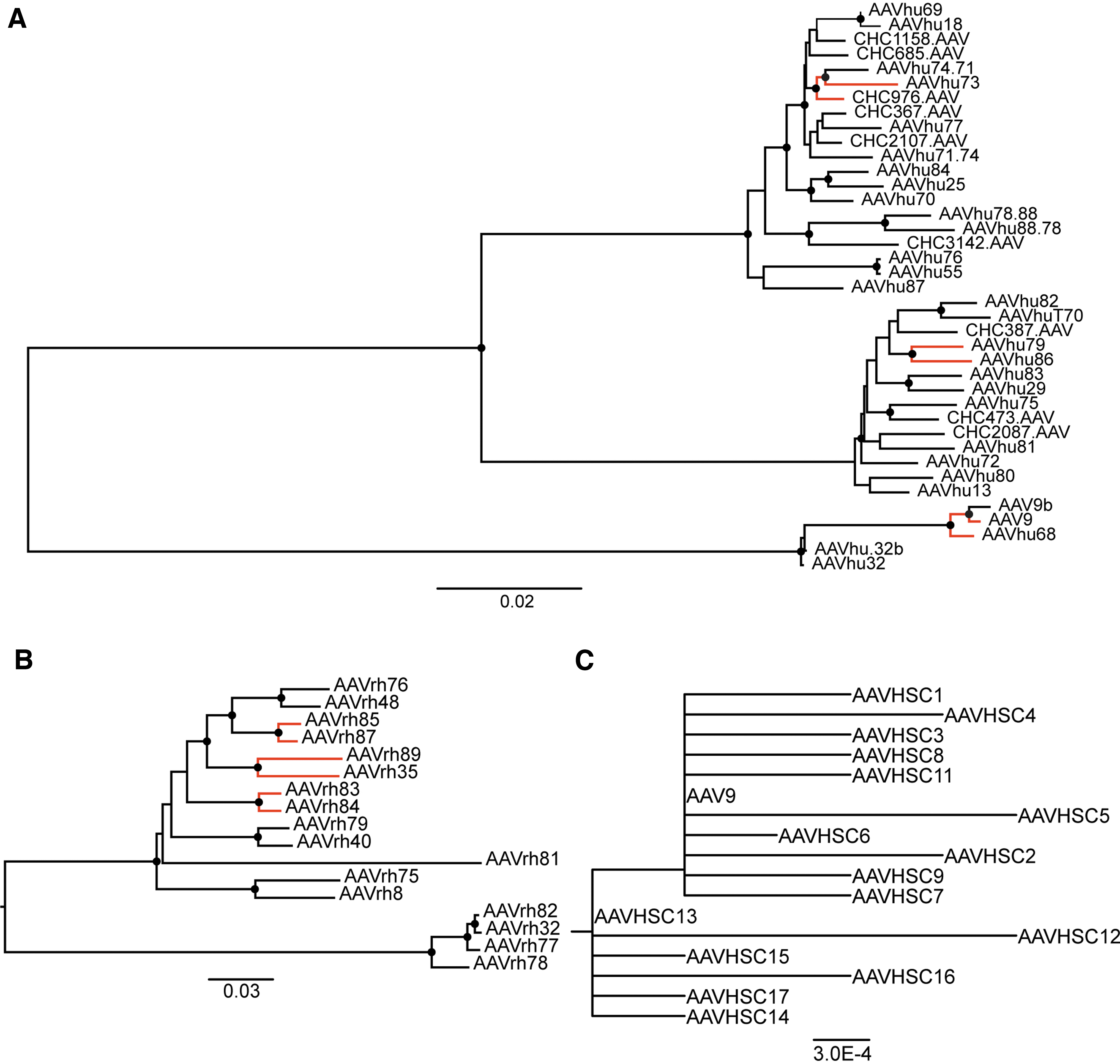
Phylogenetic analyses of positive selection in AAV VP1 genes. Neighbor-joining phylogenies of AAV VP1 DNA sequences from
Branch-site unrestricted statistical test for episodic diversification analysis of novel adeno-associated virus VP1 genes to closest natural isolate sequence
p-Values <0.05 shown in bold.
Statistical significance determined by BUSTED, Likelihood ratio test.
BUSTED, branch-site unrestricted statistical test for episodic diversification; HSC, hematopoietic stem cell.
In 3/20 cases, our human-derived AAV natural isolates were positive for diversifying selection from their closest natural isolate clade member (Fig. 2A; Table 3). In 3/9 instances of rhesus isolates, diversifying selection was apparent in at least one region across the capsid sequence (Fig. 2B; Table 3). In contrast, BUSTED analysis did not show evidence of positive, diversifying selection when we compared test branches across the entire phylogeny of sequences from a group of previously published AAV natural isolates derived from human HSCs (Fig. 2C; Table 3). 25 Similarly, the HiFi PCR mutant AAV VP1 genes did not show evidence of positive selection (Table 3; Supplementary Table S1; Supplementary Fig. S1).
In addition to performing gene-wide tests for positive selection, we assessed whether individual sites within VP1 genes for each phylogeny showed evidence of positive or negative selection. To analyze each group of AAV sequences for the presence of positively selected evolutionary hotspots, we used the MEME program due to its ability to detect episodic and pervasive selection. 33,34
MEME detected thirteen sites that displayed evidence of positive diversifying selection in the VP1 genes of the AAVs isolated from human samples (Table 4). Four of these sites are located in the hypervariable regions (HVRs) of the capsid gene (i.e., surface-exposed capsid regions that display significant sequence diversity). Six sites are located in the internal VP1 unique region (VP1u). In addition, we found 19 sites of significance in the capsid sequence dataset in samples from rhesus macaques (Table 4). Among these 19 sites, 10 are located in HVR regions, while one was located in VP1u. Both sets of sequences also showed evidence of positive selection in areas between the HVRs, which comprise the non-surface-exposed regions of the capsid structure (Table 4). MEME was unable to detect any sites that were subject to positive selection in either the AAVHSC sequences or the HiFi PCR mutant-capsid sequences.
Mixed-effects model of evolution analysis of novel adeno-associated virus VP1 phylogenies
All sites with p < 0.05 shown.
Statistical significance determined by MEME, Likelihood ratio test.
HVR, hypervariable region; MEME. mixed-effects model of evolution.
We also used the FEL program 35 to detect sites across branch pairs in the novel human and nonhuman primate AAV phylogenies that had undergone negative selection (Supplementary Table S3). Sites within 15 out of 29 novel AAV natural isolate sequences compared to their closest known AAV relatives showed evidence of negative purifying selection. In contrast, neither the AAVHSC variants nor the HiFi PCR mutants contained any site across the entire phylogeny that showed evidence for evolution by negative selection.
DISCUSSION
AAV sequence isolation techniques have greatly evolved since the discovery of AAVs in 1965. 36 In this study, we compared the DNA replication fidelity of two DNA polymerases in terms of AAV isolation: HotStar HiFidelity polymerase and Q5 Hot Start High-Fidelity polymerase. We found that using the HiFi polymerase and a protocol with a high number of PCR cycles—a method previously used to discover novel AAVs 25 —resulted in a significantly higher rate of random mutations in amplicons generated from template DNA compared to the method utilizing the Q5 polymerase. The mutant PCR isolates produced vector and transduced Huh7 cells in vitro at variable levels. These experiments highlight the variable and unpredictable impact that low DNA polymerase fidelity can exert on AAV function during capsid-genome isolation.
Tindall and Kunkel were among the first to demonstrate that DNA polymerases can generate mutations in amplified DNA. 37 Since then, researchers have isolated and engineered a variety of new polymerases to address this issue, including Q5—one of the most accurate polymerases—with a base substitution rate of 5.3 × 10−7 bp, which corresponds to an ∼280-fold higher fidelity compared with Taq polymerase. 20,38 In contrast, the fidelity of the HotStar HiFi polymerase is reported to be only 10-fold higher compared with Taq. 29 We demonstrated that optimal AAV isolation requires using the highest-fidelity DNA polymerases available, in this case, Q5.
We also used the Q5 polymerase to perform AAV-SGA to validate the sequence identity of one of the human-derived AAVs isolated in this work, AAVhu68. The replicative nature of this technique, 23 coupled with the high fidelity of Q5 polymerase, allowed us to precisely and accurately identify the capsid sequence of this isolate. Furthermore, the sequencing data of the resulting amplicons we obtained using the Q5 polymerase-based technique were congruent with the amplicons we obtained using NGS methods, thereby validating the identity of this AAV natural isolate capsid gene. AAV-SGA did recover a small minority of amplicon sequences in which 1–2 nucleotides were mismatched from the AAVhu68 genome, which may be attributed to NGS error, the low error rate of Q5, or DNA damage induced by thermocycling, as characterized by Potapov and Ong. 20 These data demonstrate that AAV-SGA is a robust tool for analyzing viral populations with very high precision and accuracy.
By utilizing the high-fidelity Q5-based AAV isolation method, we found that natural AAV variant capsid protein sequences remain relatively stable, while their DNA sequences can exhibit considerable changes in comparison to their closest relative in GenBank. This finding stands in stark contrast to our HiFi PCR mutant sequences and a subset of AAV sequences identified from human HSCs (AAVHSCs), in which many more amino acid changes correlated with DNA sequence alterations. In any viral population, one would expect host-mediated evolutionary pressure from the immune system or factors that mediate tissue tropism to promote positive, diversifying selection 33,39 in relation to processes involving host–capsid interactions such as cellular adhesion, entry, and viral trafficking. 40 –45 However, these selection pressures are absent in an in vitro replication environment, such as that used when generating PCR mutants.
We used the BUSTED program to determine whether the overall AAV capsid sequence was subjected to positive selection in its recent evolutionary lineage. Our results showed evidence of diversifying selection, even for cases exhibiting high DNA sequence variation, yet high amino acid sequence homology between two isolates. Conversely, BUSTED analysis gave no evidence of diversifying selection for the few instances in which DNA sequence variation between multiple AAVs resulted in amino acid changes (i.e., AAVHSCs and AAV HiFi PCR mutants). An unexpected finding was that a population of AAVs recovered from natural sources, such as human HSCs, showed no evidence of evolutionary pressure-mediated changes, despite having a high nonsynonymous mutation rate. Given that Smith et al. used the HotStar HiFidelity polymerase and a high-cycle-number protocol to isolate these variants, 25 a number of these amino acid differences may represent polymerase replication errors that arose during the isolation procedure.
We used MEME to elucidate patterns of site-specific evolution in the novel AAV natural variants. 34 The majority of sites exhibiting evidence of evolution mapped to the AAV HVRs; surface-exposed HVRs mediate interactions with host factors such as antibodies and cell surface receptors. 41,46 –48 In addition, a few of the sites were positioned before the start of VP3 in the VP1u region that interacts with host-cell intracellular trafficking machinery. 49 –51 The evolutionary pressure exhibited at these sites could provide a good indication of which capsid regions are amenable to modification from a vector engineering standpoint. In contrast, neither the AAVHSC isolates nor the HiFi PCR mutants contained any site that displayed significant selective pressure, further confirming that polymerase-introduced errors can significantly influence AAV sequence analysis, discovery, and function. While high-fidelity DNA polymerases are necessary for optimal PCR-based AAV isolation and characterization from natural sources, error-prone polymerases can expand and diversify the library of candidate AAVs by introducing random mutations into a given AAV capsid backbone. 52,53
These results highlight the need for accurate AAV isolation methods to reach valid conclusions about AAV evolution, genetics, and biological functions arising from genome variation. Our findings indicate that not all “high-fidelity” DNA polymerases are created equal and that one must use caution when analyzing AAV sequences generated with a lower-fidelity polymerase. Utilizing methods such as SGA in conjunction with high-fidelity polymerases enables the accurate isolation of natural AAV populations that may contain the next candidate gene therapy vector.
Footnotes
AUTHORS' CONTRIBUTIONS
Q.W.: conceptualization, methodology, investigation, visualization, writing—original draft, and writing—review and editing. K.N.: conceptualization, methodology, investigation, writing—original draft, and writing—review and editing. G.H.: investigation. X.L.: investigation. J.M.W.: conceptualization, writing—review and editing, and funding acquisition.
ACKNOWLEDGMENTS
We wish to thank Gui Hu and Xiaobin Liu for study support. We also thank the Biostatistics and Bioinformatics Core, the Nucleic Acid Technologies Core, and the Scientific Communications and Research Administration Division of the Gene Therapy Program at the University of Pennsylvania for study support.
AUTHOR DISCLOSURE
J.M.W. is a paid advisor to and holds equity in Scout Bio and Passage Bio; he also has sponsored research agreements with Amicus Therapeutics, Biogen, Elaaj Bio, FA212, Janssen, Passage Bio, Regeneron, and Scout Bio, which are licensees of Penn technology. J.M.W. and Q.W. are inventors on patents that have been licensed to various biopharmaceutical companies and for which they may receive payments.
FUNDING INFORMATION
This research was supported by the Perelman School of Medicine of the University of Pennsylvania and Amicus Therapeutics.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.