
Abstract
Advanced therapy medicinal products (ATMPs), such as gene therapies that consist of or contain genetically modified organisms (GMOs) need to comply with the European Union (EU) GMO legislation, as implemented in each EU Member State, before a clinical trial can commence. Complying with GMO requirements is complex, varies significantly across EU Member States and is leading to delays to clinical trials with ATMPs. Such delays and varying implementation of the GMO legislation makes the EU less attractive as a region to conduct clinical trials with investigational gene therapies. This is detrimental to EU patients, since their timely access to these transformative potentially curative medicines is delayed. Despite recent initiatives coordinated by the European Commission (EC) to facilitate and reduce discrepancies across the EU regarding the application of the GMO requirements, it remains particularly difficult to conduct multicenter clinical trials with ATMPs containing or consisting of GMOs involving several EU Member States. The recent decision for the EC to temporarily derogate potential coronavirus disease 2019 treatments and vaccines from some provisions of the GMO requirements was made on the basis of a clear recognition of such complexities and resulting delays to clinical development. The Alliance for Regenerative Medicine, the European Federation of Pharmaceutical Industries and Associations, and the European Association for Bioindustries call upon the EC, together with national competent authorities, to exempt ATMPs containing or consisting of GMOs from the GMO legislation. Such a simplification will eliminate the delays currently reported to occur when submitting environmental risk assessments and GMO applications to the national competent authorities. An exemption from GMO requirements will make the EU a more attractive region for clinical development of gene therapies and could accelerate European patients' access to these potentially life-saving medicines. Maintaining a system for GMO assessment that is different across countries may also prevent ATMPs from realizing the full benefits of a harmonized clinical trial approval process under the Clinical Trials Regulation. The undersigned organizations to this publication urge the EC to use its right of initiative to put forward a legislative proposal to exempt ATMPs in clinical development from the EU GMO legislation, within the timeframe proposed in the 2020 EU Pharmaceutical Strategy (by 2022). Implementation of a GMO exemption scheme before the end of the transition period for the Clinical Trial Regulation (the end of 2023) is important to avoid new Clinical Trial Application submissions for ATMPs under the Clinical Trial Regulation having to conduct the whole GMO assessment process in parallel. It is considered that ATMPs pose negligible risk to the environment. Such ATMPs include the following: human somatic cells modified ex vivo; recombinant virus-based vectors, including those containing genome editing nucleic acid sequences (which may also be delivered nonvirally); and bacterial vectors. Outside of controlled storage conditions, gene therapies cannot survive for any appreciable length of time. Upon clinical administration, any recombinant gene therapy viral vector particles that do not enter host cells are diluted within the body and if excreted are in such low multiplicity to no longer be viable or considered infectious to persons, animals, or living organisms within the environment. Any nucleic acids released into the environment are rapidly degraded.
Background And Problem Statement: The European Union Genetically Modified Organism Legislation Delays Clinical Trials and Patient Access to Innovative Medicinal Products Which Have the Potential to Treat, Modify, Reverse or Cure Serious Diseases
Advanced therapy medicinal products (ATMPs) are innovative medicinal products that have the potential to bring highly transformative value to patients, including potential cures, by either correcting the underlying cause of their disease (e.g., a genetic defect) or by modifying a function in the body to cure or significantly ameliorate their disease. Some ATMPs, such as gene therapies, consist of or contain genetically modified organisms (GMOs). Even though the European Commission (EC) Directive 2001/18/EC was enacted primarily for agricultural applications (GMO plants) with the goal to protect food consumers and the environment, the authorization procedure for clinical trials with investigational ATMPs requires additional steps to comply with the GMO legislation.
The European Union (EU) legislation prescribes that clinical trials with investigational products containing or consisting of GMOs comply with either Directive 2001/18/EC on the deliberate release into the environment of GMOs or with Directive 2009/41/EC on the contained use of GMOs. These additional requirements come on top of the requirements for authorization of clinical trials laid down in the Clinical Trial Directive 2001/20/EC and the Clinical Trials Regulation (EU) 536/20141 and hinder scientific progress in clinical gene therapy.
Since both GMO Directives are transposed with variations and applied in different manners by national authorities in each Member State, decisions made under the GMO legislation are not applied consistently, leading to different assessment processes, timelines, and outcomes across the EU, whereas the Clinical Trials Regulation will soon allow for a harmonized clinical assessment process adhering to strict timelines for decision across EU Member States. Moreover, since both regimes (GMO and Clinical Trials legislations) apply, without defining a modus operandi for how they should interoperate, different approaches may be taken by different Member States: either applying the GMO approval regime before, as part of, or in parallel with the application to national health authorities.
Data show that Europe is less successful than other regions in attracting new clinical trials with ATMPs, in particular for developing gene therapies
The Alliance for Regenerative Medicine (ARM) report Clinical Trials in Europe: Recent Trends in ATMP Development 2 of October 2019 highlighted that Europe has become less competitive than other regions of the world in attracting new ATMP clinical trials, particularly clinical trials involving GMOs. Although the number of new ATMP clinical trials has significantly grown over a 4-year period (2014–2018) on a global scale (+32%), with notable growth in North America (+36%) and Asia (+28%), this increase has not been observed in Europe where the number of new clinical trials remained constant over the time period analyzed (−2%). The proportion of new gene therapy (gene editing, gene therapy, and gene-modified cell therapy) clinical trials is also considerably lower in Europe than in other regions (Fig. 1). 2 The complexity of GMO requirements for clinical studies leading to prolonged approval timelines exacerbates Europe's lack of attractiveness and is a major cause for this lower number.
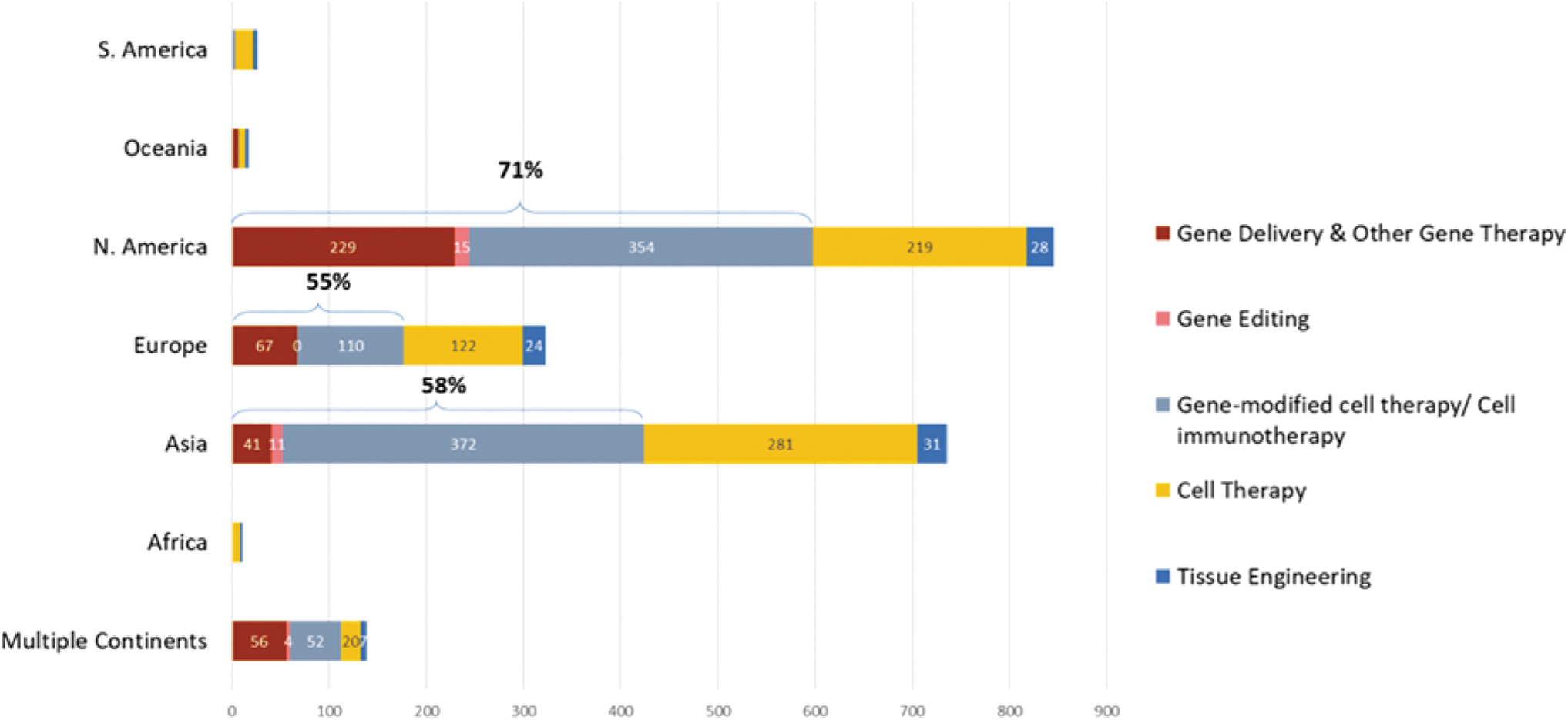
Number of New ATMP Clinical Trials started between 1st January 2014 and 30th June 2019 by Technology and Continent. ATMP, Advanced Therapy Medicinal Products
Europe's relative lack of appeal with ATMP developers may become more pronounced as the number of new gene therapies is rapidly growing. As of January 13, 2021, there are 1220 ongoing regenerative medicine and advanced therapy clinical trials worldwide. Having insufficient clinical development of advanced therapies in Europe is a missed opportunity and a major issue that needs to be addressed, with patients having limited early access to these therapies, physicians having less experience with the products when they come to market and marketing authorization applications with limited data on EU patients.
GMO requirements for investigational medicines are far more complex and cumbersome in Europe than in the United States
The most popular country for carrying out clinical trials with gene therapies is, by far, the United States. Clinical studies using GMOs in the United States are not subject to the same environmental requirements as those conducted in Europe. 3
In 2015, the U.S. Food and Drug Administration (FDA) Center for Biologics Evaluation and Research released guidance where it was recommended that a GMO environmental risk assessment is in most cases not needed for gene therapies, vectored vaccines, and related recombinant viral or microbial products (GTVVs). 4
In this 2015 guidance, it is stated how, “in 1997, the FDA amended its National Environmental Policy Act of 1969 (NEPA) regulations to, among other things, increase the efficiency of the Agency's implementation of NEPA and to reduce the number of NEPA evaluations by providing for categorical exclusions for additional classes of actions that had been identified as normally not having a significant impact, individually or cumulatively, on the quality of the environment and for which, therefore, neither an environmental impact statement nor an Environmental Assessment is required.”
A claim of “categorical exclusion” ordinarily applies to clinical studies (including GTVVs), allowing an exemption from the requirements for an environmental assessment under 21 CFR 25.31(e) for Investigational New Drugs (“unless extraordinary circumstances indicate that the specific proposed agency action may significantly affect the quality of the environment”). 4,5
Extraordinary circumstances include where a strain or vector has been identified as “pathogenic or known to be associated with animal, plant or microbial toxicities” or where a strain is “capable of survival under adverse conditions.” 4 Gene therapies such as viral vectors and genetically modified human cells do not exhibit these attributes. Thus, the EU position puts product developers at a disadvantage vis a vis the United States.
Gene therapies based on genome editing techniques are currently considered to be GMOs in the EU
On July 25, 2018, the Court of Justice of the EU decided that organisms obtained by the new techniques of directed mutagenesis are considered GMOs and are, therefore, subject to the obligations laid down by the GMO Directive. 6 Even though the background of the court ruling was an action brought in the context of crops used in agriculture, that is, the main products concerned by the GMO legislation, the ruling implies that gene therapies using genome editing techniques are also considered as GMOs under EU legislation. This decision differs from the current thinking and GMO-related expectations outside the EU.
A few months after the ruling, the EC's Scientific Advice Mechanism Group of Chief Scientific Advisors published a statement providing a scientific perspective on the regulatory status of products derived from gene editing, and the implications for the GMO Directive. 7 The advisors conclude that the GMO Directive should be revised “to reflect current knowledge and scientific evidence, in particular on gene editing and established techniques of genetic modification.”
Gene therapies using genome editing technologies, such as CRISPR-Cas9, Zinc finger nucleases or transcription activator-like effector nucleases, have the potential to radically transform the standards of care for patients who currently lack treatment options. Although the number of clinical trials based on gene therapies using genome editing technologies is now surging, 8 the ruling of the EU Court of Justice may inadvertently delay access to effective new treatments, also entailing a negative impact on the research and innovation landscape in the EU.
Recently, countries outside of the EU have undertaken consultations with a view to revising national GMO legislation, including Department for Environment Food & Rural Affairs (gene edited plants and animals) in the United Kingdom, and the Gene Technology Secretariat in Australia.
EU authorities have recognized that the EU GMO legislation is responsible for significant delays in the commencement of clinical trials
The fact that the GMO legislation causes delays in the development of novel medicines in the EU has been confirmed by the EU's actions to tackle the coronavirus disease 2019 (COVID-19) pandemic. 9 The EU strategy for COVID-19 vaccines 10 acknowledges that “There is considerable variety across Member States in the national requirements and procedures implementing the GMO Directives used to assess environmental risks of clinical trials of medicinal products that contain or consist of GMOs. This is likely to cause significant delay, particularly for multi-centre clinical trials in several Member States.” 10
To accelerate development and access to COVID-19 treatments or vaccines, the EU has adopted Regulation (EU) 2020/1043 to temporarily exempt investigational medicinal products (IMPs) for human use containing or consisting of GMOs to treat or prevent COVID-19 from complying with some provisions of the GMO legislation. 11
Recitals (8) and (9) of Regulation (EU) 2020/1043 read as follows: “(8) Experience shows that, in clinical trials with investigational medicinal products containing or consisting of GMOs, the procedure to achieve compliance with the requirements of Directives 2001/18/EC and 2009/41/EC as regards the environmental risk assessment and consent by the competent authority of a Member State is complex and can take a significant amount of time. (9) The complexity of that procedure increases greatly in the case of multi-centre clinical trials conducted in several Member States, as sponsors of clinical trials need to submit multiple requests for authorisation to multiple competent authorities in different Member States in parallel. In addition, national requirements and procedures for the environmental risk assessment and written consent by competent authorities for the deliberate release of GMOs under Directive 2001/18/EC vary greatly from one Member State to another. Whereas in some Member States a single request for authorisation concerning the conduct of the clinical trial and the GMO aspects can be submitted to a single competent authority, in other Member States parallel requests need to be submitted to different competent authorities. Furthermore, some Member States apply Directive 2001/18/EC, others apply Directive 2009/41/EC and there are Member States that apply either Directive 2009/41/EC or 2001/18/EC depending on the specific circumstances of a clinical trial, so it is not possible to determine a priori the national procedure that is to be followed. Other Member States apply both Directives simultaneously to different operations within the same clinical trial. Attempts to streamline the process through informal coordination between Member States' competent authorities have been unsuccessful. There are also variations between national requirements as to the content of the technical dossier.”
11
Moreover, as noted in the 2020 EU Pharmaceutical Strategy, 12 the GMO regulations should be fit for purpose and, therefore, are not appropriately applied to ATMPs. The need for a change of the GMO framework has also been recognized by European authorities in the European Medicines Agencies Network Strategy to 2025, which states how alignment on application of GMO requirements in the EU would also be required to support innovation and digitalization in clinical trials. 13
EU Initiatives from 2017 tO 2019 are Insufficient to Significantly Reduce Approval Timelines for ATMPS Containing or Consisting of GMOs
Regulatory authorities have previously recognized that differences in GMO requirements among different EU Member States led to clinical trial delays. From 2017 through 2019, initiatives were launched to reduce national discrepancies across the EU and facilitate the process for authorizing clinical trials with IMPs containing or consisting of GMOs.
In October 2017, the EC and the European Medicines Agency (EMA), in collaboration with the Member States' authorities, launched a joint action plan on ATMPs
14
to facilitate the development and authorization of these products in the EU for the benefit of patients. In an acknowledgment of how GMO requirements represent a major hurdle to ATMP developers, the EC has initiated dialogue with national competent authorities to address the interplay between the GMO and the medicines legislation to reduce discrepancies across the EU with regard to the application of GMO legislation to ATMPs containing or consisting of GMOs. The initiative has successfully resulted in many actions to clarify and harmonize national requirements: An online repository of national regulatory requirements
15
A questions and answers document related to the interplay between the medicinal products framework and the GMO framework regarding authorization procedures
16
Harmonized application forms for clinical research and Good Practices for assessment of GMO aspects of clinical trials: ○ A common application form (CAF) and Good Practice document
‡
for human cells genetically modified by means of retroviral or lentiviral vectors
17
○ A CAF and Good Practice document
§
for gene therapies that contain or consist of adeno-associated viral vectors
17
○ A CAF** for IMPs containing viral vectors.
17
These initiatives were welcome as they aid the better understanding of requirements by sponsors and contribute to streamlining common implementation across EU Member States.
Recent experiences indicate that 2017–2019 initiatives are insufficient
Despite the developments from 2017–2019, recent experience indicates that these initiatives have been insufficient. Administrative complexity and lengthy application timelines for ATMPs containing GMOs are still causing significant delay to clinical trials.
A 2020 survey of ATMP developers to characterize recent experiences with GMO applications for gene and gene-modified cell therapies in the EU sought to examine whether the adoption of CAFs had led to significant improvements to the process and timeframe for GMO approval, to ultimately allow commencement of clinical trials. As shown in the Supplementary Data, the survey results, based on 73 GMO applications filed since August 2018, including 17 applications using the CAFs, show that, despite improvements attributed to the EC-EMA Action Plan, the GMO approval process for clinical trials remains a significant hurdle, leading to delays in the initiation of clinical trials. Furthermore, sponsors have been selectively choosing countries where the GMO process lends itself to a speedier approval and commencement of clinical trials.
Some of the survey findings include the following:
There is a high degree of variability across EU Member States with regard to:
○ Length of time for approval (up to 12 months in some countries)
○ Decisions for Contained Use versus Deliberate Release Classification
○ Different hazard risk classifications for the same ATMP (within Contained Use)
○ Data requirements (content and format) despite the application of CAFs and following the questions and answers document. 16
The review and approval of GMO applications are not faster when applying for a second and subsequent trial with the same product.
The available data indicate that CAFs are of limited value, since in most cases, additional data, such as national application forms, are still requested by the national GMO competent authority.
Many survey respondents also indicated that the preparation and submission of the GMO package and the implementation at the clinical sites remain very time consuming and resource intensive.
Delays in clinical trials result in delayed access to potentially life-saving treatments
By temporarily lifting some GMO requirements for COVID-19 treatments and vaccines, EU authorities have recognized that the EU GMO legislation is responsible for delays in clinical trials despite the recent initiatives. In June 2020, when the EC presented a European strategy to accelerate the development, manufacturing, and deployment of vaccines against COVID-19, a press release stated that “Time is of the essence. Every month gained in finding such a vaccine saves lives, livelihoods and billions of euros.” 9
A survey conducted by European Federation of Pharmaceutical Industries and Associations (EFPIA), Vaccines Europe and Medicines for Europe in September 2020 explored the impact of the COVID-19 GMO derogation. As illustrated in Fig. 2, the evidence has confirmed the benefit to development time scales and administrative burden not only for sponsors, but also likely for all trial sites and health systems. In the collected comments, respondents noted clear savings in resources and time, which led to trials starting more expediently and efficiently. COVID-19 vaccine developers who did not make use of the derogation noted that this was because the derogation came into effect too late to benefit their clinical development.
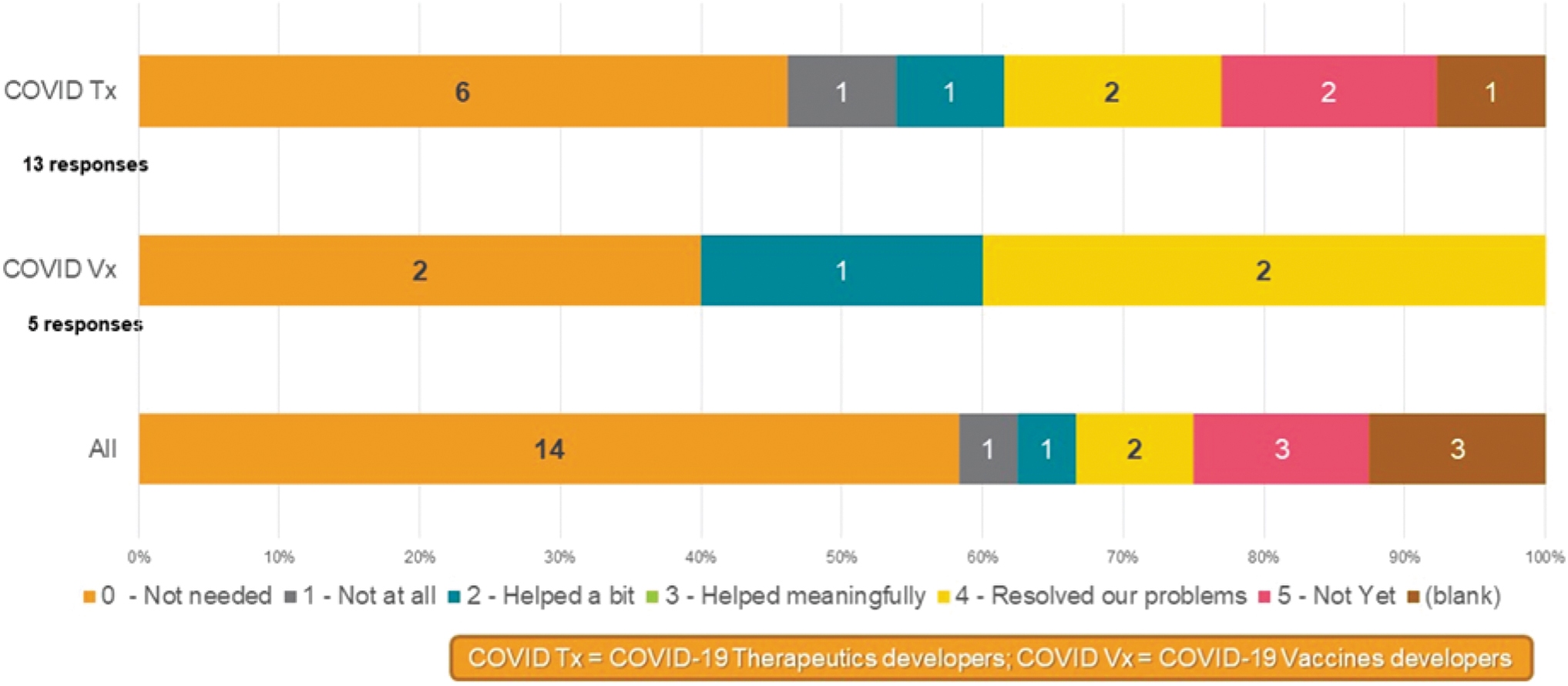
EFPIA-Vaccines Europe-Medicines for Europe Regulatory Flexibilities Survey, September 2020. Responses to Survey Question: Did the July 2020 GMO Derogation Provide Necessary Support for your Clinical Research Programme? Twenty-four member companies responded, which included 13 COVID-19 therapeutic developers and 5 COVID-19 vaccine developers up to September 2020. “Not needed”: Specific for clinical research with products that do not consist of or do not contain genetically modified organisms. COVID-19, coronavirus disease 2019.
Time is also very much of the essence for people with cancer, inherited disorders, and other life-threatening conditions. Nearly every ATMP in clinical development addresses a life-threatening condition or an unmet medical need. Every delay to the commencement of an ATMP clinical trial, due to issues with the current application of GMO legislation, could result in irreversible progression of the disease and reduce chances of survival or quality of life. The measures taken to save lives during the pandemic can also save lives in nonpandemic circumstances.
Now is the Time to Re-Evaluate GMO Requirements for ATMPs
In view of the significant efforts made to streamline the GMO process for ATMPs, where, in effect, there has been only insufficient progress thus far, it is time to evaluate the purpose and assess the impact of the GMO legislation and overhaul its application to gene and gene-modified cell therapies. Without change, the GMO requirements for ATMPs will be a direct impediment to achieving the EMA's Regulatory Strategy to 2025 for promoting innovation in the EU. The 2020 EU Pharmaceutical Strategy 12 acknowledges that the regulatory requirements for GMO products in the EU “should be fit for purpose” and are “currently hindered by the fragmentation of national requirements”). The Pharmaceutical Strategy notes that “Solutions will be explored during the evaluation of the pharmaceutical legislation. In general, consideration should be given to mechanisms for the continuous and timely adaptation of its technical requirements in light of emerging science and technologies with a view to enhance effectiveness to protect human health whilst minimizing harmful impacts on the environment.”
An effective solution is for the EC to consider the implementation of an exemption from complying with GMO requirements for ATMPs containing or consisting of GMOs that are in clinical development.
After more than 30 years of scientific progress in the development of safer gene therapy vectors and experience based on numerous clinical gene therapy trials worldwide, we can now conclude with confidence that the environmental risks associated with this therapy are negligible. 18
After evaluating the effectiveness of previous nonlegislative measures in this context, as well as the potential of the latest temporarily applicable measure set out in Regulation (EU) 2020/1043, we consider that an effective and future-proof solution can be best achieved by legislative means. Without effective harmonization and significant simplification of the GMO registration process for clinical trials with ATMPs consisting of or containing GMOs, it will be difficult for developers to leverage the advantages of the improved Clinical Trial Regulation. Ensuring a harmonized, science-based, and consistent approach on GMO requirements for ATMPs across all Member States is important to achieve the objective of the Clinical Trial Regulation and increase the EU's attractiveness as a leading region for clinical development.
ARM, EFPIA, and European Association for Bioindustries (EuropaBio) call on the EC to use its right of initiative to put forward a legislative proposal to amend the existing legal framework, or, if deemed sufficiently effective, to adopt implementing legislation to supplement the current legal framework, to implement an exemption regime for ATMPs in clinical development, that contain or consist of GMOs, from complying with GMO requirements. The trade associations who have jointly authored this publication consider that an exemption represents an ideal future state with a view to accelerating clinical development, and ultimately patient access to life-changing and life-saving gene therapies.
The undersigned organizations are willing and prepared to engage with the EC and other stakeholders to jointly find the best solutions. Without action, the EU risks falling further behind the rest of the world in the development of novel treatments for the most challenging medical conditions, and more importantly, in securing the access of patients to transformative potentially life-saving therapies.
Footnotes
Acknowledgments
ARM, EFPIA, and EuropaBio thank the following contributors: Annie Hubert (previously ARM, undertook the GMO Survey and drafted results); Stuart Beattie, Biogen; Jill Morrell, Biomarin; Jacquelyn Awigena Cook, Celgene; Michael Werner, ARM; Patrick Ginty, (previously Cell and Gene Therapy Catapult, now Handl Therapeutics); Simon Butler, Gilead; Andeleeb Dahy, Astellas; Vicki Coutinho, Gamma Delta; Neil Roberts, Gilead; Daniel Rabbie, Achilles Therapeutics; Marcia Gaido, Precision Medicine; Seoan Huh, Pfizer; Lauren Oliva, Biogen; Violeta Georgieva, EuropaBio; Nathalie Lambot, pharma.be; Tatiana Reimer, Bayer; Ine de Goeij, Astellas; Julien Romanetto, Transgene; Virginia Acha, MSD; Par Tellner, EFPIA.
Author Disclosure
No competing financial interests exist.
Funding Information
No funding was received for this work.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.