
Abstract
In the literature, there are few high-throughput screens or even methods for high-throughput screens of recombinant adeno-associated virus (rAAV) production despite potential benefits to research and production. In this study, a generalizable high-throughput relative rAAV titration method is examined within the context of an siRNA screen as siRNA knockdown is a common means of pathway engineering in bioproduction. Crude samples generated from transfected HEK293T/17 cultures were subjected to quantitative PCR (qPCR) and used to transduce COS7 cells to assess relative differences in genomic and infectious rAAV titer, respectively, at the 384-well scale, evaluating both supernatant and lysed samples. To evaluate relevant differences in titer for conditions that could be used in an actual screen, cultures subjected to an siRNA reverse transfection and subsequent rAAV forward transfection were also tested. The delayed forward rAAV triple-plasmid transfection was not seen to affect the siRNA activity of tested controls, while siRNA transfection was shown to measurably impact rAAV titer. Effective differentiation between infectious titer levels was dependent upon the choice of sample dilution, but trends between qPCR and infectious titer assays were consistent across sample sets.
Introduction
There is a paucity of high-throughput screens or even screening methods targeting the improvement of recombinant adeno-associated virus (rAAV) production in the literature. 1 This may be a missed opportunity since, despite refreshed concerns around high-dose toxicities, 2 increasing viral yields remains a pressing goal.
Higher gene therapy vector loads are needed to account for larger patient sizes, systemic diseases, or diseases in less accessible body sites. 3,4 Thus, amid constant efforts in safety, alternative host cells, and alternative viral helpers, an additional area of rAAV research has been in scale-up, moving production from laboratory scale to industrial scale. 5 –8 On the contrary, methods for evaluating small-scale product have been neglected, correlating to the lack of high-throughput screens. Current methods of evaluation used at other scales may be adapted for streamlined rAAV evaluation at microscales.
Processes for rAAV production, 9 isolation, 10 and characterization 11 –13 vary according to the scale involved. Isolation of rAAV at the laboratory scale for experiments on cell cultures and animal models has relied on gradient ultracentrifugation and affinity column-based kits, while larger scale efforts have benefitted from affinity and ion-exchange chromatography methods. 14 –18
Subsequent characterization of purified virus tends to include some combination of quantitative PCR (qPCR), droplet digital PCR, enzyme-linked immunosorbent assay, ion-exchange chromatography, transduction assays, and transmission electron microscopy. 19 –21 Larger scale efforts usually include polishing and concentration steps typical of Good Manufacturing Practices. 17,22 –28 Among quality concerns, determinations of genomic titer and infectious titer are of first order interest. However, progression from production to isolation to characterization is not well suited to the microscale.
Here, we leverage and expand upon recent work on direct qPCR assays 29 –31 and rAAV processing. 29,32 Isolation steps were eschewed and rAAV production and titer of crude material were examined on a relative basis. The presented titration methods involved minimal manipulation and relatively direct assay of samples from transfected cultures in 384-well plates. The resulting process was shown to work at microscale conditions suitable for a high-throughput siRNA screen. The titration methods should be generalizable for most high-throughput screens.
Supernatant and intracellular-derived samples from transfected HEK293T/17 cultures were tested for relative genomic and infectious rAAV2 titers. Sampling supernatant from attached cultures entailed a straightforward liquid handling step. rAAV2 from intracellular stores were derived by incubating attached cell cultures in a mild detergent solution. Importantly, remnant plasmid and detergent in crude samples did not preclude assay of genomic titer and infectious titer on a relative basis by qPCR and COS7 cell transduction, respectively.
Assays were initially characterized by testing serial dilutions of an rAAV2-EGFP (enhanced green fluorescent protein) standard, after which the assays were used to evaluate crude samples of HEK293T/17 cultures transfected with a triple-plasmid system. As a follow-up, the same methods were used to evaluate titer from HEK293T/17 cultures double transfected with siRNA and the rAAV triple-plasmid system using conditions similar to those of a high-throughput siRNA screen.
siRNA libraries targeting specific genes for knockdown with as few off-target effects as possible are frequently used in high-throughput screens. In fact, siRNA screens have been used to increase the permissiveness of target cells to transduction, 33 but this has not been extended to the production of rAAV itself. Here, siRNA transfection and changes in transfection conditions were shown to impact assay results. Further, twofold differences in titer were statistically distinguishable under some conditions. Finally, assay results supported each other insofar as trends from one assay mirrored those in the other. The overall experimental scheme is depicted in Fig. 1.

rAAV genomic and infectious titration in the high-throughput screening process. Diagram of workflow and timeline for process. Cells were first reverse transfected with siRNA or only plated for transfections without a reverse transfection. Cells were then forward transfected with rAAV plasmids 2 days thereafter. Samples were harvested 4 days after forward transfection and were evaluated for infectious titer and viral genome titer as noted. Asterisks represent the degree to which a particular step could be deemed suitable for high-throughput processes. rAAV, recombinant adeno-associated virus.
Materials and Methods
Reverse transfection
HEK293T/17 cells were transfected with 40 nM siRNA using RNAiMAX (ThermoFisher). Nonsense siRNA negative control no. 2 (Ambion) and Allstars Hs Cell Death Control siRNA (Qiagen) were used where indicated. Allstars Hs Cell Death Control siRNA is a mix of potent siRNA targeting genes required for cell survival, often used as a positive control.
Initially, siRNA diluted in OptiMEM-I (Thermo Fisher) was seeded at 2 μL per well in 384-well black-walled clear-bottom plates at 800 nM. Of RNAiMAX, 0.11 μL was diluted into 18 μL of OptiMEM-I was added to the siRNA and was incubated at room temperature for 15 min. To cultures forgoing reverse transfection, 20 μL of OptiMEM-I was added per well. After siRNA complexation, 20 μL of 293T/17 cells at 0.1e6/mL in 20% fetal bovine serum (FBS) Dulbecco's modified Eagle's MEDIUM (DMEM) was added for 2e3 cells per well.
Forward transfection
Transfection of HEK293T/17 cells with an rAAV2-EGFP-triple-plasmid system (pSR460A, pSR457, and pSR449B, courtesy of Dr. Richard Smith) was done 2 days after the initial plating. On a per well basis, 0.12 μg of total plasmid was diluted into 10 μL OptiMEM-I. Of ViaFect (Promega), 0.36 μL was then added to diluted plasmid, and the mixture was vortexed immediately and incubated for 15 min. The resulting solution of ViaFect-plasmid complexes was then added to the wells. Samples were collected 4 days after forward transfection.
Culture sampling
Commercial rAAV2-EGFP (GeneCopoeia and ATCC) was used as a standard. Otherwise, rAAV2 was assessed from crude samples of HEK293T/17 cultures. Treatment of transfected cells with 0.5% nonionic detergent Triton X-100 (Bio-Rad) and RNase has recently been shown to be an effective alternative means of releasing intracellular rAAV without damaging the viral end product. This eliminates common freeze/thaw and ultracentrifugation steps that are impractical in high-throughput settings.
On a per well basis, 5.5 μL of 5% Triton X-100 was added to the original 50 μL of transfected cell culture and incubated for 1 h. Supernatant was removed and retained. Residual attached cellular material in the well was washed with phosphate-buffered saline (PBS) and the wash was added to the original supernatant. A negative control lacking the rep/cap plasmid was used to account for potential carryover of EGFP expressed from plasmid-based ITRs in HEK293T/17 cells.
qPCR genomic titration
Inhibitor-resistant Taq polymerases allow for no-extraction qPCR from a variety of complex sample media backgrounds. 31,34,35 SsoAdvanced Universal Inhibitor Taq (Bio-Rad) was used here. For qPCR-based genomic titration of purified rAAV, reducing residual plasmid by nuclease is essential. For unpurified crude material residual plasmid levels are very high and are likely to interfere with even relative measurements of viral genomes. To reduce plasmid and inadvertently released genomic DNA, samples were treated for 1 h at 37°C DNase I (Millipore Sigma) at 10 U in 10 mM Tris-HCl pH 7.5 with 2.5 mM MgCl2 and 0.5 mM CaCl2. DNase-I was inactivated by the addition of ethylenediaminetetraacetic acid (EDTA) to a working concentration of 5 mM followed by incubation at 75°C for 10 min.
Disassociation of the capsid from the genome is also required and was accomplished via a conventional Proteinase K digestion step. Samples were treated with >11 U per sample of Proteinase K (Millipore Sigma) in 2 mM EDTA 100 mM Tris-HCl 8 pH at 55°C for 60 min. Potentially faster methods such as heat treatment and Tween have been suggested and may find utility elsewhere. 11,36 Proteinase K was inactivated by incubation of the sample at 85°C for 20 min.
qPCR was assessed using a Master Mix of the inhibitor tolerant Taq polymerase mentioned before. ΔCt was calculated by using the most relevant negative control as a baseline, which varied. For rAAV2 samples derived from cell culture, plasmid-free controls were used. For samples of reference rAAV2, dilution media alone was used as the baseline control. The primers there were used targeted the CMV promoter and were a kind gift from Dr. Richard Smith (FWD primer: TGGTGATGCGGTTTTGGCAG, REV primer: AATGGGGCGGAGTTGTTACGA). A proprietary set of primers against inverted terminal repeat (ITR) (GeneCopoeia) mirrored the results using the CMV promoter primers.
COS7 transduction infectious titration
COS7 cells were transduced with 5 μL of either crude culture sample diluted in 0.01% pluronic F68 in PBS pH 7.4. The exact nature of the crude sample depended on whether the sample was a supernatant sample or a Triton X-100-treated sample. Supernatant samples were directly sampled from well-mixed cultures and diluted in pluronic-PBS. Triton X-100 treatments have been described in the “Culture Sampling” section.
As noted throughout, the level of dilution applied to samples varied and was chosen to ensure that the signal was within an appropriate range. For lysed samples containing Triton X-100, insufficient dilution could also be expected to impact cell viability and thereby assay accuracy. On a per well basis, 45 μL COS7 cells were added at a concentration of 0.022e6 cells/mL in 10% FBS in DMEM. Expression of the EGFP transgene was assessed by fluorescence using a SpectraMax i3 (Molecular Devices). The number of positive cells was assessed by SoftMax Pro 7.0.3 (Molecular Devices) image processing.
All experiments described here were carried out in triplicate and without automation.
Results
Viral titration of serially diluted standards—method testing
Testing the screening assays with rAAV2 standards produced log-linear ranges for qPCR assessment of viral genomic titer and COS7 transduction-based assessment of infectious titer (Fig. 2).
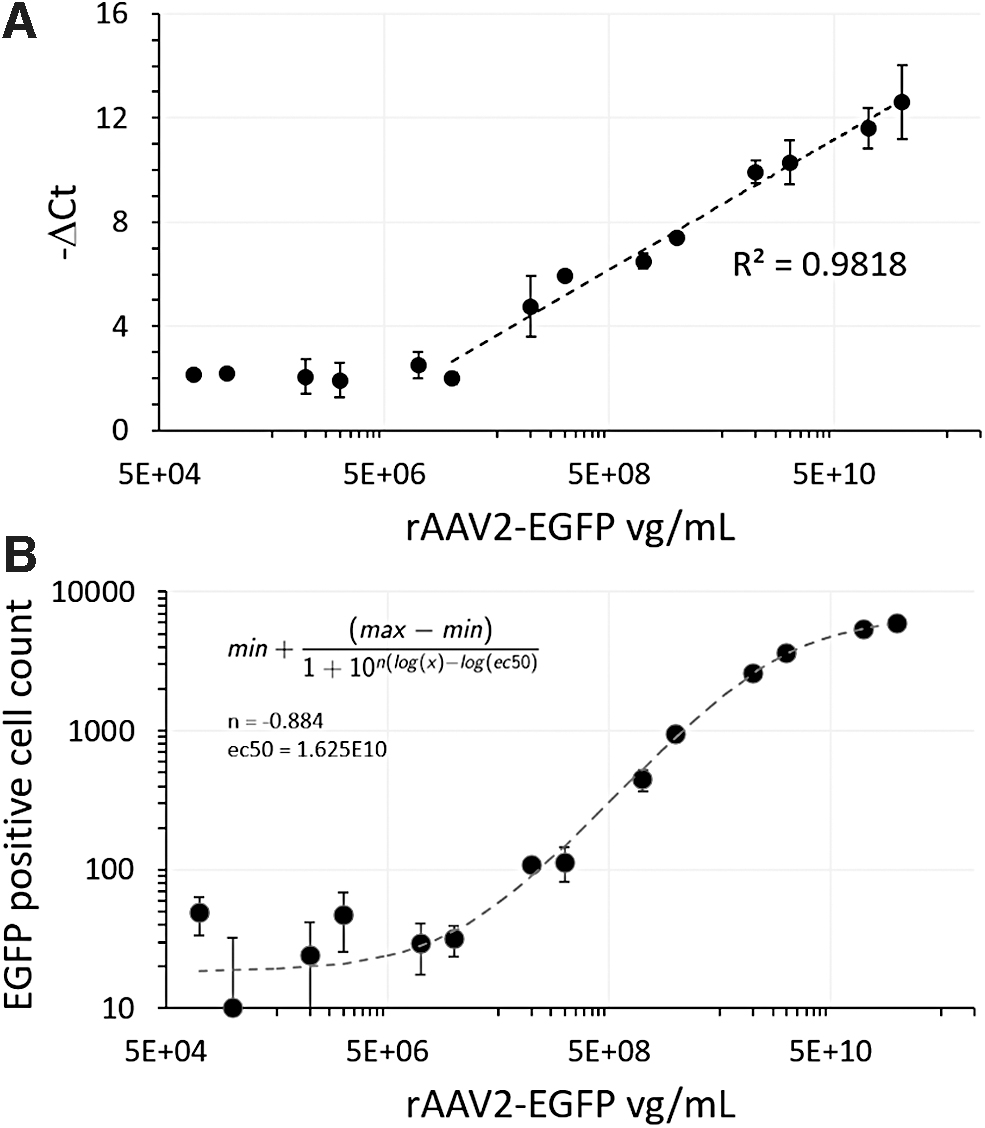
Genomic titer and infectious titer of serially diluted rAAV2-EGFP standard.
Although the qPCR assay utilized an inhibitor tolerant polymerase, Ct values remained sensitive to background media effects. Thus, background media in the sample was standardized before processing. −ΔCt values were relatively flat and consistent below certain viral titers, suggestive of an assay baseline with a limit of detection of 5.74E7 vg/mL. Above that baseline, −ΔCt values were log-linear with viral particles, a log-linearity (R 2 = 0.982) that held for values up to the maximal achievable titer given the starting material concentration (Fig. 2A). This pattern was consistent when using rAAV2-EGFP standards from different sources or with different primer sets (described in the methods).
COS7 is a monkey kidney cell line that has been shown to be among the most efficiently transduced cell lines by any rAAV serotype. 37,38 To induce transduction of COS7 cells, rAAV2 standard or putative virus from transfected cultures was mixed with COS7 cells at the time of plating. Measured fluorescence was assessed 3 days after plating, and fluorescent transgene expression was used as a proxy for infectious titer.
There was a sigmoidal response for the transduction as assessed according to EGFP-positive cell counts when plotting rAAV2 titer on a log basis, with a lag at the low end and the beginning of an asymptote at the high end of rAAV2 concentrations (Fig. 2B). Although lacking a true linear range, the sigmoidal response does offer an ability to discriminate between infectious viral levels within a range. The n value smaller than 1 (fit to |n| = 0.887, shown in Fig. 2B) suggests that while the choice of sample dilution level will be critical, that distinguishing between relative infectious titer differences is feasible.
Assessment of virus from HEK293T/17 transfection with rAAV2 triple-plasmid system
Supernatant and lysed samples from HEK293T/17 cultures transfected with a triple-plasmid system were assayed by qPCR and COS7 transduction (Fig. 3). Transfection experiments included three control conditions: (1) the omission of all plasmids (plasmid-free), (2) the omission of the pSR457 plasmid containing rep and cap elements as a transfection-EGFP positive process control (no rep/cap), and (3) a twofold dilution of the base condition after transfection complex formation (half tfect).
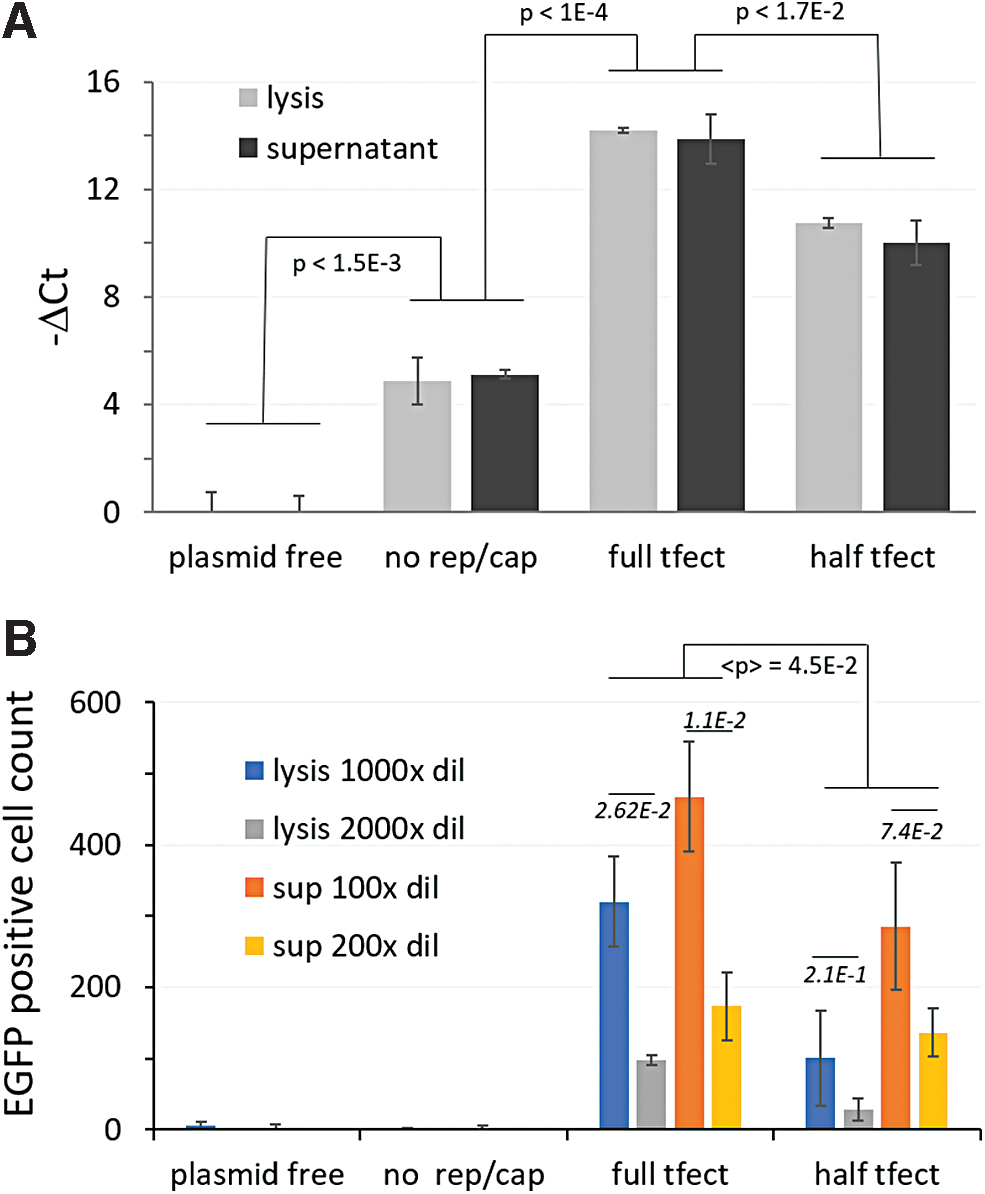
Genomic and infectious titer assessment from samples of transfected HEK293T/17 cells.
Plasmid-free control culture samples contained neither residual plasmid nor rAAV genomic material from which to generate qPCR signal and lacked any EGFP fluorescent material to carryover to transduction wells that might result in a false EGFP-positive cell. Therefore, readings from this condition were used as a baseline. Readings from the no rep/cap sample were above the plasmid-free baseline (t-test p < 0.0015), but well below either the full transfection condition (t-test p < 1E-4) or the half transfection condition by the qPCR assay (t-test p < 0.0007) (Fig. 3A). That the no rep/cap sample was so much higher than the plasmid-free baseline suggests that the DNase I treatment degraded transfecting plasmids incompletely but sufficiently, preventing the obfuscation of true viral genome signal, allowing measurement of titer on a relative basis.
More evenly matched were the infectious titer readings for the two negative control conditions, with readings below the limit of detection (5.74E7 vg/mL), eliciting no more EGFP-positive cells than untreated wells (Fig. 3B). Transfected cells strongly expressed the EGFP transgene from ITRs. If any of this EGFP-positive material carried over from the transfection to transduction wells, it was not enough to elicit a signal by the measure used, as evidenced by transduction results for the plasmid-free and no rep/cap controls, neither of which elicited signal above untreated wells.
Cultures transfected with half the concentration of lipid-plasmid complex elicited less viral genome (t-test p-value <1.7E-2) (Fig. 3A) and less transduction relative to the standard transfection condition (t-test geometric mean p-value of 4.49E-2) (Fig. 3B). Importantly, for conditions of standard transfection, dilution of samples twofold led to statistically significant differences for both lysis and supernatant samples (t-test p-values of 2.62E-2 and 1.1E-2, respectively). The differences for twofold dilutions of half transfection samples did not produce statistically significant differences due to assay noise. These differences, while present, were not as apparent on day 2 (Supplementary Fig. S1).
Assessment of virus from reserve siRNA transfected and forward plasmid transfected cultures
Direct qPCR and COS7 transduction assays were used to evaluate samples from double transfections. HEK293T/17 cultures were reverse transfected with an siRNA death positive control or an siRNA nonsense negative control upon plating and then forward transfected with a triple-plasmid system and its varied controls 2 days thereafter. Cultures were assessed and samples were collected 4 days after the rAAV plasmid transfection.
To verify that the siRNA reverse transfection worked as expected, CellTiter-Glo was used to examine reductions in cell viability associated with siRNA controls. rAAV from the forward transfection was assessed with qPCR and COS7 transduction. Results presented below indicate that the reverse transfection was effective, that rAAV from the forward transfection remained assayable, and that reverse transfection impacts on rAAV synthesis were measurable.
CellTiter-Glo assay of double-transfected cultures
Double transfection cultures were evaluated by CellTiter-Glo 6 days after the reverse transfection (4 days after forward transfection with rAAV plasmids) (Fig. 4). CellTiter-Glo readings from cultures treated with AllStars Hs Cell Death Control siRNA were severely reduced relative to the negative control and no siRNA cultures (Fig. 4A).

HEK293T/17 double transfection CellTiter-Glo Assay results 4 days after forward transfection.
Among the subset of cultures treated with nonsense negative control siRNA (Fig. 4B), plasmid-free control cultures produced the highest reading, suggestive of the burden of heterologous protein production on cell growth or viability. No rep/cap control cultures had similar activity in the CellTiter-Glo assay compared to the full transfection condition. This may be associated with rAAV helper protein expression leading to potent distortions of native cellular operations such as maintaining the cell cycle primarily in the S phase among other effects. 39,40
Viral genomic titration of double transfection samples by qPCR
qPCR on samples from double-transfected cultures of HEK293T/17 cells was done to evaluate relative genomic titer. As with the forward transfection alone, negative control signal remained low, while the no rep/cap control samples containing residual plasmid template were higher than the plasmid-free baseline (t-test p-value of 7.17E-3) (Fig. 5A), but well below the signal derived from cultures transfected with the full triple-plasmid system (t- test p-value of 1.66E-6).
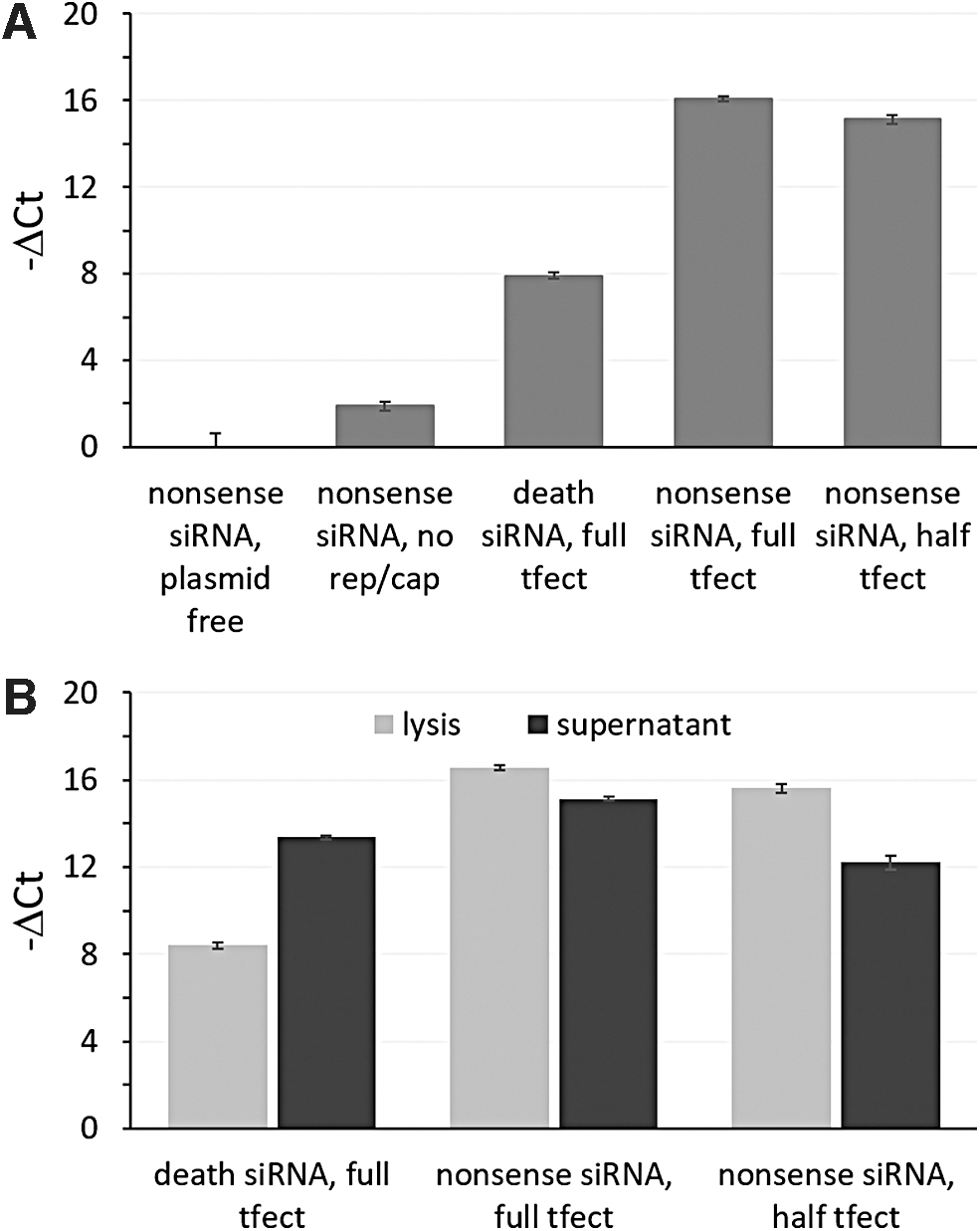
qPCR results for viral genomic titer from double-transfected HEK293T/17 cultures.
Positive control death siRNA treatment reduced titer levels relative to double transfection cultures treated with nonsense negative siRNA controls. The nonsense siRNA/half transfection complex condition consistently showed less viral genomic titer relative to the nonsense siRNA/base condition in both the lysed and supernatant samples. Supernatant showed lower rAAV2-EGFP genome signal than the lysed samples except in the case of the death siRNA/base condition where the relationship was inverted (Fig. 5B).
Infectious titration of double transfection samples by transduction of COS7 cells
CellTiter-Glo measurements indicate that COS7 culture cell number or viability were similar across transductions (Fig. 6A). The number of EGFP-positive COS7 cells varied in a manner that reflected trends in genomic titers (Figs. 5 and 6B). Samples from the no rep/cap transfection cultures showed no carryover of signal in the transduction assay, with the signal falling within the noise similar to readings from the plasmid-free control cultures.

Transduction results from double transfection samples.
Transduction with sample from siRNA cell death control treated cultures showed strongly reduced COS7 cell fluorescence, while remaining elevated above control signals in a pattern reflecting qPCR results. Also, for samples from the half transfection condition, fewer initial transfection complexes translated to less infectious titer (not statistically significant for the lysis sample p = 0.313 and statistically significant for the supernatant sample p = 3.42E-2), correlating to qPCR results and forward transfection-only results.
Minimal dilution of lysed samples led to low fluorescence signal in COS7 cell transduction (Fig. 6C). Increasing sample dilution led to a peak at 100-fold dilution (corresponding to a Triton X-100 concentration below a critical value related to viability) followed by a gradual decrease in assay signal. Lower assay signal at fold dilutions less than 100 likely reflected cell death from Triton X-100 as captured by CellTiter-Glo assay (Fig. 6D).
Discussion
Directly assessing rAAV2-EGFP titer from crude preparations of transfected cultures is a simple process and has been demonstrated using different processes and at larger scales. 29 The smaller scales involved in high-throughput are more susceptible to noise, but processes used here should be suitable for typical production levels of viral particle per cell. High-throughput evaluation of rAAV infectious titer has been pursued before on an absolute basis. 1 Previously, dot blots of transgene-specific probes measured viral genomes generated from rep-cap HeLa cells infected with serial dilution that comprised 12–30 points per sample in 96-well plates. Samples were derived from microtiter plates using freeze thaw and centrifugation to clear cellular material. Here, we focused on relative titer to eliminate the requirement for a high degree of replication and increase throughput in a 384-well format. Centrifugation and freeze thawing were also avoided.
Methods to measure rAAV2 titer from transfected cultures without purification are essential in high-throughput settings. Assays must be sufficiently robust to both signal inhibition and induction by extraneous cellular and digestive substances. Using an inhibitor resistant polymerase and evaluating viral genome amounts by −ΔCt, results indicated that rAAV2 genomic titer can be assessed on a relative basis by qPCR of crude samples from transfected HEK293T/17 cultures. Results also showed that relative infectious titer could be assessed by transduction of COS7 cells with diluted crude culture samples, where the measured output was the number of EGFP-positive cells evaluated 3 days posttransduction.
While twofold differences produced statistically significant signal for some conditions (Figs. 3B and 6B), differences were not always statistically significant but were nonetheless consistent in the direction of the effect and consistent between assays when comparing half transfection to full transfection conditions.
Differences between supernatant and intracellular accumulation were assessed despite the crude nature of the samples. Results from conditions lacking viable cells at the time of harvest suggest that the supernatant-associated rAAV are largely unstable to the addition of Triton X-100 described in the sampling procedure, making viral genomes sensitive to DNase I and viral particles incapable of transducing COS7 cells. Intracellular rAAV2 requiring Triton X-100 treatment for release was somehow protected from this effect.
In the literature, the ratio between rAAV sequestered within cells and rAAV released into the media has been reported to vary between serotypes and preparation methods. The mechanism by which rAAV is released from cells may at least be influenced by apoptosis and necrosis. Therefore, we assume that the ratio of virus in supernatants relative to that within cells may vary over screening conditions, and measures of both may be useful in capturing increases in overall rAAV production. For large screens requiring high-throughput, the simplicity of assessing supernatant and forgoing measurements of intracellular rAAV may represent a reasonable tradeoff.
Due to the DNase I and Proteinase K steps required to clear residual plasmid and genomic DNA and release the viral genome from the capsid, qPCR directly from samples may not be feasible for high-throughput.
Since measurements were considered on a relative basis, complete removal of remnant plasmid from unpurified vector was not necessary, and less time and DNase were needed for sample preparation. Indeed, complete removal of plasmid DNA was not achieved in the experiments shown. This makes the qPCR assessment of actual samples not directly comparable to standard curves using reference material, emphasizing the relative nature of the assay using crude starting materials. Although not demonstrated here, given a low volume-low error liquid handling tool, one-pot qPCR evaluation could be feasible even if DNase I and Proteinase K steps are retained.
Transduction of COS7 cells with a serial dilution of rAAV2 standard generated sigmoidal signal. This was probably impacted by a lack of infectivity at the low end of viral particles and a limit of transducible cellular material paired with increasing multiplicity of infection at the high end of viral particles.
The serotype tested was rAAV2, but this type of method should be applicable to other serotypes because of the broad susceptibility of COS7 cells, so long as the transgene used is readable in a high-throughput context. As remnant transfection complexes or plasmid alone containing the ITR and EGFP transgene alone did not result in apparent EGFP signal, results suggest that triple-plasmid transfection remnants likewise did not result in virus production in COS7 cells or generate spurious EGFP signal therein, all the while acknowledging that recombinant virus production involves different dynamics than typical heterologous bioproducts.
Additional considerations for screening will be the typical titer produced from a given system. If the typical titer is very high for a given system, additional fold dilution of crude material may be required for proper comparison between samples. In theory, this may impact throughput or automatability depending on the complexity of the available robotics. If the typical titer is very low for a given system, qPCR may be suitable for determination of viral genomic titer, but infectious unit titration will likely require assaying less dilute transfection samples. If this is not sufficient, the use of more sensitive reporter proteins may be warranted.
Results here were for rAAV2 produced from HEK293T/17 cells. This cell line is an especially productive subclone of HEK293T cells. None of the methods here is specific to HEK293T/17 cells and therefore should be applicable to any high-yielding attached HEK293 variety used for the production of recombinant AAV.
Conclusion
Streamlined titer evaluation directly from crude samples like that described here, even on a relative basis, could help speed upstream optimization of rAAV production. Such simplification is even more important for high-throughput screening where isolating rAAV before genomic and infectious unit titration is not feasible.
Screening for siRNA effecting rAAV transduction for the identification of cellular restriction factors is related and are more abundant in the literature, 41 but does not include the use of crude materials examined here and has a different overall objective. As a generality, the increased use of screens for rAAV production itself by processes like those described here could offer complementary results as well as improve production capacities for gene therapy vectors.
Footnotes
Authors' Contributions
D.N.Q. designed the studies, generated the data, interpreted the results. D.N.Q. wrote and edited the article with input and advice from J.S.
Acknowledgments
The authors acknowledge Dr. Richard Smith for kindly letting us use the triple-plasmid system and for multiple helpful correspondences along the way. We thank Dr. Ken Chih-Chien Cheng, Dr. Sirisha Chakka, and Dr. Lara El Touny for their helpful suggestions and conversations. We thank Gerardo Martinez Cardiel for his help in conducting preliminary experiments. This research was performed under the auspices of the Intramural Research Program of the NIH, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Author Disclosure
No competing financial interests exist.
Funding Information
This research was supported by the Intramural Research Program of the NIH, The NIDDK.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.