
Abstract
DNA-encoded delivery of antibodies presents a labor- and cost-effective alternative to conventional antibody therapeutics. This study aims to improve the potency and safety of this approach by evaluating various plasmid backbones and expression cassettes. In vitro, antibody levels consistently improved with decreasing sizes of backbone, ranging from conventional to minimal. In vivo, following intramuscular electrotransfer in mice, the correlation was less consistent. While the largest conventional plasmid (10.2 kb) gave the lowest monoclonal antibody (mAb) levels, a regular conventional plasmid (8.6 kb) demonstrated similar levels as a minimal Nanoplasmid (6.8 kb). A reduction in size beyond a standard conventional backbone thus did not improve mAb levels in vivo. Cassette modifications, such as swapping antibody chain order or use of two versus a single encoding plasmid, significantly increased antibody expression in vitro, but failed to translate in vivo. Conversely, a significant improvement in vivo but not in vitro was found with a set of muscle-specific promoters, of which a newly engineered variant gave roughly 1.5- to 2-fold higher plasma antibody concentrations than the ubiquitous CAG promoter. In conclusion, despite the limited translation between in vitro and in vivo, we identified various clinically relevant improvements to our DNA-based antibody platform, both in potency and biosafety.
Introduction
Monoclonal antibodies (mAbs) have become mainstay in a wide range of indications such as cancer and inflammatory/autoimmune disorders. 1 However, high treatment costs and frequent high-dose administrations limit patient accessibility. An alternative approach is antibody gene transfer, where the goal is to administer the mAb-encoding nucleotide sequence rather than the protein, allowing the patient to produce the therapeutic in a cost- and labor-effective manner for a prolonged period of time. 2
Viral vectors, mRNA, and plasmid DNA (pDNA) have all been applied for in vivo expression of antibodies, both preclinically and clinically. Each platform has its own qualities and limitations. Viral vectors exhibit an exceptional transfection efficiency, resulting in high mAb levels in vivo for a prolonged period of time, 3 but remain challenged clinically due to vector immunogenicity. 4 mRNA presents a quick onset of expression, providing peak mAb levels just hours after delivery. 5 However, expression is transient and requires repeated dosing to maintain mAb levels, thus providing minimal benefit over conventional protein delivery. pDNA as a vehicle for gene transfer is an attractive approach because of the ease of manufacturing, large payload capacity, lack of cold-chain storage requirements, and favorable biosafety profile. 2 To enhance cellular uptake following, for example, intramuscular injection, pDNA is typically administered in vivo in combination with electroporation. 6 The concept of DNA-encoded antibodies is currently being investigated in the clinic (NCT04079166 and NCT03831503). The momentum of this field is illustrated by the increasing support by large pharmaceutical companies and funding agencies. One notable example is the U.S. Defense Advanced Research Projects Agency (DARPA) Pandemic Prevention Program (P3), initiated in 2017, aimed at the rapid discovery, testing, and manufacture of antibody treatments, including pDNA- or mRNA-based delivery, to fight any emerging disease threat. The P3 Program was consequently also applied in response to the coronavirus disease 2019 (COVID-19) outbreak.
Our group has previously demonstrated proof of concept for intramuscular DNA-encoded antibody gene transfer in mice and sheep. In both animal models, mAb plasma levels peak at single- to double-digit microgram per milliliter range 2 to 4 weeks after delivery, after which levels steadily decline, but remain detectable for several months up to a year. 7 –9 Comparable mAb levels have been reported in other studies. 10 –12 Despite progress made in preclinical research and early clinical trials, translation of DNA-encoded antibodies to patients is subject to further improvement. First and foremost, current achievable in vivo mAb levels after gene transfer are relatively low, which limits the number of therapeutic mAbs applicable for this approach. Cumulative pDNA dosing, although feasible, 7,8 stands in contradiction to the cost-effective alternative this strategy aims to provide. Second, current plasmids typically contain large bacterial elements, such as an origin of replication and an antibiotic selection marker. These elements give rise to safety concerns and can result in increased transgene silencing. 13,14 Third, mAb expression is typically driven by potent ubiquitous promoters, which can lead to undesired expression in, for example, antigen-presenting cells, potentially triggering antidrug-antibody (ADA) and cytotoxic T lymphocyte responses. 15 Fourth, plasmids that contain repeats of sequences, for example, two identical expression cassettes for expression of the light and heavy chain, respectively, are prone to recombination and make production more challenging. 16
The present study aims to address these hurdles and build toward a next-generation DNA-based platform for in vivo mAb expression, with a focus on improved safety and potency. Therefore, the impact of multiple DNA-engineering strategies on mAb expression was evaluated in vitro and in vivo. 4D5, the murine equivalent of trastuzumab (Herceptin®), was used as a model, allowing prolonged expression without evoking an ADA response in immunocompetent mice. 7 First, a number of plasmid backbones with decreasing amounts of bacterial sequences were evaluated, ranging from large conventional plasmids to a minimal Nanoplasmid DNA (npDNA). 17 This plasmid has a small origin of replication (R6K) and an antibiotic-independent selection marker (RNA-out), and was selected for its potential for industrial scale-up, 18 a necessity for clinical translation. Second, a single plasmid encoding both mAb chains, with identical or different expression cassettes, was tested with swapped mAb heavy- and light-chain configurations. This single-plasmid setup was also compared with a setup with dual plasmids, each driving expression of an mAb chain (i.e., light and heavy chain, respectively). Finally, the ubiquitous CAG was compared with a set of muscle-specific promoters.
Materials and Methods
Design and production of mAb-encoding plasmid backbones
An overview of the various constructs and plasmid backbones evaluated in this study are provided in Table 1 and Fig. 1. cpDNAL is an enlarged bacterial backbone, containing F1 and ColE1 origins (1,290 basepairs—bp), ampicillin and kanamycin resistance markers (1,653 bp), and noncoding sequence for a total of 3,937 bp. cpDNA is our previously evaluated conventional backbone, 7 containing a pUC origin (715 bp), ampicillin resistance marker (861 bp), and noncoding sequence for a total of 2,322 bp. Both cpDNA and cpDNAL were produced in-house in E. coli TOP10F’ strain and purified using the NucleoBond Xtra Maxi EF kit (Machery-Nagel, Düren, Germany) following the manufacturer's instructions. RNA-out Nanoplasmids (NTC9385R; Nature Technology Corporation, Lincoln, NE) carry an antisense RNA element that blocks the translation of a host chromosome-encoded selectable marker (SacB) permitting sucrose selection (139 bp), an R6K origin (281 bp), and noncoding sequences for a total of 512 bp. All npDNA was produced by Nature Technology Corporation following an established procedure. 19 Briefly, replication was performed using proprietary Plasmid+ shake culture medium containing sucrose to select for RNA-OUT Nanoplasmid vectors. Flasks were grown with shaking to saturation at 30°C with a temperature shift to 37°C to increase plasmid copy number before harvest. Low endotoxin npDNA was purified using Nucleobond AX 2000 or AX 10000 columns (Macherey-Nagel). For each of the evaluated constructs, purity was verified via UV spectrophotometry, and the size and integrity of the constructs were validated via agarose gel electrophoresis. All constructs were formulated and stored in sterile Milli-Q H2O.

Overview of the evaluated single- and dual-plasmid backbones. Single plasmid carrying 4D5 heavy- and light-chain expression cassettes (upper) or dual plasmids each carrying one expression cassette (lower) for the evaluated plasmid backbones, cpDNAL, cpDNA, and npDNA. Promoter and polyadenylation (pA) elements of the expression cassette were variable (Table 1). AmpR (ampicillin resistance marker), KanR (kanamycin resistance marker), F1 (F1 ori), ColE1 (ColE1 Ori), pUC (pUC ori), R6K (R6K mini ori), L (light chain), H (heavy chain).
Overview of key parameters of the constructs in this study
DES (murine desmin), DES-INR (murine desmin+initiator sequence), 26 polyA (polyadenylation signal), bp (basepairs), pDNA (plasmid DNA), npDNA (Nanoplasmid), H (4D5 heavy chain), L (4D5 light chain), TK (thymidine kinase), BGH (bovine growth hormone), RGB (β-globin), NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), sk-CRM4 (cis-regulatory module), 25 CK6 (creatine kinase enhancer). 26
Plasmid setups in bold are used throughout the different experiments. CAG, CMV1, and CMV2 are ubiquitous and CRM4, MCK, and MCKI are muscle-specific promoters. Dual plasmids are a combination of a heavy- and light-chain plasmid with an identical promoter denoted as “H/L.”. In a single plasmid, the order of H and L in the name represents their order on the plasmid shown in Fig. 1.
Cell lines and reagents
293F cells (Freestyle 293-F suspension cells purchased from Thermo Fisher Scientific, Waltham, MA in 2015) were maintained in FreeStyle 293 expression medium (Thermo Fisher Scientific). Cells were cultured in T175 flasks (Sarstedt, Nümbrecht, Germany) on an orbital shaker (Thermo Fisher Scientific) at 150 rpm and 8% CO2 in a 37°C humidified incubator. Murine C2C12 myoblast adherent cells (purchased from ATCC, Manassas, VA in 2016) were maintained in Dulbecco's Modified Eagle Medium (DMEM) F12 (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific). Cells were cultured in T175 flasks in a 37°C humidified incubator at 5% CO2. Identity of the 293F cell line was confirmed in 2017 using short tandem repeat analysis at the Laboratory of Forensic Biomedical Sciences (Leuven, Belgium). Identity of the C2C12 cell line was confirmed in 2018 at the IDEXX Laboratories (Ludwigsburg, Germany). All cell lines were routinely tested for mycoplasma contamination. Early passages from expanded master cell stocks were used for all experiments.
Mice
Gene transfer experiments were performed in 8–9-week-old female mice with an approximate weight of 18–20 g. BALB/c (BALB/cAnNCrl) mice were bred at the KU Leuven Animalium or purchased at Janvier (Le Genest-Saint-Isle, France) or Envigo (Horst, The Netherlands). Blood was collected via retro-orbital bleeding, processed to plasma, and stored at −20°C until analysis. All animal experiments were approved by the KU Leuven Animals Ethics Committee (project P157/2017).
In vitro transfection assay
293F and C2C12 transfection assays were developed to assess 4D5 production in vitro. For 293F, 500,000 cells were plated into 24-well plates (Costar; Sigma-Aldrich, Saint Louis, MO) in FreeStyle 293 expression medium. After 2 h, 0.50 μg for the largest construct or equimolar amounts were transfected per well using the X-tremeGENE HP DNA Transfection Reagent (Roche, Basel, Switzerland) following the manufacturer's instructions. For C2C12, 50 000 cells were plated into 24-well plates in DMEM F12 + 10% FBS. The next day, the medium was changed to DMEM F12 without FBS. After 2 h, plasmid transfections were performed as described above. Four hours after transfection, each well was spiked with DMEM F12 containing horse serum (HS), resulting in a final concentration of 2% HS. As soon as 24 h after addition of HS, myotube formation was observed, which appeared complete after 3 to 4 days. Independent of the assay, supernatant was collected 6 days after transfection and stored at −20°C before analysis by enzyme-linked immunosorbent assay (ELISA). Of note, the substantial differences in setup between 293F and C2C12 transfection assays do not warrant direct comparison of the resulting absolute 4D5 expression levels. Assay readout should thus be interpreted relatively within a given assay and cell type.
Intramuscular DNA electrotransfer in mice
Intramuscular pDNA electroporation was performed in the right tibialis anterior muscle following a previously optimized and validated preclinical protocol. 7 Briefly, the skin was prepared using depilatory product (Veet, Reckitt Benckiser, Slough, UK), at least 1 day before pDNA injection. Intramuscular delivery sites were injected with 40 μL of 0.4 U/μL hyaluronidase from bovine testes reconstituted in sterile saline (H4272; Sigma-Aldrich), ∼1 h before pDNA electrotransfer. Intramuscular injections of 30 μL of pDNA, diluted in sterile MQ H2O at 2 μg/μL for the largest constructs or equimolar amounts at lower concentrations for other constructs, were immediately followed by in situ electroporation using the NEPA21 Electroporator (Sonidel, Dublin, Ireland) with CUY650P5 tweezer electrodes at a fixed width of 5 mm. Signa Electrode Gel (Parker Laboratories, Fairfield, NJ) was applied to the muscle to decrease impedance below 0.4 Ohm. Three series of four 20 ms square-wave pulses of 120 V/cm with a 50 ms interval were applied with polarity switching after two of the four pulses. Pulse delivery was verified using the NEPA21 readout.
ELISA for mAb quantification
4D5 levels in cell culture supernatant and in plasma samples were quantitated using a previously described HER2-coated in-house ELISA. 7
Statistics
Statistical analyses and figure drawing were done using GraphPad Prism 8.0 (Graphpad Software, San Diego, CA). Data are presented as mean + standard error of the mean. Data of two groups were compared using unpaired Student's t-test, and data of three or more groups were compared using one-way analysis of variance with Tukey's multiple comparison. Two-sided p-values below 0.05 were considered significant.
Results
Impact of plasmid backbone size
To evaluate the impact of the plasmid backbone on mAb expression, two conventional plasmids (cpDNAL: 3.9 kb backbone, and cpDNA: 2.3 kb backbone) and a minimal plasmid (npDNA: 0.5 kb backbone) (Fig. 1) were evaluated in vitro in C2C12 (murine myoblast) and 293F (human embryonic kidney cells) and in BALB/c mice. All backbones were compared as a single plasmid with identical CAG-driven expression cassettes for the 4D5 heavy and light chain (referred to as “CAG-HL”) using equimolar amounts of DNA. Differences between the plasmids thus resided only in the bacterial elements in the backbone (Table 1 and Fig. 1). The cpDNA backbone and CAG promoter were previously selected as lead configuration, and have been extensively used for DNA-based mAb expression. 7 –9,20
In both cell lines, transfection with the minimal plasmid resulted in higher 4D5 supernatant levels than with conventional plasmids (Fig. 2A, B). An inverse association was observed between the size of the plasmid backbone and the resulting 4D5 levels, providing an npDNA > cpDNA > cpDNAL potency rank order. In a first in vivo experiment, cpDNAL-CAG-HL was compared with npDNA-CAG-HL. npDNA-CAG-HL gave significantly higher 4D5 plasma levels starting from day 7 (p = 0.0003) until the end of follow-up (Fig. 2C). cpDNAL-CAG-HL yielded peak 4D5 plasma levels of 3.2 ± 0.9 μg/mL, which remained above 0.5 ± 0.3 μg/mL throughout the 20 weeks of follow-up. npDNA-CAG-HL yielded significantly higher peak 4D5 plasma levels of 6.8 ± 1.3 μg/mL (p < 0.0001), which remained above 1.8 ± 0.9 μg/mL throughout the follow-up. In a second in vivo experiment, cpDNA-CAG-HL was compared with npDNA-CAG-HL. At the 2-week time point, npDNA-CAG-HL demonstrated a significantly higher 4D5 plasma level than cpDNA-CAG-HL (p = 0.03). Despite this initial statistically significant difference, mAb pharmacokinetics were comparable for both plasmids for the remaining 8 weeks of follow-up (Fig. 2D). Overall, a limited translation between in vitro and in vivo findings was observed. The construct with the largest bacterial backbone gave the lowest mAb concentrations, both in vitro and in vivo. However, in contrast to the in vitro findings, the standard (smaller) bacterial backbone gave comparable antibody levels as the npDNA in vivo. Nevertheless, the smaller size and limited bacterial backbone of npDNA hold benefits over conventional plasmids, and it was therefore considered the lead backbone in subsequent experiments.
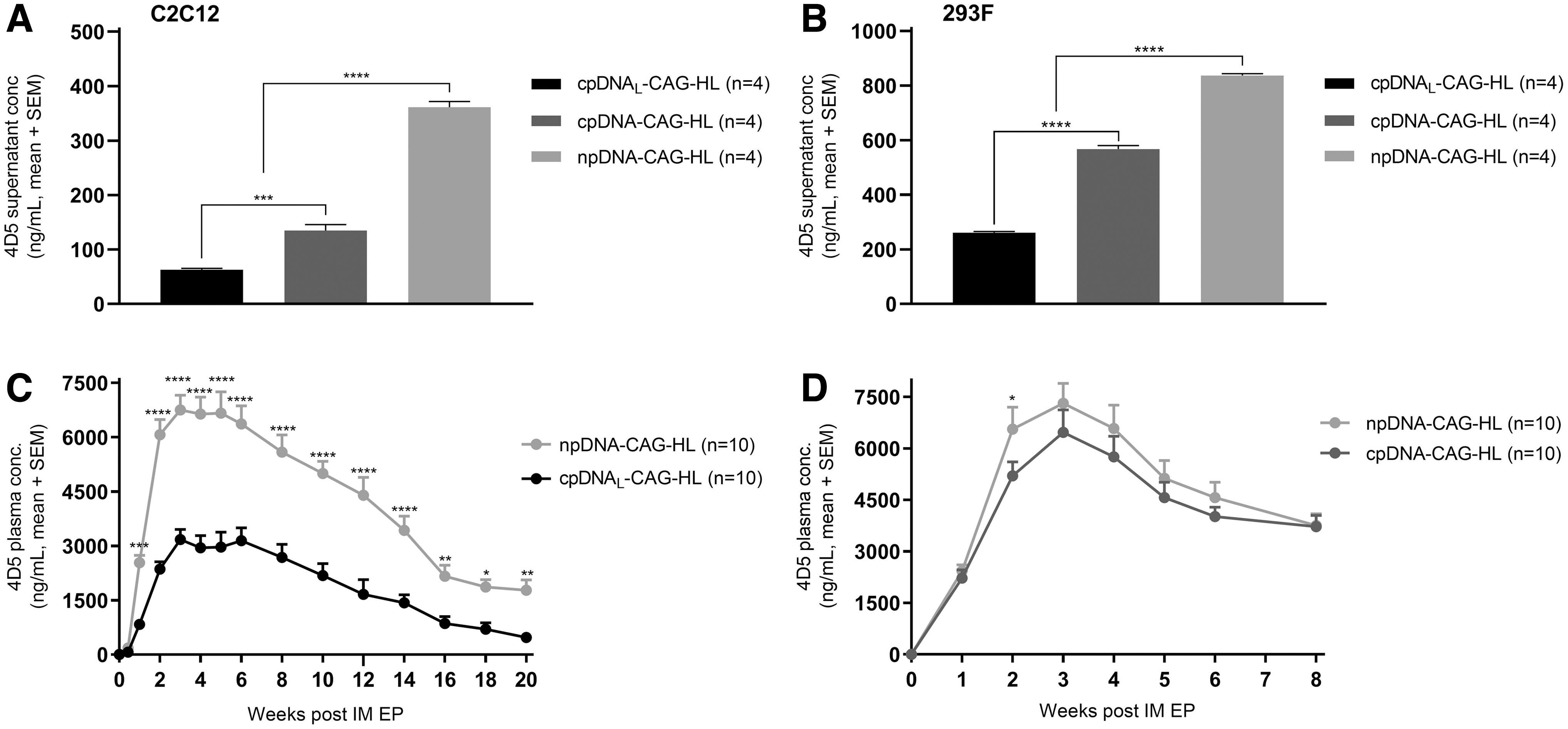
4D5 levels after delivery of conventional and minimal plasmid backbones in vitro and in vivo. 4D5 supernatant levels 6 days after transfection in
Expression cassette configurations in single-plasmid setup
The above-described single-plasmid setup contains two expression cassettes (in tandem) with identical promoters and polyadenylation signal sequences (Table 1). Such large tandem repeats are prone to recombination and complicate production. Furthermore, identical promoters in tandem could suffer from transcriptional interference due to competition for transcription factors, 21 resulting in lower mAb levels. To that end, two CMV promoters, CMV1 and CMV2, as well as a β-globin and bovine growth hormone polyadenylation signals, respectively, were cloned in the single npDNA backbone. CMV1 contains the human T lymphotropic virus type I (HTLV-I) R region, which is incorporated downstream of the promoter. 22 CMV2 contains a CMV enhancer modified to match consensus NF-κB sites as well as a small-intron region of minute virus of mice (Nature Technology Corporation). CMV2 is more potent than CMV1, which was more pronounced in 293F than in C2C12 (Supplementary Fig. S1). This could impact the expressed mAb chain ratio and resulting mAb levels. Indeed, excess light chain was previously observed to improve mAb levels in recombinant production. 23,24 To control for the difference in promoter potency, two configurations were evaluated, with each promoter driving either the light or the heavy chain, resulting in npDNA-CMV1-H-CMV2-L and npDNA-CMV1-L-CMV2-H. As reference, constructs with two identical CMV1 promoters and β-globin polyadenylation sequences, as well as matching cassette order, npDNA-CMV1-HL, and npDNA-CMV1-LH (Table 1), were included.
In C2C12, the constructs with two CMV1 promoters were outperformed by the constructs with CMV1 and CMV2 promoters, irrespective of antibody chain configurations (HL or LH) (Fig. 3A). In 293F, this was only the case for the HL configuration (Fig. 3B). The latter was confirmed in an independent repeat (data not shown). The data in C2C12 could be indicative of promoter competition, while less obvious in 293F. Irrespectively, the improvement linked to the use of two different promoters was rather limited. Indeed, the largest improvement of 4D5 levels in both cell lines was observed from swapping the order of the chains (HL to LH) (Fig. 3A, B), while maintaining the overall framework of the expression cassettes. Given the more limited effect, promoter competition between identical cassettes was not further investigated. We then compared npDNA-CMV1-L-CMV2-H and npDNA-CMV1-H-CMV2-L in vivo, to further investigate the impact of swapping chain order. At day 7 after gene transfer, significantly higher 4D5 plasma levels were observed for npDNA-CMV1-L-CMV2-H (p = 0.04). Thereafter, higher 4D5 levels were observed for npDNA-CMV1-H-CMV2-L, resulting in significant differences at weeks 5 (p = 0.0011) and 6 (p = 0.0018). However, the overall pharmacokinetic profiles were comparable, and considerably lower than previous findings with CAG (Fig. 2C, D). 7 The use of the CMV promoter was therefore not further investigated. As observed in the previous section, in vitro data did not correlate with in vivo findings, again illustrating the limited predictive value of in vitro data. Overall, these in vivo data suggest no improvement in mAb levels by swapping the mAb chain configuration.

4D5 levels after delivery of single plasmids with nonidentical expression cassettes in vitro and in vivo. 4D5 supernatant levels 6 days after transfection in
Single- versus dual-plasmid setup
Previous experiments focused on a single-plasmid driving expression of both mAb chains, considered preferable from a development and production perspective. However, mAbs can also be expressed through cotransfection of two plasmids, each driving the expression of the heavy and light mAb chain, respectively (Fig. 1). Therefore, both cpDNA and npDNA backbones were evaluated as a single (referred to as “HL”) or dual plasmid (referred to as “H/L”) (Table 1).
In vitro, the dual-plasmid setup outperformed the single-plasmid setup for both cpDNA and npDNA. However, the improvement in 4D5 concentrations was significantly higher with the minimal plasmid, both in C2C12 (p < 0.0001) and 293F (p < 0.0001) (Fig. 4A, B). We subsequently compared npDNA and cpDNA in a dual-plasmid setup in vivo. In contrast to the in vitro data, but similar to the in vivo single-plasmid data (Fig. 2D), npDNA demonstrated comparable mAb pharmacokinetics as cpDNA (Fig. 4C). Only at the 4-week time point, mAb plasma levels were significantly higher for npDNA-CAG-H/L (p = 0.03). As observed previously, in vitro data did not translate in vivo.
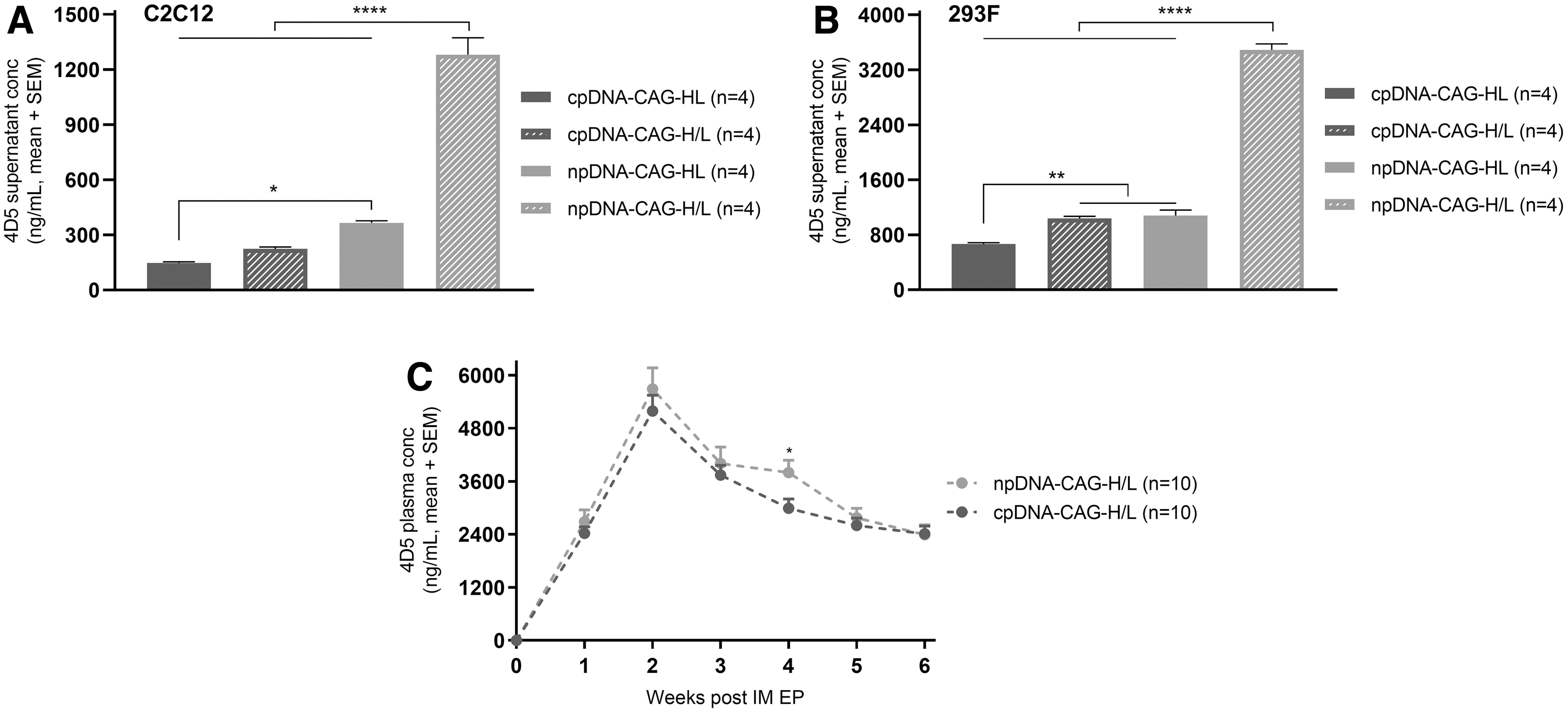
4D5 levels after single- or dual-plasmid delivery. 4D5 supernatant levels 6 days after transfection in
Muscle-specific promoters versus CAG promoter
Additional engineering focused on various engineered muscle-specific promoters in the dual npDNA setup. Sk-CRM4 (CRM4), MCK, and MCKI are all built around the muscle-specific desmin promoter (Table 1). CRM4 contains a specifically organized transcription factor binding site cluster. 25 MCK carries a set of muscle creatine kinase enhancer elements as well as a small-intron region of minute virus of mice (Nature Technology Corporation). MCKI is identical to MCK, but contains specific mutations that have been demonstrated to increase expression at the cost of reduced muscle specificity. 26
To rank order potency and muscle specificity of the promoters, transfections were carried out in C2C12 and 293F. In C2C12, npDNA-CRM4-H/L, npDNA-MCK-H/L, and npDNA-MCKI-H/L yielded similar 4D5 levels compared with npDNA-CAG-H/L (Fig. 5A). In 293F, 4D5 levels for the different muscle-specific promoters were more variable (Fig. 5B). Assuming that reduced mAb supernatant levels in 293F are reflective of improved muscle specificity, the following specificity rank order emerged: CRM4 > MCK > CAG > MCKI. Although MCKI gave a higher expression than CAG in this assay, and consequently appears less muscle specific, expression of a reporter using these promoters in various cell lines resulted in low expression for MCKI in two nonmuscle cell lines compared with CMV (Supplementary Table S1, data provided by Nature Technologies). Subsequently, all three muscle-specific promoter constructs, npDNA-CRM4-H/L, npDNA-MCK-H/L, and npDNA-MCKI-H/L, were compared in vivo. 4D5 plasma levels peaked between 12 and 16 μg/mL for each of the muscle-specific constructs (Fig. 5C), higher than observed in previous experiments with the CAG promoter (Fig. 4D). To confirm this, npDNA-MCK-H/L, containing the least leaky newly engineered muscle-specific promoter (Fig. 5B), was compared head-to-head with npDNA-CAG-H/L. Significantly higher mAb plasma levels were observed starting from week 2 after gene transfer for npDNA-MCK-H/L (p < 0.0001), which remained throughout follow-up (Fig. 5D). npDNA-MCK-H/L resulted in peak 4D5 plasma levels of 11.1 ± 2.3 μg/mL, which remained above 2.9 ± 1.0 μg/mL throughout the 8 weeks of follow-up, whereas npDNA-CAG-H/L resulted in peak 4D5 plasma levels of 6.7 ± 2.5 μg/mL, which remained above 1.3 ± 0.6 μg/mL. In contrast to the comparable promoter strength observed in C2C12 in vitro, in vivo data show that muscle-specific promoters can result in improved mAb levels over ubiquitous promoters.

4D5 levels after delivery of dual plasmids with ubiquitous and muscle-specific promoters in vitro and in vivo. 4D5 supernatant levels six after transfection in
Discussion
In the current study, we combined engineering at the level of the plasmid backbone and mAb expression cassette to generate a safer and more potent DNA-based platform for intramuscular antibody gene transfer. Improvements could strengthen the potential and broaden the implementation of DNA-encoded antibodies, and possibly be translated to other DNA-encoded therapeutics.
Conventional plasmids typically contain bacterial elements. Especially antibiotic resistance markers carry biological safety concerns, and regulatory authorities recommend avoiding them for therapeutic use. 27,28 Thereto, plasmid backbones free of antibiotic resistance markers have been developed and validated for large-scale antibiotic-free manufacturing, such as pCOR, pORT, and npDNA (NTC9385R), the format investigated in this study. 17,29,30 Other constructs with even less bacterial backbone have also been developed, for example, minicircles. The manufacturing process thereof typically involves intramolecular recombination to separate the producer vector from the minicircle. This bottleneck can result in a more labor- and cost-intensive production compared with formats that retain residual bacterial elements (reviewed in Alves et al.). 31 For Nanoplasmid production, a high-yield antibiotic-free selection system was previously established, 14,18 providing a clinically scalable manufacturing methodology in line with safety recommendations by regulatory agencies. 27,28
Overall, minimal plasmids not only provide safer alternatives but have also demonstrated improved and/or sustained transgene expression over conventional plasmids in a variety of tissues, including lung, liver, kidney, and heart. 32 –35 Indeed, smaller vectors can result in more effective cell transfection and are less prone to shear forces associated with delivery. 32 This could support improved expression levels, which is critical to the success of plasmid-based antibody gene transfer. Up until now, minicircles are the only minimal plasmids reported for antibody gene transfer, although delivered through hydrodynamic tail vein injection and with mixed results in terms of expression levels (reviewed in Hollevoet et al.). 2 Nanoplasmids have not previously been explored in this context.
In our in vitro data set, mAb levels were inversely correlated to plasmid size for each of the evaluated backbones, indicating a higher transfection efficiency for smaller backbones, which is in line with reported literature. 36 –38 In vivo, only the largest conventional plasmid (10.2 kb) gave lower mAb levels, whereas the regular conventional plasmid (8.6 kb) demonstrated similar levels compared with npDNA (6.8 kb). These data demonstrate that a reduction in size beyond the standard conventional backbone does not improve mAb levels, and further gain from backbone downsizing is likely limited. Furthermore, the conventional plasmid backbones resulted in mAb pharmacokinetics comparable to that of the Nanoplasmid, suggesting they do not suffer from increased transgene silencing. 39 As opposed to previous reports, 32 –35 our in vivo data show no improvement in transgene expression from a minimal plasmid. We believe this can possibly be attributed to differences in the expressed transgene, target tissue, and/or transfection methodology. In fact, Andrews et al., using a comparable intramuscular antibody gene transfer setup, demonstrated even higher mAb levels using a >2 kb larger backbone (gWiz > pVax), 10 suggesting that backbone-specific sequence elements and organization, rather than their size, are relevant to the achievable transgene expression. 34,40 While the bacterial backbone is reduced in the Nanoplasmid, additional reduction of CpG motifs within the antibody expression cassette could improve transgene expression. 41,42 Overall, our current data suggest that minimal plasmids as such have little impact on the magnitude of in vivo antibody expression achieved through intramuscular electroporation, apart from providing a better biosafety profile. While other minimal plasmid configurations remain of interest, 43 the ability for upscale of plasmid production should be a key criterion for plasmid selection.
Separation of the expression cassettes into two (conventional) plasmid backbones has previously resulted in improved mAb levels in vivo, 9,10 despite conflicting results in vitro. 44 In the current study, mAb levels in vitro were favored in the dual npDNA setup. In vivo, however, dual cpDNA demonstrated similar mAb levels compared with dual npDNA, confirming that size reduction had minimal impact.
To further improve the safety of our plasmid system, we explored tissue-specific expression through muscle-specific promoters, which could lead to more stable prolonged transgene expression than ubiquitous promoters. 7,25,45 In C2C12, mAb levels were comparable between the muscle-specific promoters and CAG. All muscle-specific promoters gave a similar, robust mAb expression in vivo. The newly engineered variant, MCK, yielded roughly 1.5- to 2-fold higher plasma concentrations than the ubiquitous CAG promoter, again demonstrating lack of correlation with the in vitro data. mAb pharmacokinetics followed a comparable trend for CAG and all three muscle-specific promoters, indicating that muscle-specific designs did not result in a more stable transgene expression. To the best of our knowledge, this is the first report demonstrating that muscle-specific promoters can improve in vivo mAb expression over potent ubiquitous promoters.
Overall, we explored various routes to increase mAb expression. While this multitude of parameters could complicate comparison between assay readouts, it was clear that not all modifications were successful. Additional improvements in mAb expression may be achieved through alternative delivery methodology, including adaptations of the formulation and delivery device, 10,12,46 as well as through mAb sequence optimizations. 11,12,44,47 Of note, throughout our experiments, improvements in mAb levels in vitro generally did not translate in vivo and vice versa. The available literature provides a mixed and complex picture. While several studies confirm this lack of in vitro and in vivo correlation (e.g., for modifications in cassette configurations and orientations or antibody sequence), other studies did show a correlation. 8,10 –12 To possibly elucidate the origin of the in vitro and in vivo disconnect, direct transfection of C2C12 myotubes (instead of myoblasts) might be relevant. However, in preliminary experiments, transfection efficiency in fully differentiated myotubes appeared to be very low, resulting in nearly undetectable mAb levels (data not shown). In addition, it could be relevant to explore other in vitro setups, for example, alternative muscle cell types, of both mouse and human origin 48 –50 and other means of in vitro transfection, including electroporation. For now, the lack of translation illustrates a need for in vivo evaluation to select the most potent DNA-based mAb configurations. To improve and expedite clinical translation, the most promising configurations could also be evaluated in larger animal models, for example, sheep. 8
In conclusion, we evaluated various backbone and expression cassette modifications to our DNA-based antibody platform. Even though complicated by a rather limited correspondence between in vitro and in vivo findings, these efforts provide guidance on approaches for future optimization. Indeed, we identified various clinically relevant improvements in both potency and biosafety, including the use of a scalable minimal plasmid and a potent muscle-specific promoter.
Footnotes
Authors' Contributions
G.V., N.G., J.A.W., P.D., and K.H. contributed to the study design. G.V. and E.D.S. performed the experiments. G.V., E.D.S., J.A.W., N.G., P.D., and K.H. interpreted the results. G.V. wrote the article, which was reviewed and edited by P.D. and K.H. All authors reviewed and approved the article for publication.
Author Disclosure
J.A.W. has commercial interests in Nature Technology Corporation. All other authors declare no conflict of interest.
Funding Information
This research is supported by Research Foundation—Flanders (FWO: PhD mandate 1S50617N to G.V.; research project G0E2117N to P.D. and K.H.), KU Leuven (C2 grant: C22/15/024 to P.D. and K.H.), and Flanders Innovation & Entrepreneurship (VLAIO: IWT.150743 to K.H.).
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.