
Abstract
Recombinant adenovirus vectors enable highly efficient gene delivery in vitro and in vivo. As a result, they are widely used in gene therapy, vaccination, and anticancer applications. We have previously developed the AdZ vector system, which uses recombineering to permit high-throughput cloning of transgenes into Adenovirus vectors, simplifies alteration of the vector backbone, and enables rapid recovery of infectious virus, even if a transgene is incompatible with vector replication. In this study, we adapt this vector system to enable high-throughput cloning of sequences for CRISPR/Cas9 editing. Vectors were optimized to ensure efficient cloning, and high editing efficiency using spCas9 and single guide RNA (sgRNA) sequences in a single vector. Using a multiplicity of infection of 50, knockout efficiencies of up to 80% could be achieved with a single sgRNA. Vectors were further enhanced by altering the spCas9 sequence to match that of SniperCas9, which has reduced off-target activity, but maintains on-target efficiency, and by applying modifications to the sgRNA sequence that significantly enhance editing efficiency. Thus, the AdZ-CRISPR vectors offer highly efficient knockout, even in hard to transfect cells, and enables large-scale CRISPR/Cas9 projects to be undertaken easily and quickly.
INTRODUCTION
CRISPR/Cas9
Furthermore, plasmids, mRNA, and protein transfection require cells that transfect efficiently, while lentiviruses result in permanent expression of Cas9, which may lead to increased off-target effects over time. These issues also limit use of plasmids, proteins, and lentiviruses, for in vivo CRISPR/Cas9 delivery. As a result, adeno-associated viruses (AAV) are often used for in vivo purposes, where their low immunogenicity is extremely advantageous in permitting long-term expression of transgenes for gene therapy applications. 6 However long-term expression is unnecessary, and may be undesirable, in the case of CRISPR/Cas9, and the relatively small packaging limits of AAV can necessitate the use of novel Cas9 variants. 7 –9
Adenovirus (Ad) vectors circumvent many of these issues 9 ; they transduce a wide variety of cells in vitro and in vivo with extremely high efficiency; they do not integrate; they have higher packaging limits; they have an excellent in vivo safety profile; and they give transient expression. As a result, they are widely used for gene delivery, for vaccination, and in oncolytic applications in vivo, and as highly efficient gene delivery agents in basic research. They also grow rapidly, to high titers, and are extremely stable, readily manipulated, and comparatively cheap to grow at scale. Together, these properties make them extremely useful for CRISPR/Cas9 delivery in vitro and in vivo.
The inherent immunogenicity of Ad vectors does have the potential to reduce the efficacy of in vivo delivered CRISPR/Cas9 in comparison to AAV. 6,10 Nevertheless, a number of studies have demonstrated that CRISPR/Cas9 expressing Ad vectors can successfully treat inherited disorders in animal models, 9 and genetic or chemical manipulation of the vector can be used to reduce immunogenicity further. 11
The adenovirus type 5 (Ad5) genome is the most widely used in vector applications. It comprises 36 Kb linear dsDNA, bracketed by inverted repeats. Genes are grouped into four early transcription units (E1, E2, E3, and E4), delayed early, as well as a multiple spliced major late transcript. 12 Most recombinant adenovirus (RAd) vectors are rendered replication deficient by deleting the essential E1 region, and must therefore be propagated on helper cells expressing E1 in trans. 13,14 The E3 region is nonessential for replication in vitro, and removing E3, in addition to E1, permits insertion of coding sequences up to ∼8 Kb. Additional regions of the Ad genome can be removed to provide for larger inserts; however, these vectors become more difficult to produce for routine applications.
Early RAd vector systems involved insertion of a transgene into a transfer plasmid using traditional cloning techniques, and then recombination of that transfer plasmid with the Ad genome in Escherichia coli, yeast, or mammalian cells. 15 –18 These systems were widely used, however they were relatively labor-intensive and poorly suited to cloning multiple genes simultaneously due to the requirement for sequential subcloning steps to generate the transfer vector, followed by additional steps needed to recombine the vector with the backbone. Recombination could be problematic, and could suffer from low efficiency. Finally, if expression of the transgene was toxic, or incompatible with vector replication, the final vector could not be propagated.
To address these problems, we developed a novel RAd vector (“AdZ”;
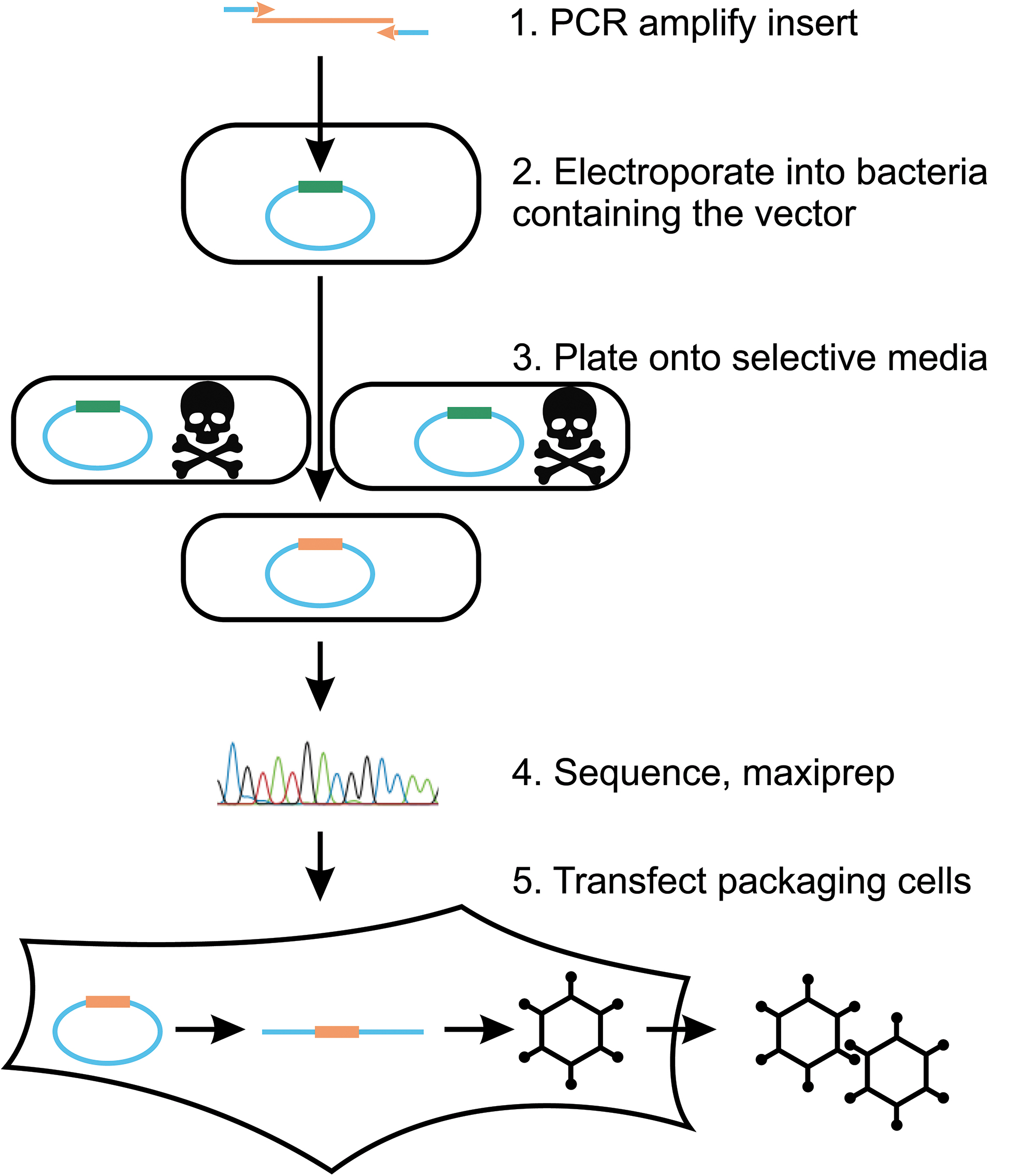
Schematic of transgene insertion in the AdZ system. Inserts are PCR amplified with sequences in the primer that are homologous to the target insertion site. Escherichia coli containing the RAd vector as a stable BAC are grown to mid-log phase, heat-shocked to induce expression of genes for recombineering, and electroporated with the PCR product. After recovery, bacteria are plated on selective media. Colonies containing the insert are identified based on blue-white screening, miniprepped, and modifications verified by Sanger sequencing. Vectors are maxiprepped and transfected into 293TREx cells. The RAd genome must be linearized to be infectious; this occurs through I-SceI sites engineered into the genome termini, which are cut by I-SceI expressed from the vector backbone when in mammalian cells. In addition, in 293TREx cells, expression of the cloned transgene is silenced by expression of TetR, but is constitutively on in the absence of TetR (i.e., most other cell types). BAC, bacterial artificial chromosome.
An additional advantage of generating a RAd vector compatible with recombineering is that this technique is not restricted to insertion of transgenes. It can also be used to modify any region of the Ad genome, in a completely scarless manner. As a result, this vector system has been used by our laboratory and others to generate oncolytic viruses, 27,28 to enhance their infection efficiency, 29,30 to expand the tropism of adenovirus vectors, 31 to alter tropism specificity of the virus to become tumor selective, 32 –36 to shield virus from neutralizing antibodies, 37 to enhance immunogenicity, 38 and to dissect adenovirus gene function. 39
In this work, we expand the use of the AdZ system to generate a CRISPR/Cas9 vector that is capable of performing CRISPR-Cas9 gene editing with high efficiency and accuracy, for use in vivo, or with hard to transfect cells in vitro.
MATERIALS AND METHODS
Cells and viruses
293TREx cells were purchased from Invitrogen and RPE-1 cells were purchased from Clontech. HFFF-hCAR are human fetal foreskin fibroblasts (HFFF) that have been immortalised with human telomerase, and express the human coxsackie-adenovirus receptor (hCAR), and have been described before. 39 All cells were grown in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal calf serum, at 37°C, in 5% CO2.
The AdZ vector has been described before. 19 Vector was recovered from AdZ constructs by midiprepping DNA (Nucleobond Xtra Midi, Machery-Nagel), and transfecting 293TREx cells using effectene, according to manufacturer's instructions. Vector was passaged in 293TREx cells, and titrated by infecting 293TREx cells with serial dilutions of the preparation, followed by staining for adenovirus proteins as previously described. 40 In all cases, multiplicity of infection (MOI) was calculated on the basis of spot-forming units, which are equivalent to plaque-forming units. 40
Recombineering
All recombineering steps were carried out as previously described. 19 In brief, SW102 bacteria containing the AdZ bacterial artificial chromosome of interest were grown at 32°C until OD600 = 0.55, and then incubated at 42°C for 15 min to induce expression of recombineering genes. Bacteria were washed twice with ice-cold water, resuspended in a small volume, and DNA constructs to be recombined with the vector were added. Mixtures were electroporated using the “EC3” program (Micropulser, Biorad), then 1 mL LB media was added. After recovering for 1 h at 32°C, bacteria were plated onto selective media. For modifications to the vector backbone, an initial positive selection step using a cassette encoding ampicillin resistance, lacZα, and sacB was performed.
This cassette was PCR amplified using primers containing 80 bp homology to the target insertion site in the 5′ region of both primers, gel purified, and recombineered into the target site, and bacteria were plated on media containing X-gal, IPTG, chloramphenicol (12.5 μg/mL), and ampicillin (50 μg/mL) to select for recombinants. In a second, negative selection step, sequences to be inserted were PCR amplified using 80 bp arms of homology, and gel purified, before being inserted in place of the selection cassette, again using recombineering. This time, bacteria were recovered for 3 h in 5 mL of LB after electroporation, and plated on LB Agar lacking NaCl, but containing 5% sucrose. This efficiently selects against colonies in which the original selection cassette remains. Full protocols are available on our website, AdZ.cf.ac.uk.
Primers used in recombineering are listed in Table 1, all constructs were verified by Sanger sequencing. To insert spCas9 under the control of the HCMV major immediate early promoter (MIEP), the entire ORF was gene synthesized and inserted into the AdZ vector pAdZ5-CV5. 19
Primers used to construct AdZ-CRISPR vectors
To insert the sgRNA sequence, the ampr/lacZ/sacB cassette was inserted either immediately after the polyadenylation sequence for spCas9 or in the E3 region. The cassette was then replaced with a sgRNA sequence that was PCR amplified from pX459 (Addgene), using primers gRNAF-1/gRNAR-1 (after the polyadenylation sequence, forward orientation), gRNAF-2/gRNAR-2 (after the polyadenylation sequence, reverse orientation), gRNAF-3/gRNAR-3 (E3, forward orientation), and gRNAF-4/gRNAR-4 (E3, forward orientation). To enable insertion of spacers targeting different genes into the sgRNA constructs, the ampr/lacZ/sacB cassette was inserted in place of the original gene-specific spacer sequence, using primers sacbF gRNA and sacbR gRNA.
To modify spCas9 to carry HypaCas9 modifications, the ampr/lacZ/sacB cassette was amplified using primers SB-HypaCasF and SVB-HypaCasR, and then the cassette removed and replaced with HypaCas modifications using oligo HypaCas. To modify spCas9 to carry the SniperCas sequences, the ampr/lacZ/sacB cassette was amplified using primers Sniper-SacBF and Sniper-SacBR, before being replaced with a gene-synthesized DNA fragment carrying the appropriate modifications. To introduce enhancing modifications into the sgRNA sequence, the ampr/lacZ/sacB cassette was amplified using primers gRNA_optSacBF and gRNA_optSacBR, and then replaced using the paired oligos gRNA_optF and gRNA_optR.
To introduce spacers targeting additional genes by CRISPR/Cas9, into vectors carrying the original sgRNA, the primers listed in Table 2 were used. To introduce the same spacers into vectors carrying the enhanced sgRNA, the primers listed in Table 3 were used.
Primers used to introduce spacers targeting different genes
Primers used to introduce spacers targeting different genes into an enhanced single guide RNA
Plasmid transfection
gRNA were cloned into the pX459 vector according to the guide at Addgene (
Flow cytometry
Cells were dissociated with TrypLE, stained with antibodies targeting HLA (W632), PDGFRα (BD Pharmingen), or CD155 (D171; Invitrogen) followed by anti-mouse AF647 (Thermo), before being washed and fixed in 4% PFA, and then run on a Accuri C6 cytometer (BD) and analyzed in FlowJo. All gates were set using a nonbinding isotype-matched control antibody (for the negative gate) and cells that had been transduced with a control RAd vector lacking expression of Cas9 (for the positive gate).
Analysis of genetic editing by sequencing
DNA was extracted from cells using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions, and then a 151 bp region surrounding the target site for the beta-2-microglobulin (B2M) gRNA was PCR amplified using primers AGAGACTCACGCTGGATAG and CTGGGCACGCGTTTAAT, and the Expand HiFi PCR kit (Roche). Amplified DNA was gel purified, TOPO™ cloned into pCR4.1-TOPO according to manufacturer's instructions (Thermo), and then sequenced by Sanger sequencing (Eurofins Genomics).
RESULTS
Generation of an AdZ vector encoding CRISPR/Cas9 sequences
To determine whether the AdZ vector system could be converted into a vector capable of mediating gene editing with CRISPR-Cas9, we inserted the complete coding sequence for a human optimized spCas9, along with two nuclear localization signals, and a FLAG tag, downstream of the tetR-regulated HCMV MIEP in the AdZ vector. We then inserted an expression cassette encoding the U6 promoter, along with an sgRNA capable of targeting Cas9 to B2M. 42 Where two expression cassettes are incorporated into a single RAd vector, two sites are commonly used for the second cassette, either immediately following the first (in the E1 region) or in the E3 region.
To determine whether there was any variation in expression efficiency between these sites, we inserted the sgRNA expression cassette either directly after the Cas9 expression cassette or in E3, in both forward and reverse orientations (Fig. 2A). Vectors were then grown, and knockout efficiency was compared. The vector with the sgRNA in the reverse orientation within E3 grew to 20-fold lower titers than the other three vectors, and therefore was not tested further (Fig. 2B). However, the remaining three vectors all grew to similar titers. To determine the relative efficiency of knockout, a cleavage detection assay can be used. However, cleavage does not necessarily correlate with loss of protein, due to reinitiation or exon skipping. 43

Construction of a CRISPR/Cas9 RAd vector in the AdZ system.
We therefore assessed knockout by flow cytometric staining for HLA-I, which is lost in the absence of functional β2m. No major difference was observed between the vectors when HLA-I knockout was tested by flow cytometry (Fig. 2C). No cells were observed with an intermediate level of HLA-I expression. This implies that in the majority of cells where gene editing occurred, both alleles were altered. This effect also occurred when using spacer sequences targeting other genes, when using lentiviral or plasmid-based vectors, and in other cell types (not shown), and is consistent with published data. 44
AdZ vectors support rapid, high-efficiency cloning of sgRNA sequences
We next replaced the 20 bp spacer within the sgRNA that targets B2M, with a selectable cassette encoding amp r, lacZα, and sacB, in each vector (Supplementary Fig. S1). This cassette enables the selection of correctly recombined colonies once spacers targeting genes of interest have been inserted. For any gene to be targeted, a 100 bp oligo encoding the 20 nt spacer that targets that gene is synthesized, along with 40 bp on either side that has homology to the sgRNA cloning site. Recombineering is then used to replace the cassette with the oligo, followed by plating on selective media containing sucrose and X-gal, and selecting white colonies (Supplementary Fig. S1).
We then cloned 16 different spacer sequences targeting a variety of genes, into vectors containing sgRNA in either E1 or E3. Cloning efficiency was consistently higher in the vector containing the sgRNA in E3 (Fig. 3A); this vector was therefore used in all experiments moving forward. When white colonies were sequenced, all contained the intended spacer insertion; however, single base changes or deletions were occasionally observed either within the spacer insert or in the region where recombination had occurred, presumably as a result of errors during recombination.
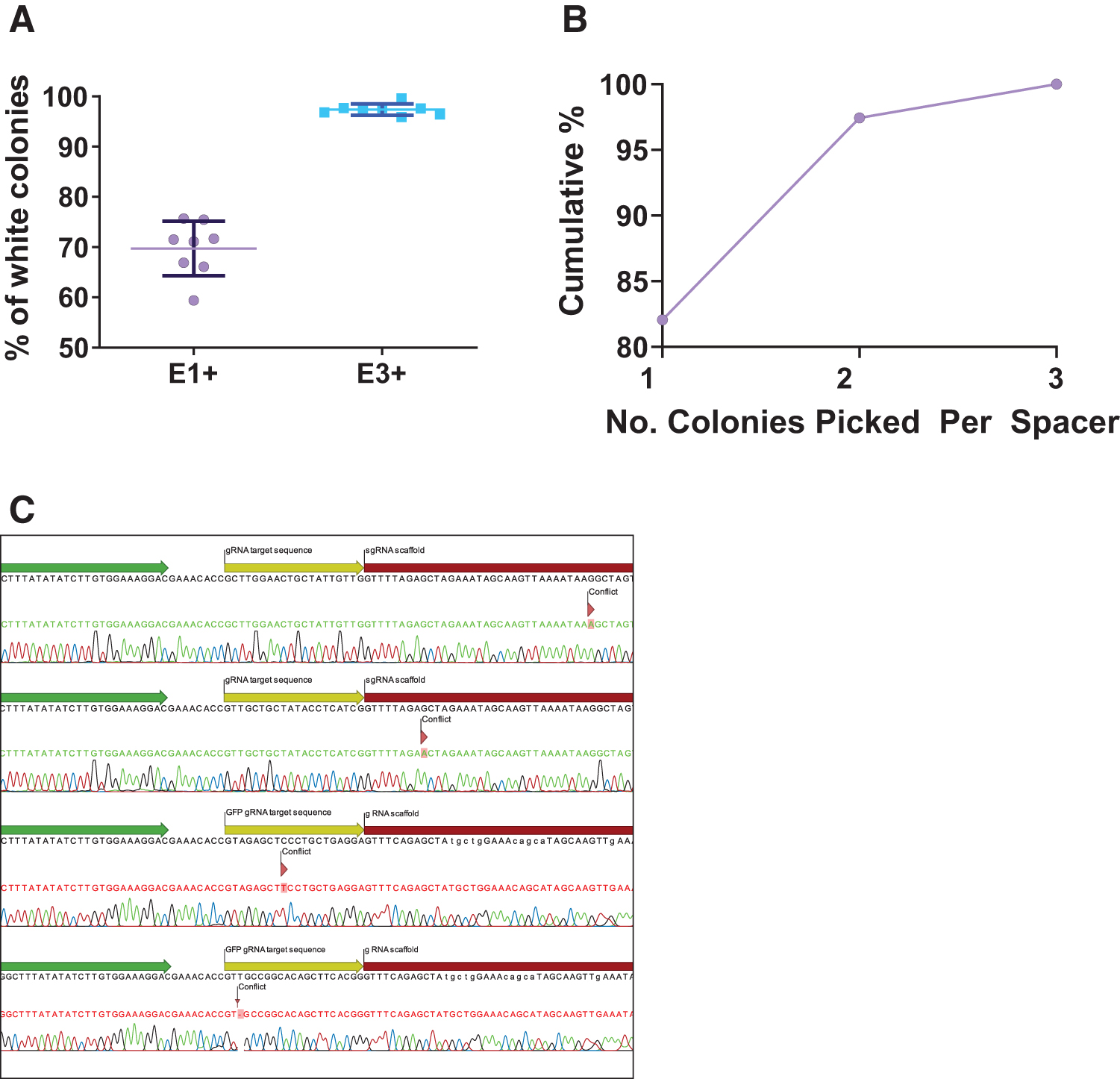
Cloning efficiency of different spacers into sgRNA within the AdZ-CRISPR vector.
When 39 different spacers were inserted, 32 colonies had the correct sequence after a single clone was picked, a further 6 after a second colony, and the final one after the third colony was sequenced (Fig. 3B, C). Thus, cloning efficiency is extremely high, and sequencing a single colony from each plate is sufficient to obtain sequence-verified clones for the majority of inserts.
Higher MOI enables higher rates of editing
Having established a RAd vector that was compatible with high-throughput cloning of sgRNA, we determined the parameters that affected knockout efficiency. HFFF-CAR (human fetal foreskin fibroblasts expressing the CAR) were transduced with the CRISPR RAd targeting B2M, at MOI = 5. We did not observe strong knockout at 4 days post-transduction; however, when cells were left to grow, a population containing lower surface levels of HLA-I was apparent by day 9, and by day 13, HLA-I signals in this population were reduced to the level of negative control isotype staining (Fig. 4A). Thus, in this system, it was important to wait 10–14 days to clearly define knockout populations.
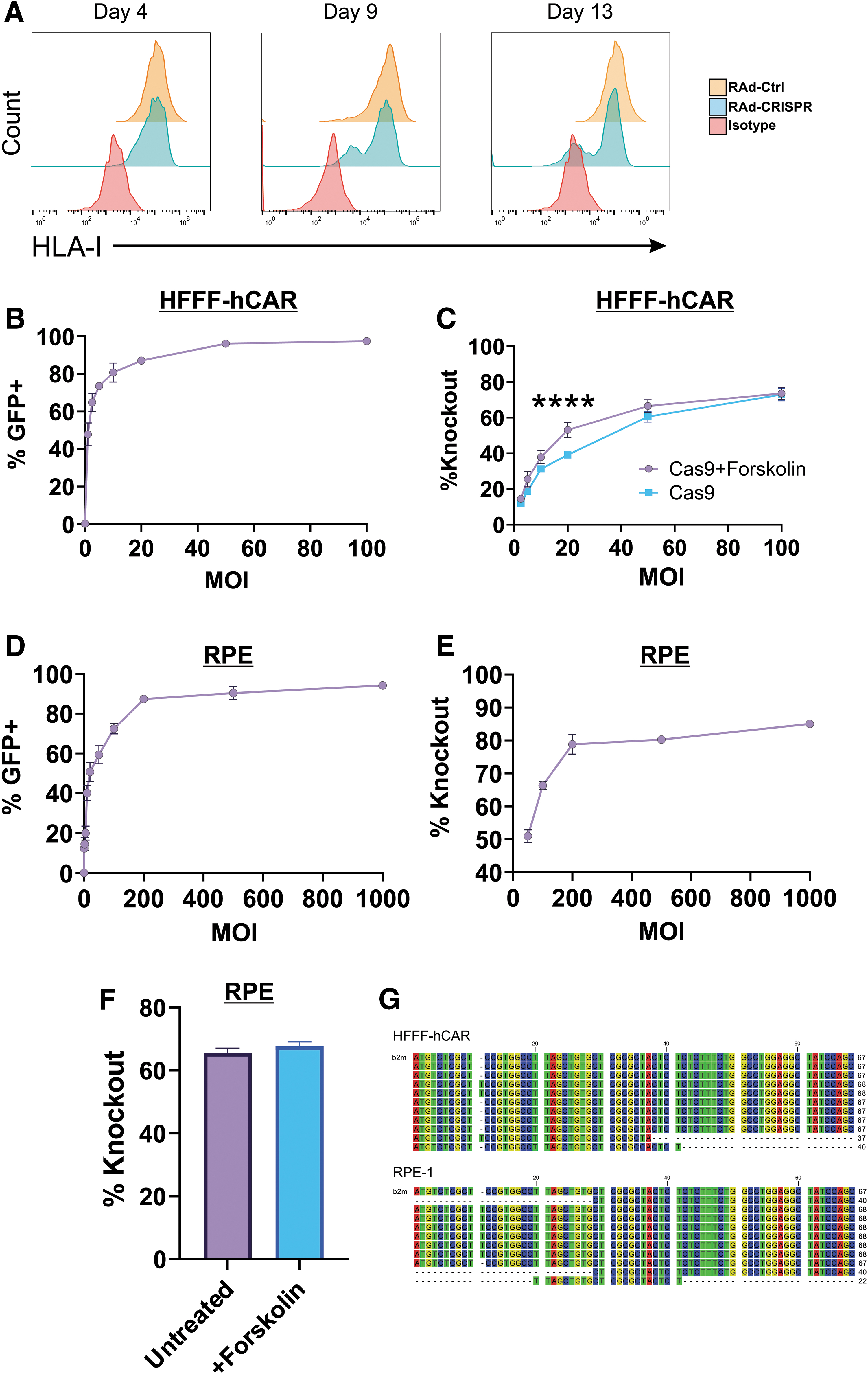
CRISPR/Cas9 editing efficiency.
Although the ability of the CRISPR RAd to give complete knockout in 50% of cells was impressive (Fig. 2C), especially given that the low transfection efficiency of these cells meant that transfection of the same sgRNA in the popular PX459 plasmid vector did not result in detectable knockout (data not shown), we investigated whether it was possible to improve knockout efficiency by adding more vector. Using a GFP expressing RAd vector, ∼70% of HFFF-CAR are transduced at MOI = 5, but this can be increased to >95% by MOI = 50 without observation of toxicity (Fig. 4B).
In accordance with this, knockout efficiency increased at higher MOI (Fig. 4C). We also investigated whether knockout efficiency could be improved by increasing expression levels from the promoter that drives Cas9 expression, by the use of forskolin. 45 The addition of forskolin increased knockout efficiency slightly, at lower MOIs, resulting in knockout of HLA-I in almost 80% of cells (Fig. 4C).
HFFF-hCAR transduce relatively well with RAd, due to high levels of CAR expression. 39 To determine whether the same result occurred with cells that express lower amounts of CAR, we used the epithelial cell line RPE-1. These cells require MOI = 200 to achieve >90% transduction (Fig. 4D). Increasing the MOI to 200 gave a significant increase in knockout efficiency without obvious toxicity; however, addition of further vector did not lead to increases in knockout (Fig. 4E). Unlike in HFFF-CAR, the addition of forskolin did not increase knockout efficiency (Fig. 4F).
Thus, the optimum knockout efficiency is largely dependent on using an appropriate MOI for the cell type under investigation, and reached a maximum of ∼80% knockout. Enhancing expression levels of Cas9 with forskolin can sometimes lead to small improvements in knockout efficiency at lower MOI. To provide direct evidence that the AdZ-CRISPR vector resulted in genome editing, DNA was extracted from transduced cells, a 151 bp region surrounding the recognition site for the B2M spacer was PCR amplified, TOPO cloned, and individual clones sequenced.
The proportion of clones demonstrating editing would not be expected to exactly match the proportion of cells demonstrating knockout of protein expression; edited clones can still express protein due to re-initiation or exon skipping, 43 while cells in which Cas9 editing has resulted in larger deletions surrounding the target site (a well-known phenomenon with CRISPR editing 46,47 ) will lack protein expression, but would not be detected due to loss of one or both PCR primer binding sites. Nevertheless, we observed clear evidence of genetic editing; following transduction of cells with an MOI sufficient to infect >90% of cells, 4/10 (HFFF-hCAR, expressing high CAR levels) or 9/10 (RPE-1, expressing low CAR levels) PCR clones demonstrated edits (Fig. 4G).
AdZ vectors carrying improved Cas9 and sgRNA sequences support optimized editing
One of the risks with CRISPR-Cas9 is that gene edits can be made at sites other than those intended. As a result, attempts have been made to increase the fidelity of Cas9 edits, through mutating the Cas9 protein. 48 –53 To investigate whether higher fidelity spCas9 worked in the context of our RAd vector, we took advantage of the ability of recombineering to introduce seamless modifications anywhere in the genome; introducing N692A, M694A, Q695A, and H698A mutations into Cas9 within the RAd, generated HypaCas9 (Supplementary Fig. S2). 51 This has been reported to show high genome-wide specificity without compromising on-target activity in human cells. However, using the B2M sgRNA, we consistently observed a lower knockout efficiency with HypaCas9, and this could not be enhanced with the addition of forskolin (Fig. 5A).

Optimizing CRISPR/Cas9 expressing AdZ vectors.
As an alternative, we therefore introduced the mutations F539S, M763I, and K890N, collectively termed SniperCas9 (Supplementary Fig. S2). 52 These mutations have also been reported to reduce off-target effects without affecting on-target editing by Cas9. They offer a further advantage; however, in that, SniperCas9 is compatible with the addition of an extra “G” base at the beginning of the sgRNA targeting sequence. That is, the sgRNA must begin with a G for efficient expression from the U6 promoter; where a sgRNA does not naturally begin with a “G” one can be added, but this modified sequence cannot normally be used with enhanced Cas9 variants.
SniperCas9 is equally as effective when using such modified sgRNA sequences, giving greater flexibility in sgRNA selection. The introduction of SniperCas9 into our vector had a small impact on knockout efficiency (Fig. 5B); however, this was much less dramatic than seen with HypaCas.
Finally, we also introduced two enhancing modifications to the sgRNA scaffold. These mutations extend the sgRNA duplex by 10 bp and remove a potential RNA pol III pause signal in the sgRNA, increasing the efficiency of CRISPR/Cas9 editing (Supplementary Fig. S2). 54 The addition of this alteration had no effect on editing efficiency with the B2M sgRNA (Fig. 5C); however, it did enhance the activity of a vector carrying SniperCas9, such that it gave knockout efficiencies identical to Cas9 (Fig. 5D). The B2M sgRNA already enabled very efficient knockout. To determine whether this modification enhanced the rate of knockout with lower efficiency sgRNA, we cloned multiple spacers targeting PDGFRA or CD155 into the original vector, or the vector containing both SniperCas9 and the enhanced sgRNA scaffold.
Using these optimized vectors, all 8 sgRNA showed enhanced activity, including three that showed no activity at all in the original vector, but clear activity in the enhanced vector (Fig. 5E, F). Although these vectors contained two modifications (both sgRNA and Cas9 were modified), given that the SniperCas9 showed equal or slightly reduced activity to wild-type Cas9 (Fig. 5B), it is likely that this enhancement was due to the sgRNA sequence. Thus, the “SniperCas9/sgRNA+” vector offers both more accurate editing and significantly improved editing efficiency.
DISCUSSION
A number of systems have aimed to improve on adenovirus vectors that required traditional cloning into a transfer vector, followed by recombination with the adenovirus backbone. Systems exist to use gateway cloning from an “entry” vector, directly into the Ad genome, 55 or to use traditional cloning into a subgenomic fragment of the Ad vector, followed by re-assembly in a Gibson reaction. 56 These offer speed, ease-of-use, and throughput advantages over earlier systems. However, recombineering directly into the genome offers further advantages. There is no requirement for an “entry” vector, and there is no requirement for re-assembly of the genome after construction.
The first application of recombineering to Ad vectors was to allow manipulation of an Ad19-based vector, 57 although this system requires an additional re-transformation to select colonies in which the desired modification has occurred. The AdZ system applied a simpler recombineering approach to Ad5 vectors deleted for E1 and E3, 19 and the same technology has also been subsequently applied to gutless adenovirus vectors. 58
A number of CRISPR-based adenovirus vectors have been generated in adenovirus vector systems; however, all require an initial cloning step for the gRNA, in a shuttle vector that is then recombined with the Ad backbone in subsequent steps. 59 –61 The AdZ system therefore offers significant advantages in terms of time and effort for the insertion of gRNA sequences, especially if multiple different sgRNA expressing RAd are to be cloned simultaneously – there is not even a requirement to anneal oligonucleotides before recombineering, they are simply ordered and electroporated directly into the competent bacteria containing the intact RAd genome.
The ability to edit the vector backbone is another advantage of the AdZ system, exemplified by our modifications to the components of the CRISPR/Cas9 system, including (1) a Cas9 variant that offers lower off-target editing, while keeping on-target efficiency, (2) is tolerant of the addition of an initial “G” for efficient transcription, (3) a sgRNA scaffold structure that improves editing efficiency.
As CRISPR/Cas9 technology improves further, it will be simple to adapt these vectors. Furthermore, the use of a promoter that is silenced in packaging cells means that vectors carrying sgRNA targeting genes that interfere with adenovirus replication can be grown to high titers, ensuring success when growing vectors. 19 Finally, the relatively high packaging limit of RAd vectors allows for the incorporation of homology-directed repair templates if gene modifications, rather than gene knockouts, are required.
A major advantage of adenovirus vectors is their ability to transiently deliver genes to a high proportion of target cells, without any requirement for drug selection. Where a cell type does not express high levels of CAR, it is still possible to transduce many cells by simply using higher MOI, although this needs to be empirically tested to ensure there is no particle-associated toxicity.
Alternatively, a variety of genetic approaches can be used to retarget RAds to use novel receptors, 62 or Factor X can be added to the virus to enable entry through heparan-sulfate proteoglycans. 63,64 The transduction efficiency is demonstrated in this study by our ability to knock genes out in a cell line (HFFF) where plasmid transfection failed, with knockout in over 80% of cells achievable. It is also possible to co-deliver vectors targeting multiple sites within the same gene.
If each sgRNA works efficiently, this can result in knockout of a gene in essentially all cells, with no requirement to sort or single-cell clone. 59 The biggest issue with CRISPR/Cas9 systems remains the selection of an efficient sgRNA sequence, as demonstrated by the disparate knockout efficiency seen with sgRNA targeting B2M, as opposed to those targeting PDGFRA and CD155. A large number of computational tools are now available to design efficient sgRNA with minimal off-target effects, which can reduce this problem. 65,66 The main limitation of our system, in comparison to other vector types, is that the RAd vector must be grown up over the course of ∼2 weeks, and titrated before use.
In summary, the combination of a vector system (1) carrying an enhanced sgRNA sequence, (2) with a more accurate Cas9, (3) that supports rapid and efficient cloning of sgRNA sequences from oligonucleotides directly into an intact Ad vector genome, (4) that is automatically excised from the prokaryotic vector in mammalian cells, (5) that suppresses Cas9 expression during vector propagation, (6) that enables high efficiency editing even of hard to transfect cells, (7) without any need for drug selection, and (8) that uses transient Cas9 expression, means that CRISPR/Cas9 editing projects can now be approached with confidence in almost any cell type.
Footnotes
AUTHORS' CONTRIBUTIONS
E.S.: methodology, validation, formal analysis, investigation, writing—review and editing, and visualization. E.C.Y.W.: writing—review and editing, and funding acquisition. R.J.S.: conceptualization, methodology, validation, formal analysis, investigation, writing—original draft, visualization, supervision, project administration, and funding acquisition.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by funding from the MRC (MR/S00971X/1) and Wellcome Trust (204870/Z/16/Z).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.