
Abstract
Hemophilia A (HA) is a monogenic disease characterized by plasma clotting factor 8 (F8) deficiency due to F8 mutation. We have been attempting to cure HA permanently using a CRISPR-Cas9 gene-editing strategy. In this study, we induced targeted integration of BDDF8 (B-domain-deleted F8) gene into the albumin locus of HA mice by hydrodynamic tail vein injection of editing plasmid vectors. One week after treatment, a high F8 activity ranging from 70% to 280% of normal serum levels was observed in all treated HA mice but dropped to background levels 3–5 weeks later. We found that the humoral immune reaction targeting F8 is the predominant cause of the decreased F8 activity. We hypothesized that hydrodynamic injection-induced liver damage triggered the release of large quantities of inflammatory cytokines. However, coinjection of plasmids expressing a dozen immunomodulatory factors failed to curtail the immune reaction and stabilize F8 activity effectively. The spCas9 plasmid carrying a miR-142-3p target sequence alleviated the cellular immune response but could not deliver therapeutic efficacy. Strikingly, immunosuppressant cyclophosphamide virtually abolished the immune response, leading to a year-long stable F8 level. Our findings should have important implications in developing therapies in mouse models using the hydrodynamic gene delivery approach, highlighting the necessity of modulating the innate immune response triggered by liver damage.
Introduction
Hemophilia A (HA) is a monogenic disorder caused by mutation of the coagulant factor 8 (F8) gene that resides on chromosome X. 1 The World Federation of Hemophilia (WFH) estimates that there are 1,125,000 male hemophilia patients. HA accounts for 80–85% of all hemophilia cases. 2 The severity of clinical manifestation is parallel with bioactive F8 levels. 3 Hemophilia patients are defined as having an F8 level lower than 40 IU dL−12. Patients with severe conditions (F8 < 1 IU dL−1) experience symptoms of recurrent and spontaneous bleeding, especially internal bleeding into joints, muscles, or internal organs that pose a tremendous challenge for long-term management. 2 The current treatment is mainly regular IV injection of recombinant F8. 2 However, these approaches have inevitable shortcomings, including high expenditure and increased risk of antibody development, and subsequent immune-related adverse effects due to the need for repetitive administration. Gene therapy has been considered the only cure for hemophilia. AAV-based gene therapy has been making remarkable progress in clinical trials. 4,5 However, the nonintegration nature of AAV renders the first generation of hemophilia gene therapy trials limiting to adult patients, and also with dwindling clinical benefits over time. 6 The advent of CRISPR gene-editing technology offered a novel strategy for treating HA.
Recently, CRISPR-Cas9 technology has galvanized the field of gene therapy and gene editing due to its convenience and affordability. This bacteria-derived system depends on an engineered single-guide RNA (sgRNA) binding with the target DNA site adjacent to the protospacer adjacent motif (PAM) sequence, and Cas9 nuclease then makes a double-strand break in the DNA 3–4 nucleotides upstream of the PAM sequence. 7 On most occasions, double-strand breaks can either be repaired through the more mutagenetic nonhomologous end joining (NHEJ), which may induce gene disruption, or a less frequent but highly accurate homologous directed repair, leading to gene insertion. These methods have been exploited to treat Duchenne muscular dystrophy, 8 hereditary tyrosinemia type 1, 9 and various types of hemophilia. 10 –12
Considering its monogenic cause and the precise biochemical indexes and clinical manifestations, hemophilia A has been an ideal disease model for gene-editing therapy. Nevertheless, the length of F8 protein is 2,332 amino acids, 13 rendering a much higher challenge for the efficient delivery than the F9 gene in hemophilia B models. Consequently, researchers found that the B domain deleted F8 (BDDF8) or F8-SQ is also functional. As such, BDDF8, with its size of 1,457 aa, has been widely used in gene therapy of HA. 14
The efficient delivery of transgenesis is another concern for developing gene therapies. AAV is the most popular delivery vector in clinical gene therapies. However, AAV is less affordable since it demands significant resources to produce AAV. In comparison, hydrodynamic tail vein injection of plasmids can be conducted with great ease. This approach is still used as an efficient in vivo delivery method of naked DNAs for rodents to identify new therapies or create mouse models, 15 with the highest efficiency in liver tissues. 16 Theoretically, high-volume bolus injection (single-dose volume ≥10% body weight) will promptly increase the hydrostatic pressure in the blood vessels directly associated with the inferior vena cava of mice. The high pressure creates transient pores on the hepatic sinusoidal endothelium and dilates the sinusoidal fenestrae, allowing plasmid vectors to enter hepatocytes. 16,17
We have previously demonstrated a CRISPR-Cas9-based approach of inserting BDDF8 into the albumin (Alb) locus through hydrodynamic injection of editing plasmids to treat HA mice. 12 The edited mice showed high-level F8 expression in the first week after treatment, followed by a dramatic decrease of F8 activity in the next couple of weeks due to transgene-specific humoral immune response. This phenomenon has been repeatedly reported in other gene therapies concerning ectopic gene expression, 18 –21 posing a barrier for gene therapies.
Hydrodynamic injection has been used in over 2,000 studies, 22 but its influence on tissue damage and subsequent immune reaction has not been fully characterized. 22 We attempted to investigate this issue by cytokine array assay to simultaneously measure the relative expression levels of 40 mouse cytokines involved in immunity and inflammation, providing a treasure trove of information for guiding immune modulation strategies. Moreover, antigen presentation of the transgene by hematopoietic cells triggers a cellular immune response, which can be curtailed by incorporating the target sequence of miR-142-3p, a hematopoietic-specific microRNA. 23 This strategy has been used successfully to circumvent the expression of F9 in hematopoietic cells, enabling sustained transgene expression after injecting the lentiviral vectors. 24 As such, we are interested in assessing if this strategy also works in our system. Moreover, it is still unanswered whether large amounts of immunosuppressants developed in past decades can be harnessed to prevent hydrodynamic injection-associated immune rejection of transgenes.
In this study, we identified a dozen cytokines whose levels in the liver were strikingly increased after hydrodynamic injection, which might have triggered a robust immune response to transgenes. In attempts to curtail the immune reaction, we codelivered ∼10 immunomodulatory factors, only to find unimpressive effects. In addition, although miR-142-3pT decreased cytolytic response against Cas9, we did not observe appreciable effects on maintaining F8 levels. Strikingly, we found that transient cyclophosphamide (CTX) administration effectively controlled F8 levels. These findings have important implications for hydrodynamic injection-based studies.
Methods
Vector construction
The design of sgRNA targeting Alb stop codon in exon 14 and the construction of plasmids that encode sgRNA-Cas9 and BDDF8 have been described before.
12
In brief, the sgAlb (GTTGTGATGTGTTTAGGCTA) expression was designed to target the Alb locus driven by the U6 promoter. The transcription of SpCas9 was driven by EF1 promoter, or cytomegalovirus (CMV) and hepatocyte-specific promoter. For knockin of the BDDF8 gene, the left and right homology arms of 600 bp were amplified from the mouse genomic DNA and flanked by the sgAlb targeting sequence (GTTGTGATGTGTTTAGGCTA
Hemophilia A mice
HA mice containing an F8 exon 16 knockout on a 129 × B6 background were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice have been bred and housed at the State Key Laboratory of Experimental Hematology (SKLEH, Tianjin, China). This study was approved by the Ethics Committee of SKLEH.
Hydrodynamic tail vein injection
All vectors were produced using the EndoFree MaxiPrep Kits (Qiagen) or the ZymoPURE II Plasmid Maxiprep Kit (Zymo Research). To ensure no significant endotoxin contamination, we inspected the endotoxin level in our produced plasmids using Lyophilized amebocyte lysate LAL/TAL reagent (Xiamen Bioendo Technology). For BDDF8 knockin, 5 μg of sgAlb, 5 μg of SpCas9, and 10 μg of double-cut donor BDDF8 plasmid were used. 25 To examine the effects of immunomodulatory factors, we coinjected 5 μg of a transgene overexpression plasmid. For hydrodynamic tail injection, the plasmids were diluted to a volume of 10% of mouse body weight using Ringer solution (China Otsuka Pharmaceutica) and injected through the mouse tail vein within 5–6 s. To prevent excessive bleeding, each mouse was injected with 0.5 IU F8 protein (Xyntha; Wyeth Pharmaceuticals).
Flow cytometry analysis of tdTomato+ liver cells
We assessed the knockin efficiency 1 and 3 weeks after hydrodynamic injection by examining the proportion of tdTomato+ liver cells by flow cytometry. A small portion of the liver was fixed in 10% formalin solution overnight, grounded by mortar and pestle in phosphate-buffered saline (PBS), then filtered through 70-μm cell strainers. The cellular nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) and then analyzed on a BD fluorescence-activated cell sorting (FACS) Aria III flow cytometer. The percentage of tdTomato+ cells among the gated DAPI+ cells was reported as editing efficiency. In negative controls, omitting either vector led to the detection of 0% of tdTomato+ cells.
Separating plasma and serum from whole blood
For blood collection, the mouse's lateral tail vein vessel was incised with a razor blade, and blood droplets would accumulate on the tail, which can then be collected using a tube. For plasma preparation, 100 μL of blood from the tail vein was collected into a microtube containing 10 μL of 3.2% sodium citrate, then briefly applied pressure and styptic powder to stop bleeding. For serum preparation, the citrate anticoagulant was omitted, and the whole blood was allowed to clot at room temperature for 30 min, followed by spinning down at 2,000 g for 15 min at 25°C. Plasma was obtained by centrifugation at the same condition. Next, the supernatant was extracted into a new tube and then stored at −80°C and instantly thawed at 37°C before analysis.
Measurement of F8 coagulation activity and F8 inhibitors
The one-stage activated partial thromboplastin time (aPPT)-based clotting assay was used to determine the F8 coagulation activity and Bethesda assay to detect the F8-neutralizing inhibitors as described before. 12 The mouse plasma samples were diluted 4-fold with Dade Owren's Veronal Buffer (Siemens; B4234-25) to a final volume of 100 μL for detecting the F8 activity with a Sysmex CA1500 system (Sysmex, Kobe, Japan). For measuring F8 inhibitors, 50 μL mouse serum samples were treated at 56°C for 30 min to inactivate F8 activity, followed by incubating with 50 μL normal plasma at 37°C for 2 h. The residual coagulation activity was measured to titrate F8 inhibitors, following the standard Bethesda assay instructions.
Assessment of liver function
We collected serum at 6, 12, 24, 72 h, and 7 days after hydrodynamic injection. Serum ALT (alanine aminotransferase), AST (aspartate aminotransferase), and albumin were measured by the Olympus AU5400 (IDEXX Memphis, TN) as described before. 12
Cytokine profiling array
To analyze cytokines/chemokines secretion, we harvested liver tissue at 2, 6, 12, 24, 72 h, and 7 days after hydrodynamic injection. In brief, 30 mg of frozen liver tissue was homogenized in 500 μL RIPA lysis buffer (Beyotime, P0013B, mainly including 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl/sulfate, 1% Triton X-100, 100 mM phenylmethylsulfonyl fluoride, and a cocktail of protease inhibitors), and centrifuged at 4°C with 18,000 g for 20 min. The supernatant was collected, and protein concentrations were measured using the bicinchoninic acid (BCA) assay. The liver tissue lysate was stored at −80°C before detection. Protein lysates (500 μg) were analyzed using the Proteome Profiler™ Mouse Cytokine Array Kit (mouse cytokine array, panel A, ARY006, R&D Systems). The chemiluminescence reaction was measured with a ChemiDoc Imaging Systems from Bio-Rad, and pixel densities were determined for each spot on the membranes by ImageJ. The background signal was subtracted to obtain the intensity value of each spot. Last, we compared the relative signals on different arrays to determine the fold change in cytokine levels.
Enzyme-linked immunosorbent assay
The SpCas9 antibody titers were determined by ELISA (enzyme-linked immunosorbent assay), following the published protocols. 26,27 In brief, a transparent flat-bottom Immuno 96-well plate (Thermo Fisher Scientific) was coated with the SpCas9 protein (IDT) (0.5 μg/well in PBS) overnight at 4°C. The plate was washed three times with phosphate-buffered saline with Tween 20 (PBS with 0.05% Tween 20) and then blocked with 1% bovine serum albumin (BSA) for 1 h at 37°C. The SpCas9 monoclonal antibody (Thermo Fisher Scientific) with concentrations ranging from 1 to 500 ng/mL was used to generate a standard curve. Mouse serum samples were diluted 10–20 times in 1% BSA blocking solution. To titrate antibodies, 100 μL of diluted standard or serum samples were added to the SpCas9-precoated plate. After being incubated at 37°C for 1 h, the plate was washed three times with the blocking solution. Horseradish peroxidase (HRP)-conjugated protein G (Abcam) was then applied and incubated for 1 h at room temperature. Last, SureBlue™ TMB 1-Component Microwell Peroxidase Substrate (SeraCare) was added to develop color. Fifteen min later, TMB Stop Solution (SeraCare) was added to discontinue the reaction. The absorbance at 650 nm was read using a SpectraMax M3 microplate reader (Molecular Devices) to determine the antibody titers.
Enzyme-linked immune absorbent spot assay
Mouse splenocytes were separated by density gradient centrifugation using Ficoll®-Paque PREMIUM 1.084 (Sigma). The mouse interferon gamma (IFN-γ)-precoated enzyme-linked immune absorbent spot (ELISPOT) Kit (Dakewei, China) was used to detect antigen-reactive T cell immune response, following the manufacturer's instructions. Splenocytes of 2 × 105 cells in 100 μL were inoculated in each well. After adding 0.5 μg antigen, including Cas9, F8, or tdTomato protein, in triplicate, the ELISPOT plate was incubated in a 37°C 5% CO2 incubator for 16–18 h. Cells were lysed with ice-cold double-distilled water at 4°C for 10 min and then the cellular debris was discarded. Following three washes, the plates were incubated with biotinylated IFN-γ-specific detection antibodies for 1 h at 37°C. For signal detection, streptavidin-conjugated alkaline phosphatase was used to catalyze substrate to generate insoluble precipitate forming spots in the wells. Spots were enumerated using an ELISPOT Reader.
Determination of plasmid copy number by quantitative polymerase chain reaction
We collected 30 mg liver tissue from multiple groups of HA mice 3, 7 days, 3 months, or 1 year after gene-editing therapy. Mice without plasmid injection served as a negative control. The liver tissue DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen). The following primers were used to amplify Cas9 (Cas9-F, CAGACAGCAACTGCCTGAGA; Cas9-R, TGTCGAAAGTGCGCTGTTTG), sgAlb (sgRNA-F, AGCTAGAAATAGCAAGTTAAAATAAGG; sgRNA-R, GACTCGGTGCCACTTTTTCA), F8 (F8-F, GCATGAGGTGGCATACTGGT; F8-R, GGTCATGCCTCTGTTCCGAA), and tdTomato (tdTomato-F, GCAGAAGAAGACCATGGGCT; tdTomato-R, GATGTCCAGCTTGGTGTCCA). Actb, which has two copies in each diploid cell, was used to normalize plasmid copy number data. The quantitative polymerase chain reaction (qPCR) analysis was conducted using the KAPA Fast qPCR Kit on the Applied Biosystems Real-Time PCR Instruments (Thermo Fisher). The cycling conditions were 98°C for 2 min, followed by 35 cycles of 98°C for 10 s, 60°C for 15 s, and 72°C for 15 s. The plasmid copy number was calculated using the ΔΔCt method.
Assessment of transgene expression by reverse transcription-quantitative polymerase chain reaction
The liver RNA was extracted as described previously. 12 In brief, frozen liver tissue of ∼30 mg was ground into a fine powder using precooled mortar and pestle while adding liquid nitrogen. RLT (Qiagen RNeasy Mini Kit, Valencia, CA) lysis buffer was added immediately to prevent RNA degradation. Contaminated DNA was depleted by DNAse I treatment. The integrity of extracted RNA was assessed by running an agarose gel. The TransScript First-Strand cDNA Synthesis SuperMix Kit (TransGen Biotech) was used for reverse transcription. The cDNAs were quantitated by qPCR, as detailed previously. 12
Immunosuppression
To transiently suppress the immune response, multiple agents were tested in our study.
Treated HA mice were intraperitoneally injected with CTX (50 mg/kg per injection) and/or methylprednisolone (50 mg/kg per injection), on the day of vector injection (Day 0), followed by biweekly for 1 week (three times in a week), or once a week for 3 weeks (four times in 3 weeks), or biweekly for 3 weeks (seven times in 3 weeks). Retinoic acid (RA; 200 μg per mouse), Rapamycin (Rapa; 20 μg per mouse), or RA+ Rapa (200 and 20 μg, respectively) was administrated by i.p. injection every 2 days up to 2 weeks after plasmid injection. Aminooxyacetic acid (AOA; 750 μg per mouse), AOA+ RA (750 and 200 μg), or AOA+ Rapa (750 and 20 μg, respectively) was administered i.p. at days 0, 2, 4, and 6. Infliximab (IFX) (100 μg) was intraperitoneally injected weekly for 3 weeks. Etanercept (20 μg) was given twice weekly by subcutaneous injection with an interval of 3–4 days up to 3 weeks. Bortezommib (10 μg) was administered i.p. at days 3, 6, and 12. Ruxolitinib (600 μg) was given daily for 1 week.
Statistics
GraphPad Prism 7.0 (GraphPad Software, San Diego, CA) was used to analyze data. All data are shown as the mean with standard deviation. Statistical analysis between two groups was performed using a two-tailed Student's t-test. For comparing three or more groups, a one-way analysis of variance (ANOVA) test with Tukey's post-test was used for significant differences. In all cases, significance was assigned if p < 0.05.
Results
CRISPR editing of hepatocytes after plasmid delivery transiently restores F8 activity
As detailed previously, 12 we hydrodynamically injected the Cas9-sgRNA and promoterless F8 plasmids to treat hemophilia A. The expression of Cas9 and sgAlb were driven by EF1 and U6 promotors, respectively (Fig. 1A). The editing template donor BDDF8 also expressed tdTomato to facilitate cell tracking by FACS (Fig. 1A). Successful targeted integration of F8 right before the Alb stop codon led to the expression of tdTomato and BDDF8 under the control of Alb transcriptional machinery (Fig. 1B). At 1 week after hydrodynamic tail-vein injection of Cas9 and sgRNA plasmids together with a donor template, 1–3% of liver cells expressed tdTomato, and 100–300% F8 serum activity was observed (Fig. 1C, D). This result suggests that as little as 1% of editing efficiency is sufficient to restore the hemostatic activity of HA mice to normal levels. We also injected mice with donor BDDF8 plasmid only as control. No tdTomato signal was detected on dissociated liver cells, suggesting that only CRISPR-mediated insertion of BDDF8 at Alb leads to the expression of the transgenes. However, we observed ∼5-fold drop in tdTomato+ liver cells and F8 serum levels 3 weeks later (Fig. 1C, D).

Schematic of our genome editing system and loss of edited cells and F8 expression shortly after treatment.
Hydrodynamic injection of plasmids induces transient liver damage and inflammation
The procedure of hydrodynamic injection results in considerable tissue damage, 28 triggering the production of proinflammatory cytokines, which recruit or stimulate immune cells like neutrophils and macrophages 29 (Fig. 2A). Thus, we hypothesized that the rapid decline of edited cells and F8 activity might result from a liver damage-induced immune reaction.
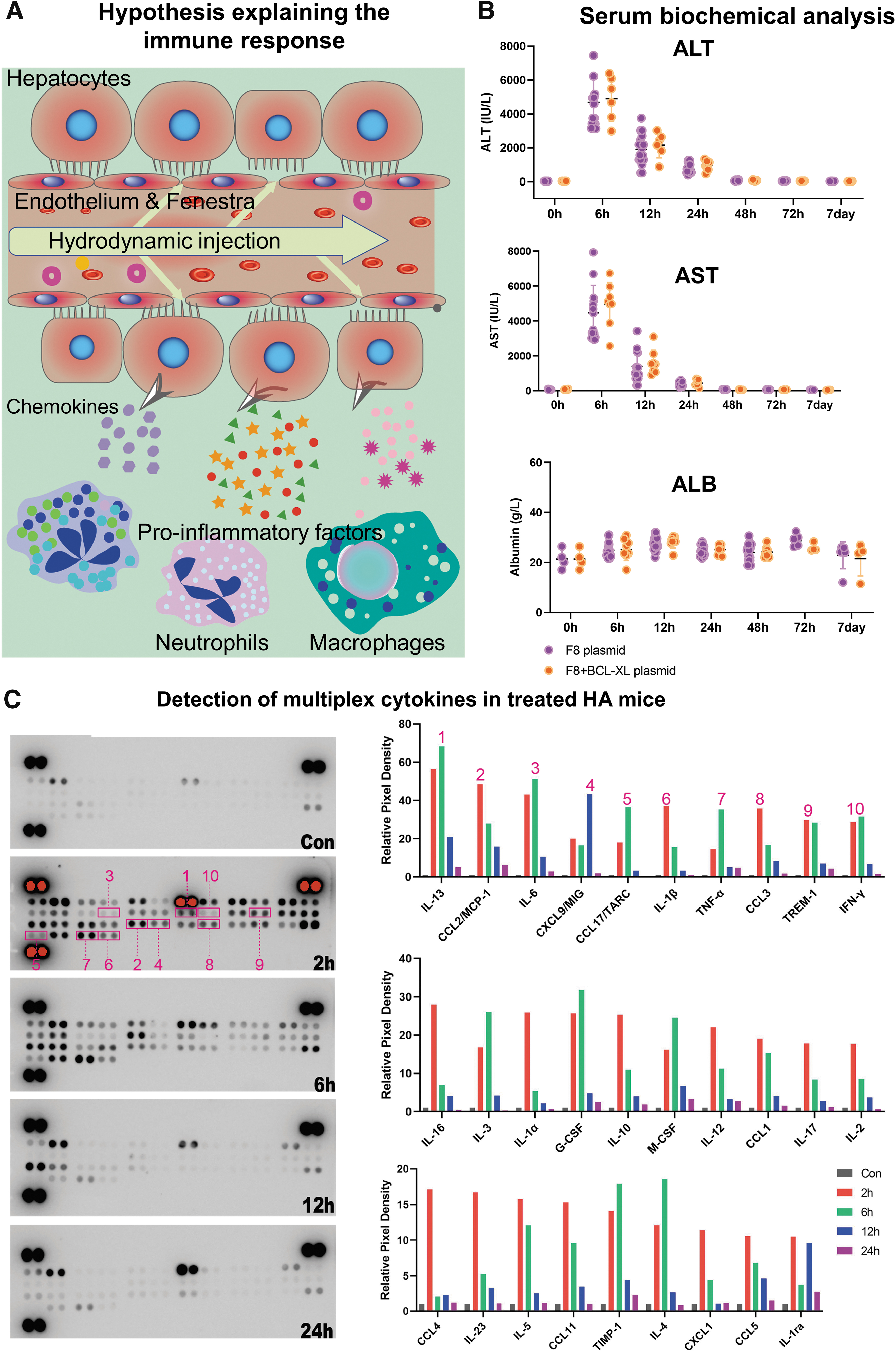
Hydrodynamic injection induces liver damage and proinflammatory reactions.
We monitored the levels of liver damage markers in the treated HA mice 6, 12, 24, 48, 72 h, and 7 days after treatment. Six hours after pressurized injection, we found a ∼50-fold increase in serum ALT and AST levels. However, this transient elevation subsided 18 h later (Fig. 2B), consistent with previous studies. 16,29 Further assessment of liver functionality in the following days indicated a complete recovery from the liver damage. Of note, albumin levels in circulation remained unchanged (Fig. 2B), suggesting that transient liver damage does not negatively affect albumin production.
To assess the immune activity following tissue damage, we collected liver tissue for cytokine analysis. We found an average of 20- to 60-fold increase in 17 proinflammatory cytokines, among which IL-13 exhibited the strongest and fastest response with a nearly 70-fold rise within 6 h after treatment, followed by CCL2/MCP1, interleukin (IL)-6, and IL-1β (Fig. 2C). IL-13 is a dominant profibrotic cytokine in mice and a potent inducer of chemokines CCL2, CCL3, CCL4, and CCL11 during tissue injury and remodeling. 30 IL-13 also protects hepatocytes and endothelial cells against ischemia/reperfusion-induced injury. 31 CCL2/MCP1, IL-6, and IL-1β are proinflammatory cytokines involved in the recruitment of T and B cells and myelogenous cells. 32 –34 Echoing the rapid drop of liver damage markers 24 h later, the serum levels of proinflammatory factors also returned to baseline levels, suggesting the transient nature of the acute immune response induced by liver damage. These results indicate that the prompt release of proinflammatory factors in response to hydrodynamic injection-induced tissue injury might activate a network of immune cells.
The humoral immune response is the predominant contributor to F8 loss
Although the innate immune response largely disappeared 2 days after hydrodynamic injection, it can elicit T cell or B cell-mediated adaptive immune response. After successful gene editing, hepatocytes will transcribe the Alb-F8-tdTomato fusion mRNA, which translates into three proteins due to the linker peptide E2A-induced ribosome skipping. Antigen-presenting cells might process the secreted F8 to stimulate the generation of plasma cells that produce specific antibodies (Fig. 3A). Likewise, surface antigen presentation of transgene products may induce T cell-mediated immunity (Fig. 3A).
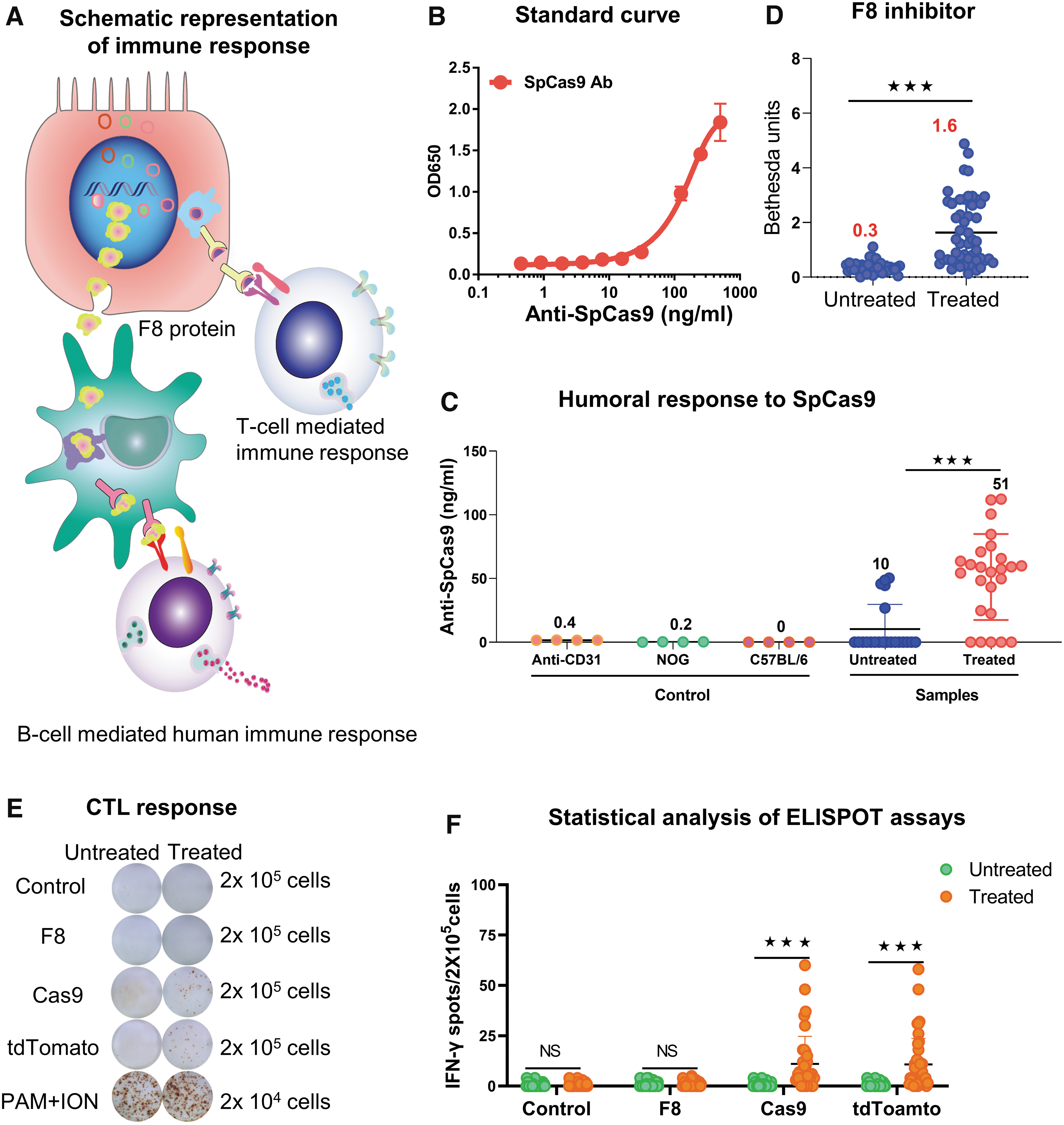
Hydrodynamic injection of editing factors induces a humoral and cellular immune response.
Previous researches have demonstrated the high prevalence of preexisting humoral and cell-mediated immune response against the bacteria-derived spCas9 during CRISPR-Cas9 gene editing, which may compromise the editing efficiency. 35,36 For this reason, we performed the ELISA analysis to detect antibodies against spCas9. First, a standard curve was generated using multiple dilutions of anti-SpCas9 (Fig. 3B). Then, we used anti-CD31, serum from NOD/Shi-scid/IL-2Rγnull immunodeficient mice, and wild-type C57BL/6 mice as negative controls and detected no antibodies against SpCas9 (Fig. 3C). Last, we assessed serum from HA mice, and found that only 23% of untreated mice (n = 22) displayed a low level of <50 ng/mL anti-SpCas9. In contrast, the injection of Cas9-expressing plasmid led to a significant increase in antibodies against SpCas9 in 80% mice (n = 25) (Fig. 3C), suggesting that the gene-editing therapy elicits a humorous response to the expressed Cas9 protein.
Neutralizing antibodies against F8 are often developed after repeated protein administration or gene therapies, which significantly compromise the efficacy of the treatments. 37,38 We used the standard Bethesda assay to measure neutralizing antibodies against coagulation factors such as F8. Moderate antibody response against F8 was observed in the treated HA mice 3 weeks after plasmid injection. This result may partially explain the declined F8 activity in the treated mice (Fig. 3D).
In addition, cell-mediated immunity may lead to the elimination of the F8-overexpressing hepatocytes by the activation of IFN-γ-secreting Th1 cells. Hence, we performed an ELISPOT assay to evaluate the presence of cellular immunity against F8, Cas9, and tdTomato. We observed a negative cellular immune response against F8 (Fig. 3E), suggesting that the ectopic expression of BDDF8 does not induce T cell-mediated killing of the edited hepatocytes. However, cellular immunity against SpCas9 was significantly increased 3 weeks after SpCas9 plasmid injection (Fig. 3F).
Interestingly, we also detected a positive cellular immunity against tdTomato protein (Fig. 3F). We noted that T cell response against SpCas9 or tdTomato was highly variable among the treated HA mice, suggesting that at least in a fraction of mice, depletion of SpCas9- and/or tdTomato-expressing liver cells might have led to decreased F8 levels.
Delivery of immunomodulatory factor-expressing plasmids failed to sustain F8 activity
Cytokine array analysis has demonstrated a massive increase of proinflammatory cytokines shortly after hydrodynamic injection. We then asked whether coinjection of immunomodulatory cytokines can attenuate the immune reaction. This study selected 10 genes for the following reasons and cloned them in an overexpression plasmid vector driven by the EF1 promoter (Fig. 4A). TGFB may inhibit immune cell proliferation and control pathogen-related tissue damage. 39 IL-6 affects inflammation, immune response, and hematopoiesis mainly associated with the JAK/STAT3 activation pathway. 40 IL-10 restrains excessive immune response and promotes tissue repair after injuries. 41 IFNG may inhibit T and B cell proliferation and the production of proinflammatory cytokines. 42 CTLA-4 and PDCD-1 (PD-1) negatively regulate T cell activation and tolerance. 43,44 ENTPD1 (CD39) produces adenosine monophosphate, which is used by NT5E (CD73) to synthesize the immunosuppressive adenosine, 45 mitigating immune response. B18R, a type I IFN inhibitor, can reduce inflammatory response. 46 DNASE1 functions to clear the damaged nuclear DNAs that may induce inflammation through the STING cytosolic DNA-sensing pathway. 47
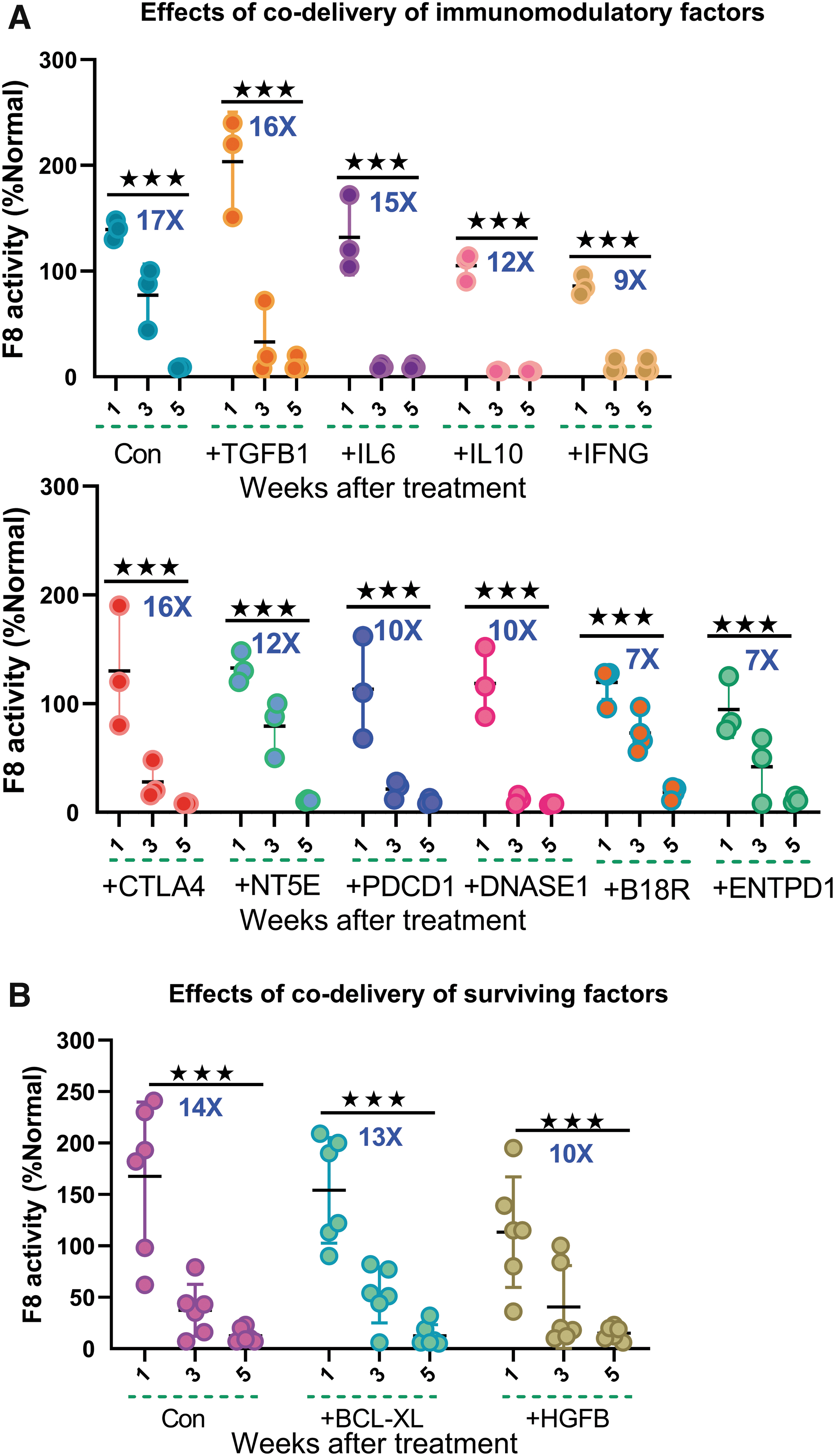
Codelivery of immunomodulatory and liver-protective factors failed to sustain the F8 activity after treatment.
We hypothesized that the concomitant expression of one of the abovementioned factors and the editing components would suppress the immune reaction, increasing F8 activity. However, unexpectedly, all of the 10 overexpressed cytokine genes failed to sustain the therapeutic F8 levels (Fig. 4A). However, these results need to be interrogated using other approaches since two factors might confound the interpretation of our data. First, we used a transient expression system for delivering immune-modulating factors. Second, there is a delay in protein expression after plasmid delivery.
Liver protective factors had little impact on maintaining F8 levels
We then asked whether cell survival factors can ameliorate liver damage and thus improve gene therapy efficacy. Hepatocyte growth factor (HGF), which is encoded by the HGFB gene, promotes hepatocyte growth and liver regeneration. 48,49 Since hydrodynamic injection-induced tissue injury elicits a network of immune reactions, we hypothesized that incorporating HGFB plasmid into the vector system would expedite the recovery process and alleviate the immune response, thus bringing a higher F8 activity. Nevertheless, we observed an identical decreasing trend during the first 5 weeks after treatment (Fig. 4B). BCL-XL, an antiapoptotic factor, was speculated to improve the longevity of hepatocytes and subsequently reduced the loss of transduced cells. However, F8 levels dropped indistinguishably with or without the presence of BCL-XL (Fig. 4B). Altogether, these results demonstrate that promoting the survival of hepatocytes after hydrodynamic injection is insufficient in sustaining F8 serum levels.
Hepatocyte-specific expression of SpCas9 is unable to maintain the therapeutic potency
A previous report showed that tissue-specific and sustainable transgene expression would reduce the host's immune response. 50 For instance, liver-specific promotors (LSPs) like CRM8-TTR can prevent antibody response by reducing antigen presentation in immune cells, 51 thus enhance F8 production. As such, we tested the effects of the LSP-Cas9 vector. Since vectors carrying the CMV promoter 52 were used in previous studies, we also assessed the CMV-Cas9 plasmid as a control.
No significant differences in serum F8 levels were observed among the three groups using different promoters for Cas9 expression 1 week after plasmid injection (Fig. 5B). We observed an 18- to 29-fold reduction in F8 activity 5 weeks later, close to baseline (Fig. 5B). In addition, the ELISPOT assay showed an identical cell-mediated immunity against Cas9 (Fig. 5C). These results suggest that simply altering Cas9 expression levels by choosing different promoters is not a promising strategy for attenuating immune reactions in the hydrodynamic injection system.

Liver-specific expression or detargeting Cas9 in hematopoietic cells failed to sustain the F8 activity after treatment.
We continued to test the miR-142-3p target approach. Tagging multiple copies of the miR-142 recognition sequence downstream of the vector leads to post-transcriptional silencing of transgene expression in hematopoietic cells. 23 Theoretically, this would prevent antigen presentation by immune cells and the subsequent immune induction against SpCas9. We observed that incorporating the miR-142-3p target sequence mitigated the T cell immune response (Fig. 5E). However, adopting miR-142-3pT did not show significant changes in F8 activity throughout 5 weeks after hydrodynamic injection (Fig. 5F).
Altogether, these data demonstrate that cellular response against SpCas9 plays a minor role in the unpronounced therapeutic effectiveness after hydrodynamic delivery of CRISPR plasmids.
Targeted immunosuppressive drugs have only transient effects on sustaining F8 activity
Small molecules or antibody immunosuppressive therapies have been used in clinical treatments. Therefore, we evaluated several agents with demonstrated immune regulation capacity: (1) Retinoic acid (RA), 53 which is crucial for equilibrating immunity and tolerance; (2) Rapamycin, 54 the inhibitor of mTOR that induces immune suppression; (3) Bortezomib, 55 one of the standard treatments in multiple myelomas, which is thought to exert inhibitory effects on IL-6, human tumor necrosis factor alpha (TNF-α); (4) Transaminase inhibitor AOA, which suppresses immature myeloid cell generation; (5) IFX, 56 an antibody against TNF, which is efficient in treating Crohn's disease (CD), and is associated with an increase in Treg cells; (6) Etanercept, 57 a TNF inhibitor, which has been approved to treat rheumatoid arthritis and chronic immune diseases; (7) Ruxolitinib, 58 an inhibitor of both JAK2 and JAK1 protein kinases that plays a central role in signal transduction in hematopoietic and immune cells.
Compared with the above-studied immunomodulatory or surviving factors, these small molecules or antibodies performed significantly better. We observed a 2- to 6-fold reduction in F8 levels during the 5 weeks instead of an over 10-fold decrease for plasmid-delivered factors. Among these agents, AOA was the least effective in maintaining F8 levels (Fig. 6B), followed by Bortezomib (Fig. 6D), IFX, and Etanercept (Fig. 6C). RA and Rapa appeared to be the most effective suppressants fighting against F8 activity drop, leading to the persistence of an appreciable amount of F8 activity in the circulation (Fig. 6A). However, the F8 levels slowly but steadily reduced during the first 9 weeks after editing.

Using targeted immunosuppressants reduces the speed of F8 decline. Dynamic changes of F8 activity from 1 to 9 weeks after editing therapy and administration of indicated immunosuppressants.
CTX maintains F8 activity by controlling both humoral and immune responses
The above results suggest that more potent immunosuppressants are necessary. We then tested CTX, an alkylating agent that kills cycling cells. Strikingly, we observed sustained F8 activity during a 9-week follow-up. Coadministration of CTX with dexamethasone (DEX) or RA showed no additional impact, suggesting that CTX alone is sufficient for abrogating the immune response (Fig. 7A). Consistent with long-lasting F8 expression, we observed a tdTomato marking of ∼1% in the liver, similar to the editing efficiency shortly after injection, suggesting that the number of edited liver cells persisted (Fig. 7B). In further support of this conclusion, the ELISPOT assay did not detect any cellular immune response against Cas9 or F8 (Fig. 7C). In addition, we observed no development of F8 inhibitors in treated mice, and antibody titers of anti-SpCas9 were not significantly changed before and after treatment, suggestive of no induction of humoral immune (Fig. 7D, E). Altogether, CTX is a potent immunosuppressant in applications that involve the hydrodynamic injection of drugs.
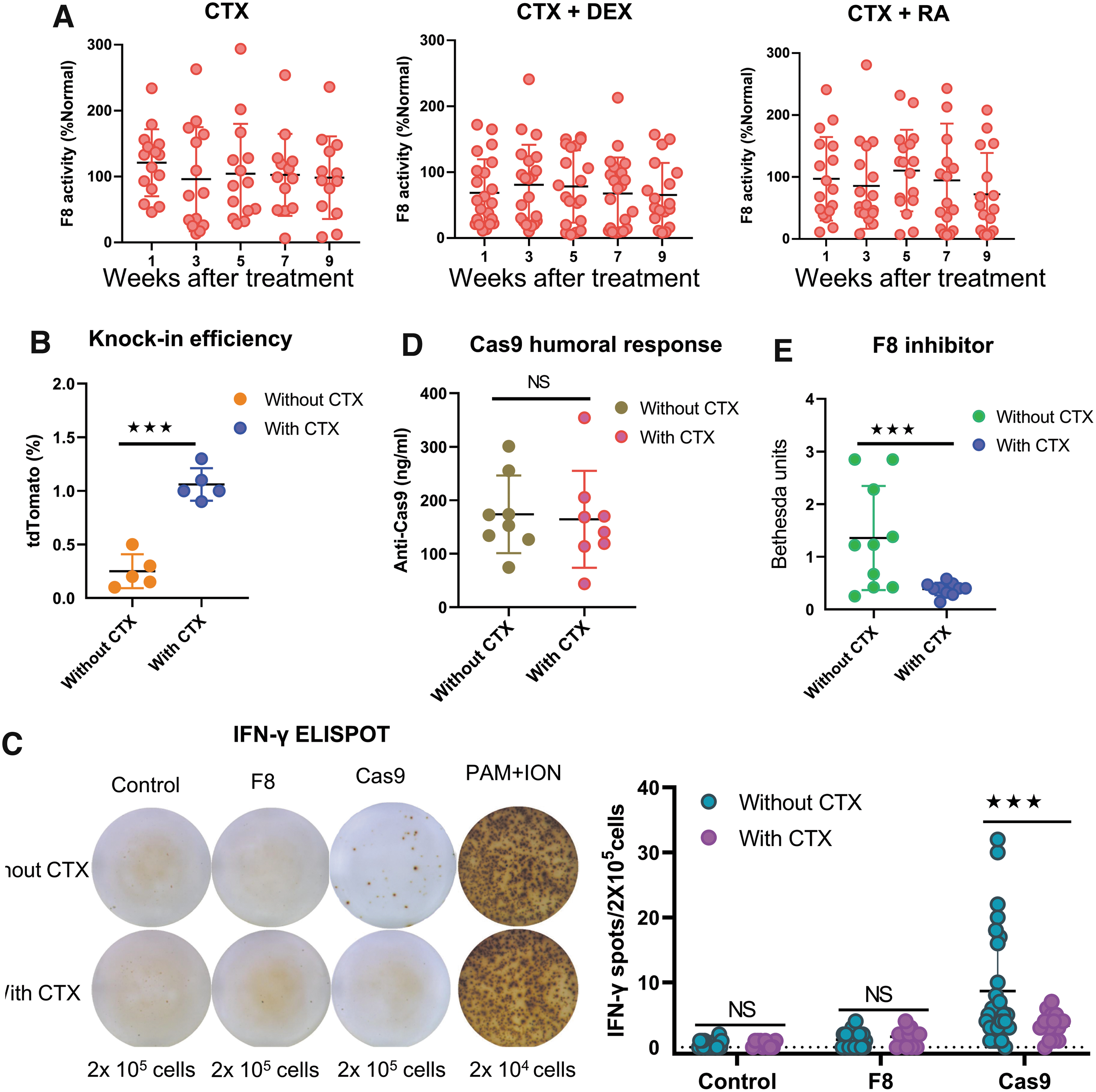
CTX treatment leads to stable F8 levels by controlling cellular and humoral immune reactions to transgenes.
The CRISPR-Cas9-mediated BDDF8 integration editing tool was efficient and safe
We followed up on the CTX-treated mice and observed a persistent F8 activity over 1 year (Fig. 8A). We then assessed the dynamic changes of injected plasmid vectors. We detected ∼20 copies of Cas9 and sgAlb plasmid during the first week after injection. In comparison, 5–10 copies of the F8 donor template entered liver cells after hydrodynamic injection (Fig. 8B). As expected, the plasmids were gradually lost during 1 year of liver turnover. We also measured mRNA expression levels of editing components. High levels of Cas9 and sgAlb expression were observed (Fig. 8C). However, their transcripts dropped to almost zero, in contrast to an appreciable amount of residual plasmids, suggesting their expression is largely silenced. F8 and tdTomoto will only be expressed after targeted integration. As expected, their expression was steadily increased during the first week and sustained at the same level for 1 year. The dynamic change of F8 expression echoed that of tdTomoto, reflecting the fact that they are encoded in the single fusion transcript.
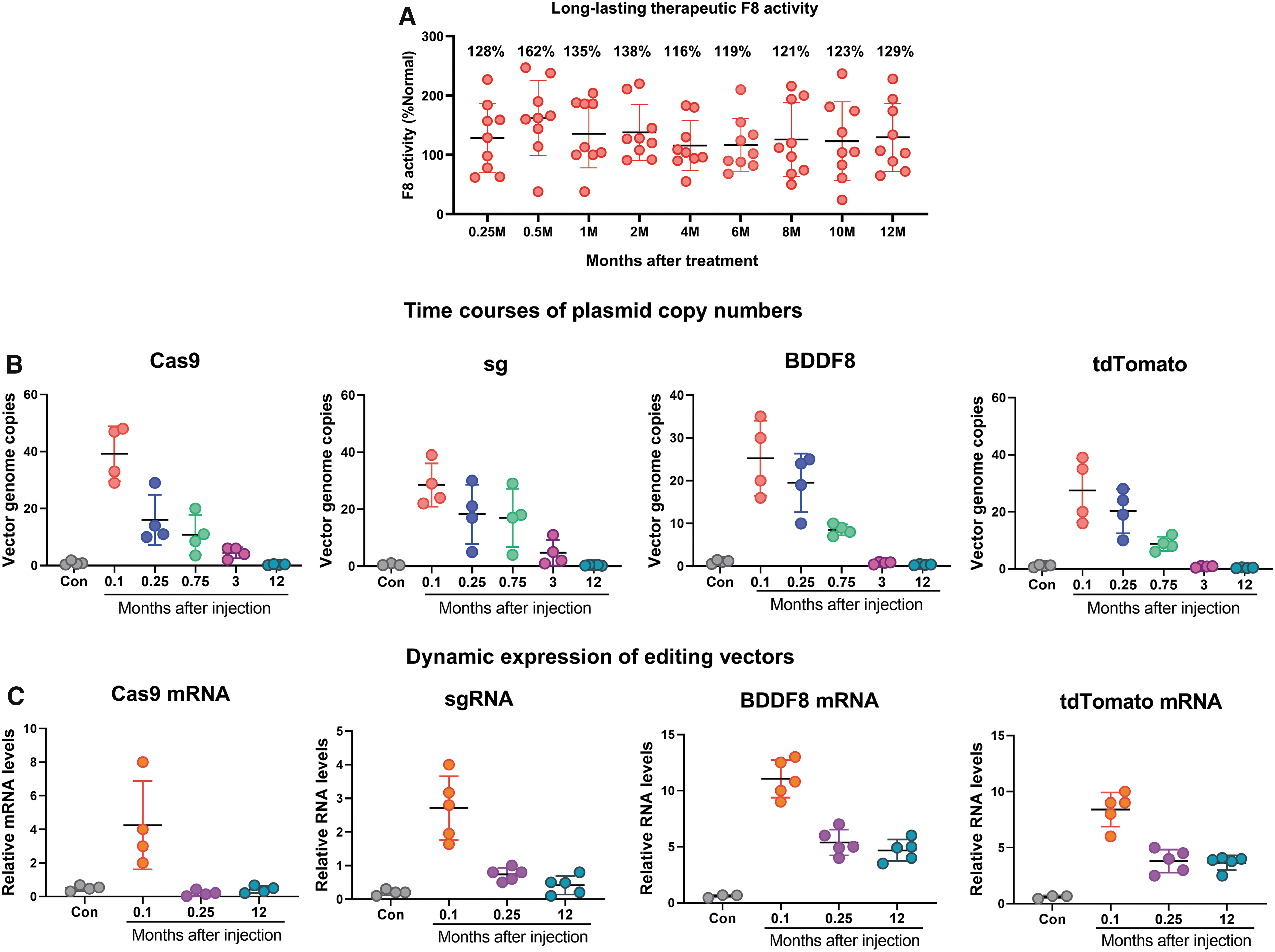
Hydrodynamic injection-mediated gene-editing strategy is efficacious and safe.
Discussion
This study used the CRISPR-BDDF8 editing plasmids to treat hemophilia A in a well-established mouse model. A therapeutic F8 level was obtained in the first week after hydrodynamic delivery but followed by a drastic decrease to baseline. We observed the involvement of both innate and adaptive immune responses, among which the humoral response against F8 is the most predominant and persistent cause of the steady loss in F8 activity. Transient liver damage was observed in the first 24 h after injection, accompanied by the prevailing upregulation of various inflammatory cytokines. We demonstrated that CTX alone or in combination with DEX or RA leads to a robust and persistent F8 activity over 1 year in hemophilia A mice. These findings also provide insights into the underlying cause of the low efficiency in gene editing when using plasmid vectors in CRISPR-Cas9-based gene therapies.
Gene therapy for hemophilia has convincingly shown dramatically raised F8 protein levels that prevent bleeding in clinical settings. 59 –63 However, the exact duration of such therapeutic effect is unsettled during disease progression. This study applied the simple and powerful CRISPR-Cas9 gene-editing platform to mediate BBDF8 gene integration at Alb locus in hemophilia A mice. Transgenes are knocked in predominantly by the NHEJ mechanism in the context of hepatocytes, as we previously reported, 12 and the expression was driven by the Alb promoter. 12
Hydrodynamic delivery with naked DNA has been considered safe and efficient for in vivo gene editing. 64,65 However, high-pressure injection affects the liver morphology and hepatic function, and induces immune activation and organ damage. 28,66 Using a mouse cytokine array to detect 40 cytokines, we observed a spike in the levels of multiple cytokines in the liver shortly after treatment, which returned to normal within 24 h. This dynamic change reflects the rise of innate immunity after traumatic injection, 67 –70 which then activates adaptive immunity. When plasmid DNAs are introduced into cells, they are recognized as foreign elements, triggering the proinflammatory cGAS–STING–IRF3 response. 71 –73 Activation of this pathway triggers the production of type I Interferon cytokines (IFN-I). As such, we saw a nearly 40-fold increase in IFN.
The most significant elevation (∼70-fold) after injection was IL-13, synthesized by Th2 cells and critical in developing T cell-mediated humoral immune responses. 74 In comparison, Miao et al. reported that IL-10 had the most significant increase, followed by IL-2 and IFN-γ. 19 Considering the absence of cellular immune reaction against the ectopic F8 protein, we speculate that the immune response against the transgene is predominantly regulated by B cells and Th2. This deduction is partly in line with a previous study showing that transgene-specific antibodies are primarily developed in response to Th2-mediated cell transduction in HA mice. 19
IL-6 is crucial in the induction of acute-phase protein synthesis and the simultaneous inhibition of albumin production, which is promptly produced in response to infections and tissue injuries. 75 Accordingly, we observed a striking rise in IL-6 levels after the traumatic injection. Similarly, serum IFN-γ and IL-6 peaked on day 1 after hydrodynamic gene transfer. 76
One of the significant hurdles for gene therapy is an immune response against the delivery vectors and the therapeutic gene products. 77 –79 Our system confirmed that the expression of Streptococcus pyogenes Cas9 protein (SpCas9) and F8 protein in mice would evoke cellular and humoral immune responses, respectively, compromising the efficiency of gene therapy. In addition, we also detected cellular immune response to tandem dimer Tomato (tdTomato), a coexpressed marker gene, to assess gene-editing efficiency. Previous studies have demonstrated that GFP protein could induce T cell immune responses against GFP+ cells. 80,81 Extending this discovery, we identified tdTomato as a source of the immune response. This result will guide the selection of appropriate marker genes in preclinical studies.
So far, we have explored the main reasons for the decrease of F8 activity in our treatment system. Hydrodynamics-based liver damage and plasmid transfection could trigger an innate immune response, leading to the release of various cytokines within 24 h, such as IFN-I, IFN-γ, IL-2, IL-6, activating the adaptive immune response. Moreover, foreign SpCas9, tdTomato, and F8 protein expression evoked a cellular and humoral immune response detected by ELISPOT and ELISA (or F8 inhibitor assay). These results highlight the importance of managing immune response in CRISPR-mediated gene therapy.
To achieve a robust and long-lasting therapeutic benefit, we adopted a series of strategies to counteract the immune response. First, we found that codelivery of plasmids that express a surviving or immunosuppressive factor showed unimpressive effects on enhancing therapeutic efficacy. Second, to avoid immune response against Cas9 protein, we incorporated a hematopoietic cell-targeting miR-142 sequence 82 –84 in the Cas9-expressing vector to prevent Cas9 expression in antigen-presenting cells. As a result, cellular response to Cas9 protein was inhibited, but the decrease of F8 activity remained unchanged. Last, we found CTX was able to maintain F8 activity. Miao et al., had a similar attempt to prevent the robust humoral response after delivering the pBS-HCRHPI-hFVIIIA plasmid. Using immunosuppressants RAP, MMF, CSA+MMF, RAP+MMF, and the MR1 antibody with or without murine Ctla4Ig, they managed to sustain the F8 expression with an undetectable level of anti-hF8 antibodies. 19 Our findings add new weapons to the arsenal in the fight against immune reaction after the pressurized injection. This result may also have implications in the clinical settings, as this transgene-induced immune response is species independent. 38
This study has limitations. Hydrodynamic injection of plasmids is harmful to patients, preventing its clinical translation. In future clinical trials, AAV or nanoparticles will be used for the delivery of editing components.
In summary, we observed a massive increase of inflammatory factors after hydrodynamic injection-induced liver damage, which triggered immune reactions against transgenes. However, administrating CTX for a short period overcomes this adverse effect. This strategy can be employed in other settings that involve the hydrodynamic injection of plasmids or other agents.
Footnotes
Authors' Contributions
J.-P.Z. and X.-B.Z. supervised the study. M.Z., Y.-D.S., M.D.Y., J.-J.Z., S.-A.L., G.H.L., F.Z., J.X., F.-Y.M., X.-Y.S., and J.-P.Z. conducted the experiments. M.Z., J.-P.Z., and X.-B.Z. analyzed the results. M.Z., and Y.-D.S. composed the first draft. J.-P.Z., T.C., and X.-B.Z. wrote the article. B.Z. edited the article.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (81870149, 81770198, 82070115, 81700184, 81570164, 81861148029, 81700183, 81890990, 81730006, and 81421002); the National Key Research and Development Program of China (2019YFA0110803, 2019YFA0110204, 2016YFA0100600, 2020YFE0203000, 2017YFA0103400, and 2018YFA0107801); Ministry of Science and Technology of China (2015CB964902 and 2015CB964400); CAMS Initiative for Innovative Medicine (2021-1-I2M-040, 2017-I2M-B&R-04, 2019-I2M-1-006, and 2017-I2M-2-001); CAMS Key Laboratory of Gene Therapy for Blood Diseases (2017PT31047, 2018PT31038); The Nonprofit Central Research Institute Fund of Chinese Academy of Medical Sciences (2020-pt310-011, 2018PT31004); Discipline Construction project of Peking Union Medical College (201920101401).