
Abstract
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare disease caused by recessive mutations in the TYMP gene, which encodes the enzyme thymidine phosphorylase (TP). In this study, the efficient integration of a TYMP transgene into introns of the Tymp and Alb loci of hepatocytes in a murine model of MNGIE was achieved by the coordinated delivery and activity of CRISPR/Cas9 and a TYMP cDNA. CRISPR/Cas9 was delivered either as mRNA using lipid nanoparticle (LNP) or polymeric nanoparticle, respectively, or in an AAV2/8 viral vector; the latter was also used to package the TYMP cDNA. Insertion of the cDNA template downstream of the Tymp and Alb promoters ensured transgene expression. The best in vivo results were obtained using LNP carrying the CRISPR/Cas9 mRNAs. Treated mice showed a consistent long-term (1 year) reduction in plasma nucleoside (thymidine and deoxyuridine) levels that correlated with the presence of TYMP mRNA and functional enzyme in liver cells. In mice with an edited Alb locus, the transgene produced a hybrid Alb-hTP protein that was secreted, with supraphysiological levels of TP activity detected in the plasma. Equivalent results were obtained in mice edited at the Tymp locus. Finally, some degree of gene editing was found in animals treated only with AAV vectors containing the DNA templates, in the absence of nucleases, although there was no impact on plasma nucleoside levels. Overall, these results demonstrate the feasibility of liver-directed genome editing in the long-term correction of MNGIE, with several advantages over other methods.
Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive, multisystem disorder caused by pathogenic mutations in the TYMP gene. TYMP encodes the cytoplasmic protein thymidine phosphorylase (TP), which in humans is widely expressed in many different cell types and tissues. The enzyme catalyses the phosphorolysis of the pyrimidine nucleosides thymidine (Thd) and deoxyuridine (dUrd) to yield their respective bases, thymine and uracil. 1 The absence of TP causes an imbalance in dNTP pools, which has mutagenic effects on the mitochondrial DNA. 2,3 Affected individuals usually develop gastrointestinal dysmotility, ptosis, and ophthalmoparesis, weakness of the arms and legs, and progressive cachexia, which is ultimately lethal.
Currently, the management of MNGIE is primarily supportive, with a reduction in the systemic accumulation of nucleosides as the main goal of therapy. To this end, several therapeutic approaches have been tested, including haemodialysis, 3,4 ambulatory peritoneal dialysis, 5 platelet transfusions, 4,6 allogeneic hematopoietic stem cell transplantation, 7,8 orthotopic liver transplant, 9 and autologous erythrocyte-encapsulated TP, 10 but their efficacy is either short-lived or the risk of morbidity and mortality is high.
Gene therapy is particularly promising for MNGIE for several reasons. First, as a monogenic disease, MNGIE can, at least theoretically, be treated by the replacement or correction of a single gene. Second, there is no need to target a specific organ, as TP substrates diffuse across plasma membranes through dedicated transporters and accumulate both intracellularly and extracellularly. Third, in carriers of TYMP variants that reduce TP activity, an overall TP activity 26–35% of the normal level is sufficient to maintain normal homeostasis of Thd and dUrd. Accordingly, the correction of a relatively small cell population will likely be sufficient to provide a significant therapeutic effect. 11 Finally, the availability of a murine model of MNGIE that mimics the biochemical abnormalities observed in patients enables in vivo testing of therapies. 12
We assessed several ex vivo and in vivo gene therapy strategies in preclinical studies, including hematopoietic stem cell gene therapy using lentiviral vectors and liver-targeted AAV vectors. When tested in the MNGIE murine model (dKO Tymp −/− Upp1 −/−), both approaches resulted in a biochemical and functional correction. 13 –19 However, hematopoietic stem cell gene therapy requires myeloablation, which is highly toxic in MNGIE patients. Furthermore, because AAV vectors are not integrative, transgene expression mediated by these vectors may decrease over time. 17,20 In addition, there are concerns regarding the potential oncogenicity of both vector systems. 21,22
Recent advances in the molecular tools that allow modifications of specific genomic sequences with very high efficiency are revolutionizing gene therapy and have raised interest in gene editing. 23 Mammalian cells have two natural pathways to repair chromosomal double-stranded breaks (DSBs): homology-directed repair (HDR) and nonhomologous end joining (NHEJ). Artificial genome-editing tools can be used in both to obtain sequence-specific DNA editing in mammalian cells. 24 One such tool, CRISPR/Cas9, has already shown therapeutic efficacy in a variety of preclinical models and in human clinical trials. 25,26
We hypothesized that, in a murine model of MNGIE, long-term TP expression by mouse liver cells could be achieved using CRISPR/Cas9 and an rAAV2/8 vector carrying a TYMP cDNA. The cDNA template tested in this study was designed to enable its integration in a region downstream of the genomic promoter of the endogenous Tymp locus or in the highly active Alb locus in hepatocytes. CRISPR/Cas9 was delivered either as RNA, using nanoparticles, or as DNA, using rAAV2/8 vectors.
Materials and Methods
Design and generation of DNA template plasmids
The sequences of all elements are shown in Supplementary Table S2, and the primers used in the experiments are shown in Supplementary Table S3. All polymerase chain reactions (PCRs) were performed using the Phusion DNA polymerase (F530; Thermo Scientific, Waltham, MA), following the manufacturer's instructions. All primers were purchased from Thermo Fisher Scientific.
Tymp locus DNA template (pM-TYMP-TYMP plasmid)
The template for the Tymp locus contained the mTymp partial cDNA (exons 3–11), flanked by a splice acceptor (SA) at the 5′ end, the polyadenylation signal (pA) SV40 at the 3′ end, and the Tymp gRNA targets at both ends. Generation of the pM-TYMP-TYMP plasmid required the construction of an intermediate plasmid (pINT-1).
The first step was the generation of the SA-mTymp-pA construct, obtained by overlapping PCRs. The mTymp cDNA was amplified from the pCMV3-mTymp-HA plasmid (MG50923-CY; Sino Biological, Beijing, China), and the pA SV40 element from the pCMV/Bsd plasmid (V51020; Invitrogen, Carlsbad, CA). This construct was then cloned into the pC backbone (derived from the pCMV/Bsd plasmid) through XhoI and EcoRI restriction sites, yielding plasmid pINT-1. An additional step was necessary to introduce the flanking gRNA targets. The pINT-1 plasmid was used as the template to generate the pM-TYMP-TYMP construct in a simple PCR with specific primers. Finally, this construct was cloned into the pC backbone through the XhoI and EcoRI restriction sites (Supplementary Fig. S3).
Alb locus DNA template (pM-ALB-TYMP plasmid)
The template for the Alb locus contained a cropped form of hTYMP cDNA, in which the first 15 nucleotides (5 codons) were lacking, thus allowing the fragment to remain in-frame with the Alb exon 1. This cropped cDNA was flanked at the 5′ end by a SA, at the 3′ end by pA SV40, and on both sides by Alb intron 1 homology arms (HAs) and then the Alb gRNA targets.
Generation of the pM-ALB-TYMP plasmid also required the construction of an intermediate plasmid (pINT-2). In the first step, the cropped hTYMP cDNA was amplified in a simple PCR using pEX-ALB-TYMP as the template and specific primers. This construct was cloned into the backbone of pC-ALB-TYMP through the SpeI and AgeI restriction sites, yielding plasmid pINT-2. This intermediate step allowed introduction of the cropped hTYMP cDNA between the HAs, containing the two mutated in-frame ATGs. The pM-ALB-TYMP construct was generated in a simple PCR using pINT-2 as the template and specific primers. This construct was cloned into the pC backbone through the XhoI and EcoRI restriction sites (Supplementary Fig. S4). Both pEX-ALB-TYMP and pC-ALB-TYMP were previously generated in our laboratory.
The pEX-ALB-TYMP plasmid was obtained using plasmid pEX-A2-Alb, purchased from Eurofins Scientific (Luxembourg City, Luxembourg) and custom-designed to include the 840-nucleotide HAs of the Alb locus. The right and left HAs were separated by the SpeI and AgeI restriction sites. The SA-cDNA-pA construct was obtained in an overlapping PCR and then cloned between the HAs at the SpeI and AgeI restriction sites. The hTYMP cDNA was amplified from p305-TP, 13 and the pA SV40 element from pCMV/Bsd (V51020; Invitrogen).
The pC-ALB-TYMP plasmid was produced as follows. First, the SA-cDNA-pA construct was PCR-amplified using pEX-ALB-TYMP as the template together with specific primers. The resulting construct, with shortened Alb HAs, was inserted into a pC plasmid backbone at the XhoI and EcoRI restriction sites. The obtained plasmid was then modified through directed mutagenesis, using the GeneArt site-directed mutagenesis system (A13282; Invitrogen) according to the manufacturer's instructions, to eliminate two in-frame ATGs of the left Alb HA.
Design and generation of CRISPR/Cas9
In the first gene-editing approach, designed to deliver CRISPR/Cas9 as RNA molecules using NP, Cas9 mRNA (L-7606; Trilink Biotechnologies, San Diego, CA) was used in all experiments. The gRNAs for the preliminary experiments were generated using the Precision gRNA Synthesis Kit (A29377; Invitrogen) according to the manufacturer's instructions. This required custom-designed specific primers, which were purchased from Thermo Fisher Scientific. Later, synthetic gRNAs containing chemical modifications that enhanced their stability and efficacy (Supplementary Fig. S6) were purchased from Axolabs (Kulmbach, Germany). The gRNA sequences used for both loci are presented in Supplementary Table S4.
In the second gene editing approach, in which rAAV2/8 vectors delivered CRISPR/Cas9 as DNA molecules, two vectors carrying the DNA templates and the coding sequences of the Cas9 and gRNA scaffolds were administered for each locus. The three required vectors, pAAV-Cas9, pAAV-TYMP5, and pAAV-ALB2, were custom-designed and ordered as synthetic plasmids from ATG:Biosynthetics (Merzhausen, Germany) (Supplementary Fig. S7). The sequences of all elements carried by the AAV vectors are shown in Supplementary Table S5.
rAAV2/8 vector production
All rAAV2/8 vectors were generated at the Viral Vector Production Unit, located at the Centre de Biotecnologia Animal i de Teràpia Gènica of the Universitat Autònoma de Barcelona (Barcelona, Spain). The vectors were produced by cotransfection of HEK-293 cells and purified by polyethylene glycol precipitation and iodixanol gradient separation.
Polymeric nanoparticle production
Oligopeptide end-modified poly-(β-amino esters) (OM-pBAEs) were synthesized at GEMAT-IQS following a two-step procedure, as described previously. 27 –33 First, an acrylate-terminated polymer was synthesized by the addition of primary amines to diacrylates (molar ratio of 1:1.2 [amine:diacrylate]), after which OM-pBAEs were obtained by modification of the resulting acrylate-terminated polymer via end-capping of the thiol-terminated oligopeptide at a 1:2.1 molar ratio with dimethyl sulfoxide. The mixture was stirred overnight at room temperature and the resulting polymer was obtained by precipitation in a mixture of diethyl ether and acetone (1:1) (Supplementary Fig. S8).
These OM-pBAEs were mixed with the desired RNAs and incubated at 25°C for 30 min to induce the formation of electrostatic bonds. A further step involving a pH change allowed the precipitation of small particles. The polymeric nanoparticles (PNPs) were lyophilized and stored at −20°C. At the time of their use, they were resuspended in sterile water.
KH-ret PNPs were obtained in an optimization process. This preparation consisted of three polymers with a common backbone but differing in either their end oligopeptides or their lateral chains by the presence of: (1) a lysine (K) triplet, (2) a histidine (H) triplet, both as end-oligopeptides, and (3) a K polymer with retinol functionalization in the lateral chain (Kret); all three contained a cysteine (C) to enable the acrylate reaction. The respective preparations were mixed in a 55:5:40 ratio (K:Kret:H polymers) and used as a carrier of Cas9 mRNA and gRNAs (at weight ratios of either 1:2 or 1:6 [gRNA:Cas9 mRNA]).
Lipid nanoparticle production
Lipid nanoparticles (LNPs) were generated at Acuitas Therapeutics® as described previously. 34 –40 In brief, gRNAs and Cas9 mRNA in aqueous solution at pH 4 were mixed 1:1 (mass ratio) and then added to a mixture of four lipids: (1) cholesterol, (2) 1,2-distearoyl-sn-glycero-3-phosphocholine, (3) a PEG2000 lipid with a C14 anchor, and (4) a proprietary ionizable lipid. 34 The proprietary lipid and the LNP composition are described in patent application WO 2017/075531A1.
The resulting LNPs were dialyzed overnight in aqueous solution to remove residual ethanol. The diameter of the LNP was in the range of 69–70 nm, with a polydispersity index <0.055 (as measured by dynamic light scattering using a Zetasizer Nano ZS; Malvern Instruments Ltd., Malvern, United Kingdom) and encapsulation efficiencies of 88% (determined by RiboGreen analyses; Molecular Probes/Invitrogen, Eugene, OR).
Animal procedures
All animal procedures were performed in accordance with European recommendations and were approved by our Institutional Ethics Committee (Comitè Ètic d'Experimentació Animal). The genetic background of both the wild-type (WT) and the dKO Tymp −/− Upp1 −/− mice 12 was C57BL/6J. WT mice were purchased from Charles River Laboratories (Wilmington, DE). Some of the dKO mice were kindly provided by M. Hirano (Columbia University, New York, NY) and used to generate a colony that was maintained until the end of the study.
The mice were kept in the conventional housing facility of our institution under controlled humidity and temperature (22°C) conditions and 12-h light-dark cycles. Food (2018 Teklad global 18% protein rodent diet) and water were available ad libitum. Female and male mice 6–10 weeks old were used in the in vivo experiments. The weight and physical condition of the treated mice were monitored periodically throughout the study. The different experimental procedures are summarized in Supplementary Fig. S9. At the end of the experiments, the mice were euthanized with CO2. Necropsies were performed immediately thereafter, at which time the mouse livers were surgically extracted.
Blood sampling
Blood samples were obtained from mice injected with both the CRISPR/Cas9 elements and the DNA templates. The first sample was collected 1 week before treatment, followed by additional sampling for up to 1-year postinjection. Blood was collected by manually restraining the mice and then puncturing the submandibular vein using a 20-gauge needle. The overflowing blood was collected in Microvette tubes (10780044; Sarstedt, Nümbrecht, Germany) by capillary action. The plasma fraction was diluted and stored at −80°C until the analysis.
Intravenous injections
All injections were performed through the lateral tail vein of the mice, with volumes of up to 200 μL administered using a 30-gauge needle. Mice in the negative control group were injected with physiological saline solution. Before their administration, the PNPs were resuspended in sterile water to a concentration of 250 ng RNA/μL. Each animal was injected with 50 μg of RNA (200 μL). Two doses were tested: dose 1 required one injection, and dose 2, two injections administered on consecutive days. Two different weight ratios of gRNA:Cas9 mRNA (1:2 and 1:6) were also tested.
The LNPs were prepared in phosphate-buffered saline (PBS) (1 × ) supplemented with a cryoprotectant and delivered at a concentration of 1 μg RNA/μL. Before their administration, the LNPs were diluted in saline solution to a concentration of 250 ng RNA/μL (200 μL). Each mouse was injected with 50 μg (200 μL) of gRNA:Cas9 mRNA at a 1:1 weight ratio. The rAAV2/8 vectors were delivered in PBS-MK +40% iodixanol buffer. Before their administration, the vectors were diluted in saline solution to obtain the desired concentration. The mice were then injected with 200 μL of the diluted vectors.
Nucleic acids and protein extraction
Genomic DNA was extracted using the GenElute Mammalian Genomic DNA Miniprep Kit (G1N70; Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. RNA was extracted using the RiboZol RNA Extraction Reagent (97064; VWR Life Science, Radnor, PA) according to the manufacturer's instructions. Protein was extracted by homogenizing the samples in lysis buffer (0.02% 2-mercaptoethanol, 1% Triton X-100, 2 mM PMSF, 50 mM Tris-HCl, pH 7.2) followed by centrifugation at 20,800 g for 30 min at 4°C. The resulting supernatant contained the extracted proteins.
Sequencing
Sanger sequencing was performed at Macrogen (Seoul, South Korea) through a service provided by our institutional Unitat d'Alta Tecnologia. Next-generation sequencing (NGS) was performed at the genomic platform of Banc de Sang i Teixits (Barcelona, Spain). The samples were processed on an Illumina MiSeq sequencing system (Illumina, San Diego, CA) and the results were then analyzed using the software QIAGEN CLC Genomics Workbench version 20 (Qiagen, Hilden, Germany). PCRs were performed using the genomic DNA extracted from the mouse livers as template, Ex Taq DNA polymerase (RR01AM; Takara), and the primers listed in Supplementary Table S6. The nuclease cutting efficiency was calculated based on the proportion of reads with mutations near the gRNA target sequence.
Detection of gene editing by PCR
The presence of edited cells was determined using PCR and specific primers to detect the inserted DNA templates (Supplementary Table S7). Edited and nonedited DNA molecules were distinguished according to the size of the observed bands, which also allowed differentiation of the insertion mechanism (HDR or NHEJ). If gene editing was undetectable using a simple PCR, due to its low efficiency, nested PCRs with specific primers were performed (Supplementary Fig. S10). The sequences of the PCR products were verified by Sanger sequencing.
Quantification of TP mRNA
The RNA samples were reverse transcribed using the high-capacity cDNA reverse transcription kit (4368814; Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The amount of TP mRNA was quantified in a real-time PCR using the TaqMan fast advanced master mix (4444556; Applied Biosystems) as recommended by the manufacturer. This methodology required the use of two custom-designed TaqMan gene expression assays (4448892; Applied Biosystems): one to quantify hTYMP cDNA (Hs00157317_m1) and the other to quantify mGapdh cDNA (Mm99999915_g1), which served as an internal housekeeping expression control (calibrator).
For each cDNA sample, two different tests, each run in duplicate, were performed, one using hTYMP cDNA and the other using the calibrator. The threshold point (Ct) was determined for all samples and used to calculate the normalized ratio of human TP (hTP) mRNA expression as follows:
Western blot
The proteins were separated by polyacrylamide gel electrophoresis (PAGE), performed using premade NuPAGE 4–12% Bis-Tris gels (NP0321; Invitrogen), and then transferred onto a PVDF membrane using iBlot 2 PVDF mini stacks (IB24002; Invitrogen).
After the membrane had been blocked with TBS-T (1 × ) supplemented with 5% dry milk, mouse TP was stained using a 1:2,000 dilution of a sheep anti-TP IgG (AF7568; Research and Diagnostic Systems, Minneapolis, MN) and then with a 1:10,000 dilution of a secondary anti-sheep IgG conjugated with horseradish peroxidase (HRP, A16041; Invitrogen). hTP was stained first with a 1:500 dilution of anti-TP monoclonal antibody (biotin-conjugated mouse IgG1; clone P-GF.44C, NB100-2737B; Novus Biologicals) and then with a 1:5,000 dilution of HRP-conjugated streptavidin (21130; Thermo Scientific).
The reacted proteins were revealed by incubating the membrane with Pierce ECL western blotting substrate (32109; Thermo Scientific). β-Actin, used as a loading control, was first incubated with a mouse anti-β-actin monoclonal antibody (IgG2A clone AC-74 from mouse, A5316; Sigma-Aldrich) and then with an HRP-conjugated goat anti-mouse polyclonal IgG (A90-105P; Bethyl Laboratories, Montgomery, TX). For each sample, the intensity of the TP band was normalized against that of the respective β-actin band. TP expression was calculated as:
Quantification of plasma Thd and dUrd
Plasma Thd and dUrd levels were quantified using high performance liquid chromatography (HPLC) with ultraviolet spectrophotometric detection as described previously. 19
Determination of TP activity
TP activity was determined as described previously. 19 In brief, tissue homogenate or plasma was incubated for 1 h in the reaction buffer in the presence of Thd, as detailed elsewhere, 41 and the amount of Thd converted to Thy in the presence of TP was quantified using HPLC and ultraviolet spectrophotometric detection.
Statistical analysis
All statistical analyses were conducted using the software GraphPad Prism 6 (GraphPad Software, Inc., California City, CA). For most comparisons, nonparametric tests were applied, as the sample size was small and a normal distribution could not be assumed. A Kruskal–Wallis test was used with unpaired values, and a Friedman test with paired values. Both were followed by a Dunn's multiple comparisons post hoc test. For comparisons of multiple groups with repeated time measures, a mixed model with group, time, and their interaction was used. The statistical significance of the interaction term was used to assess the differences in the change over time between groups. This analysis was performed with SAS® 9.4 (SAS Institute, Inc., Cay, NC). A p-value <0.05 served as the threshold for statistical significance.
Results
Successful design and generation of CRISPR/Cas9 and DNA templates
For each loci, one CRISPR/Cas9 target was selected. For the Tymp locus, the gRNA targeted the Tymp intron 2 (Tymp5 gRNA), while for the Alb locus the gRNA targeted Alb intron 1 (Alb2 gRNA) (Supplementary Figs. S1 and S2). The template for the Alb locus contained the hTYMP cDNA flanked by two homology arms (HAs), and the template for the Tymp locus the mTymp cDNA without additional homologous sequences. In both cases, the cDNAs were preceded by a SA, since introns were targeted in this gene editing strategy. Template production is explained in the Materials and Methods section and in Supplementary Figs. S3 and S4. The detailed structure of these templates and the gene-editing strategies are shown in Supplementary Figs. S1 and S2.
Efficient gene editing at the Tymp and Alb genomic loci by LNP carrying CRISPR/Cas9 mRNA
In the in vivo experiment, the first step was to assess the delivery efficiency of the two nanoparticle formulations. PNPs in a KH-ret formulation and carrying gRNA:Cas9 mRNA at different dose and weight ratios were injected into the mice. However, while in some of the mice, the level of on-target cutting was higher than the level measured in negative control mice (injected with saline), the difference at either locus was not statistically significant (Fig. 1). With LNP administered in a single fixed dose and weight ratio, the cutting efficiencies were 12.5% ± 1.4% for the Tymp locus and 47.6% ± 7.9% for the Alb locus, but the difference (relative to the control group) was significant only at the Tymp locus (Fig. 1).

In vivo hepatic CRISPR/Cas9 cutting efficiency measured by NGS. CRISPR/Cas9 efficiency (%) was determined for Tymp5 gRNA
Reduction in Thd and dUrd plasma levels in vivo using CRISPR/Cas9 mRNA delivered by LNP
The second step was to assess the treatment efficacy, by quantifying plasma Thd and dUrd levels in the mice before and 2 and 4 weeks after treatment. Two strategies were tested: in the first, NP were used to deliver the CRISPR/Cas9 and AAV vectors to the templates; in the second, these elements were delivered using two different AAV vectors.
In the first approach, the mice were treated with different doses of the KH-ret PNP formulation and the AAV templates under different conditions. Although in some cases, Thd and dUrd levels decreased slightly after treatment, the differences compared to the pretreatment values were not significant at either of the targeted loci (Fig. 2). However, in mice treated with the LNP formulation and AAV templates, the plasma levels of Thd and dUrd decreased significantly (Fig. 2).

Nucleoside plasma levels: gene editing using NP to deliver CRISPR/Cas9. The levels of
The second gene editing approach tested four different doses. While slight decreases compared to the pretreatment values were observed under all conditions, only in the Tymp locus dose 2 treatment were dUrd levels reduced significantly, from 16.9 ± 2.6 to 12.1 ± 1.9 μM at 4 weeks after AAV administration (p = 0.05) (Fig. 3).
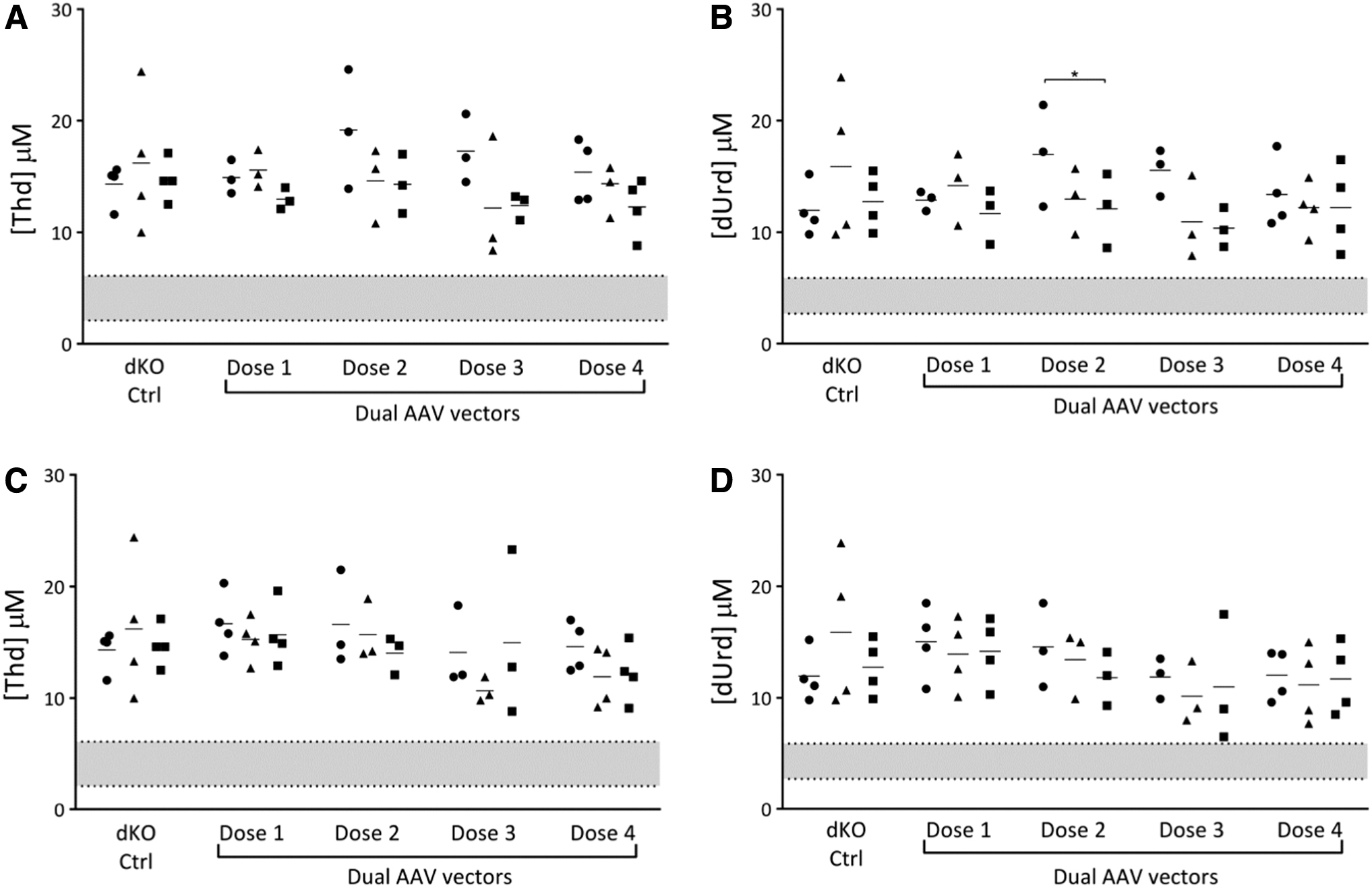
Nucleoside plasma levels: gene editing using AAV vectors to deliver CRISPR/Cas9 (dual AAV vector approach). The levels of
In addition, two cohorts of animals treated with the LNP formulation and AAV templates were monitored for 1 year. In these mice, there was a consistent reduction in nucleoside levels, in some cases reaching the range of WT mice. These reductions were similar in the two groups but progressed more steadily in mice edited at the Alb locus.
The untreated controls were followed for a shorter period of time, but a statistical analysis using the mixed model (comparing group, time, and their interaction) showed highly significant differences in the reduction of nucleoside levels between the two treated groups compared with the untreated controls (p < 0.01 in all cases) (Fig. 4). Reductions in plasma nucleoside levels remained stable in mice up to 58 weeks of age. This sustained decrease had not been observed in previous cohorts of untreated dKO mice followed long term at our institution. 16,18,19
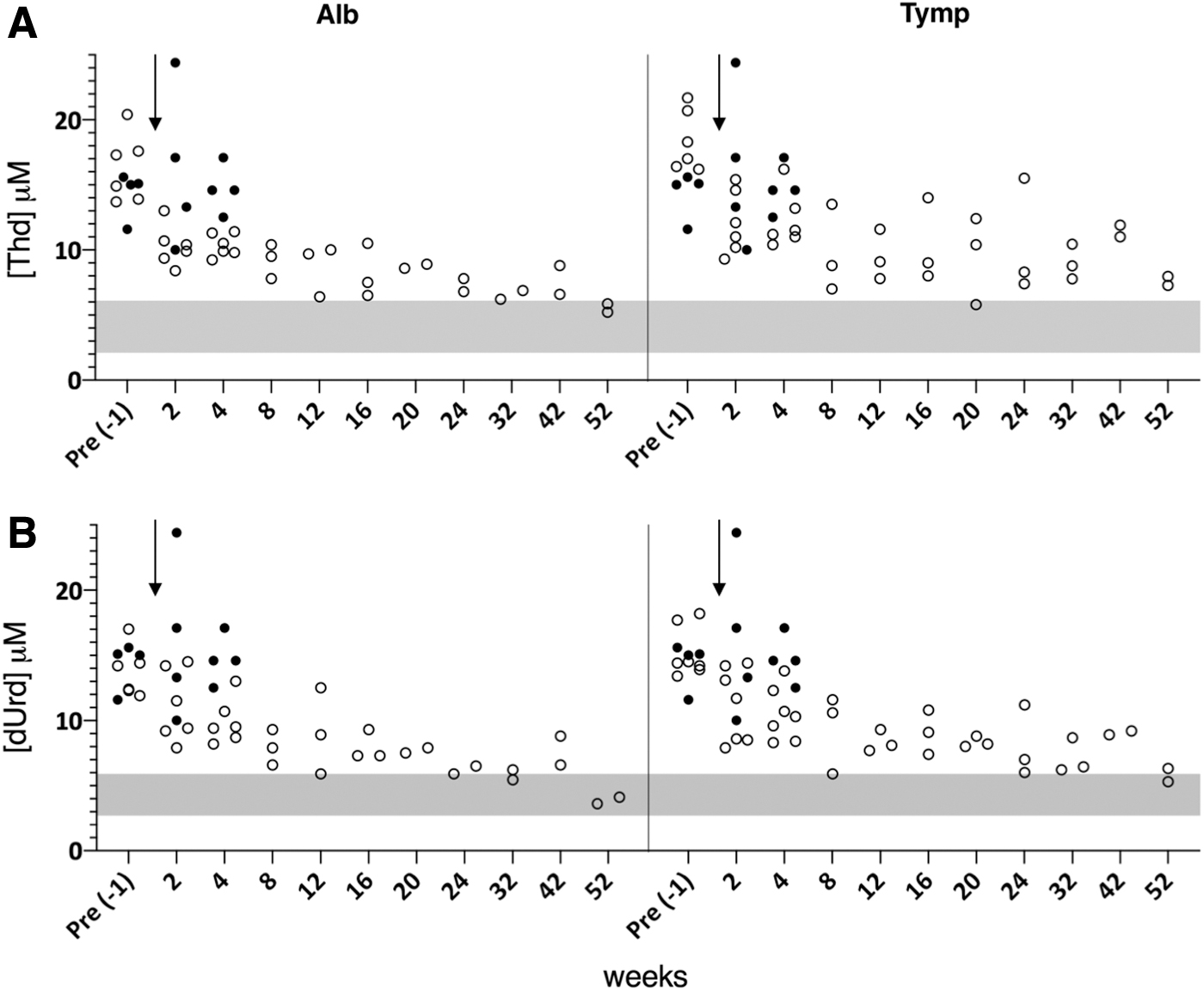
Nucleoside plasma levels during the long-term follow-up of mice treated with LNP and AAV vectors. The levels of
Successful insertion of DNA templates at the genomic Tymp and Alb loci
The livers of the mice were extracted 5–10 weeks after treatment. First, the efficacy of DNA template insertion at the target loci in murine liver cells was assessed by PCR using specific primers. Insertion through HDR was readily detected, based on the stable size of the resulting PCR product, whereas insertion through NHEJ was more difficult to detect, due to the variable size of the PCR product.
In the gene editing approach consisting of CRISPR/Cas9 delivered by NP, the best results were obtained in mice injected with the LNP formulation, with correct editing detected in all of the treated animals. In the gene editing approach that delivered CRISPR/Cas9 using AAV vectors, the best results were those of mice injected with the highest doses. These results are presented as the proportion of animals in which template insertion was confirmed by PCR and subsequent sequencing (Supplementary Table S1).
LNP-induced TP expression in the liver and increased plasma TP activity
The extracted livers were also used to measure hTP mRNA expression of mice with an edited Alb locus. In the negative controls, hTP mRNA levels were undetectable, whereas in all groups of treated mice hTP mRNA was detected in varying amounts. Specifically, hTP mRNA levels in mice treated with KH-ret PNP and the AAV templates were very low and, surprisingly, even lower than in animals treated with the AAV templates alone. In mice treated with LNP and the AAV templates, hTP mRNA levels were significantly higher (p < 0.01) than in the negative controls (Fig. 5). A dose-dependent response was observed in mice treated with CRISPR/Cas9 delivered by the AAV vectors, but only at the highest dose (dose 4) were hTP mRNA levels significantly higher than in the negative controls (Fig. 5).
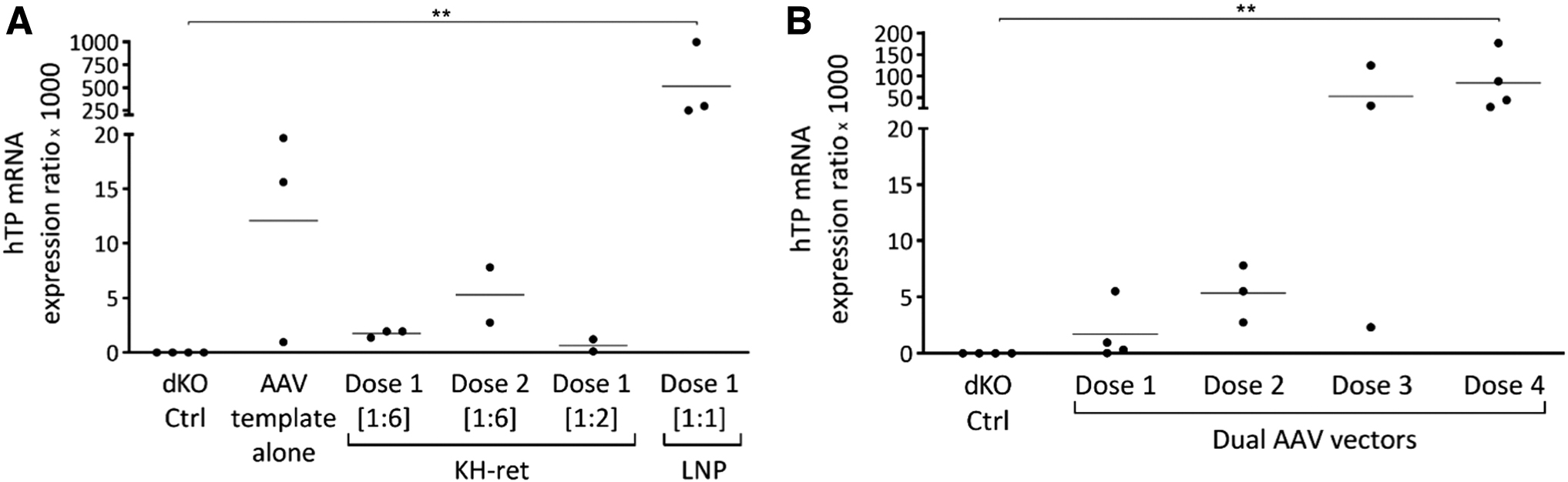
In vivo relative quantification of liver hTP mRNA from the Alb locus. CRISPR/Cas9 delivered by
The expression of TP in the liver extracts was assessed by western blot. TP was detected only in mice injected with the LNP formulation and AAV templates (Supplementary Fig. S5A). In mice edited at the Tymp locus, TP protein levels were 7% ± 2.5% of those of WT mice, while in mice edited at the Alb locus, they were 6.4% ± 1.2% (Supplementary Fig. S5B). Finally, TP activity was measured in liver extracts and in plasma. In the extracts, the highest activity of both loci was detected in mice treated with LNP and the AAV vectors (Fig. 6A, B). In the plasma samples, TP activity was significantly higher in LNP-treated mice with an edited Alb locus (125 ± 51.7 nmol Thy/h × mL plasma) than in either untreated dKO mice (undetectable) or untreated WT mice (6.1 ± 1.4 nmol Thy/h × mL plasma) (Fig. 6C).
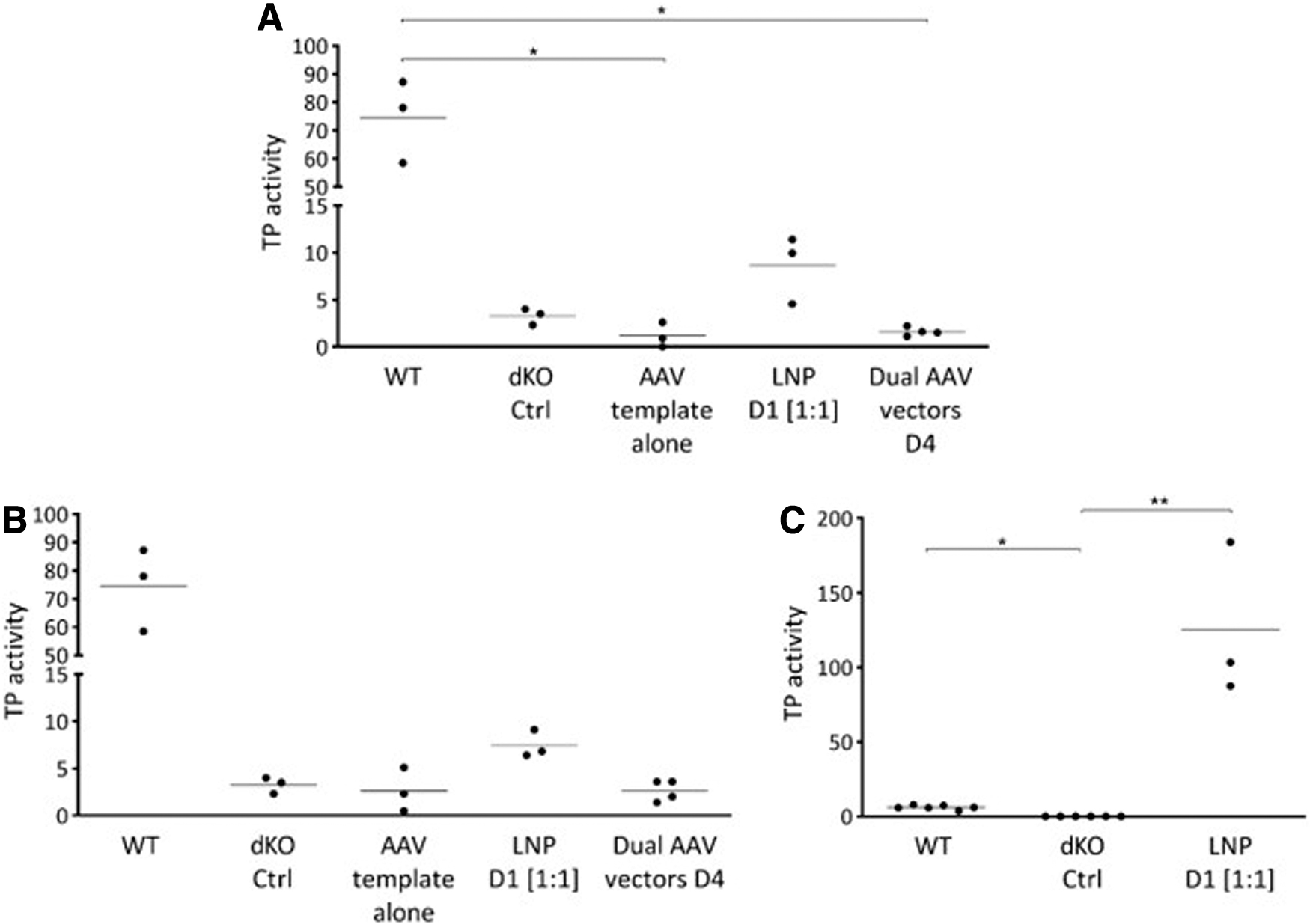
In vivo TP activity.
Discussion
Gene editing overcomes several of the limitations of current gene therapy strategies and is thus the focus of considerable research interest. In this work, we demonstrated that a metabolic disease such as MNGIE can be treated using two different hepatocyte-directed gene-editing approaches. Both rely on insertion of the transgene downstream of a genomic promoter, thus allowing cellular expression of the cDNA only when correct insertion is achieved. The edited hepatocytes become a source of TP (either intracellular or secreted), the levels of which will presumably remain relatively stable over time.
In our study, two genomic sites were targeted, the endogenous Tymp locus and the Alb locus, whose strong promoter allows potent transgene expression in hepatocytes. 34,42,43 Our gene editing approach requires efficient delivery to the same cells of two elements: a DNA template that must reach the hepatocyte nucleus, and CRISPR/Cas9, which can be delivered either as DNA or RNA. Although multiple vector platforms are currently available, 44,45 in this study, only two strategies were tested for CRISPR/Cas9 delivery.
In the first, different formulations of NP were used to deliver CRISPR/Cas9 in RNA form, thus reducing the toxicity of the Cas9 nuclease by restricting its expression to a very short period of time. 46 Several studies have reported the successful delivery of CRISPR/Cas9 RNAs via different NP formulations. 47 The DNA templates were provided using AAV2/8 vectors, which are highly efficient at targeting the liver in mice and other species, including humans. 48
In the second strategy, CRISPR/Cas9 was encoded in an AAV vector (dual AAV vector approach). Of the two types of NP tested in combination with the AAV2/8 vectors, LNPs were more efficient than PNP for gene-editing purposes. LNP are already optimized for mRNA delivery to the liver, 34,35 –37 including formulations with natural tropism for liver tissue upon systemic administration. 49 –51 PNPs, by contrast, were not initially designed to target the liver, 27 –29 and while the KH formulation was specifically modified to increase hepatic targeting (by the addition of retinol molecules), in murine liver, these particles were designed to preferentially transfect nonparenchymal cells. 30
The success of our strategy depended on the coordinated activities of the cDNA template and the gene-editing molecules. These two elements had to be efficiently delivered not only to the liver but also to the same hepatocytes, which are the only cell type with strong Alb (and probably also TP) expression. However, not all hepatocytes are metabolically equal (differing, e.g., in albumin and perhaps also in TP expression) and they likely also differ in their susceptibility to different delivery systems. In this regard, liver zonal heterogeneity may have contributed to the results obtained in this work. 52 Optimization of the delivery systems and the strategies for gene editing in the liver may require taking liver zonation into account.
In the dual AAV approach, use of the same vector type to deliver the different genetic elements theoretically ensured the even transduction of the same subset of hepatocytes. Nonetheless, the poor results obtained in this experimental arm may have been related to a suboptimal timing of the injections. While both of the gene editing elements were functional in the tested setting, either the least efficient of them or their poorly coordinated activity may have limited the overall efficiency.
This problem was addressed by the delivery of CRISPR/Cas9 RNAs using LNP, previously shown to allow expression during the first 24 h, with a peak ∼4 h postadministration. 34,35 With this approach, the templates relatively quickly become available inside the cells as ssDNA molecules. The delayed expression that occurs when AAV vectors are used is irrelevant in our system because the templates do not need to be expressed. However, it is important to note that the two strategies cannot be directly compared, as many factors might have influenced the respective results.
Among the conditions tested in vivo, the best results were obtained using LNP. Only these mice attained a significant reduction of Thd and dUrd plasma levels lasting for at least 1 year after treatment. These mice had the highest levels of TP mRNA as well as detectable levels of TP protein in the liver, and the plasma of LNP-treated mice carrying an edited Alb locus contained the highest levels of TP enzymatic activity.
The therapeutic effect was associated with a reduction in nucleoside plasma levels starting from week 2 and continuing for up to 1 year after LNP administration. This was the case in mice edited at either locus, although in mice edited at the Alb locus the reduction tended to be progressive, in contrast to the gradual decrease in transgene expression described in previous gene-therapy studies. 16 This intriguing temporal pattern of the therapeutic effect may have reflected the slow, progressive accumulation of circulating TP in peripheral tissues, but an eventual selective advantage of the gene-edited hepatocytes cannot be ruled out.
A limitation of this study was the lack of an untreated control group followed long term. However, in previous studies at our institution, in which different cohorts of untreated dKO mice of similar ages were followed for 24, 18 34, 19 and up to 88 weeks, 17 nucleoside plasma levels remained stable over the lifetimes of the mice.
Basal Thd and dUrd plasma levels in our dKO mice were slightly higher than those reported in previous studies, 13,14,16 –18 despite the fact that all of the mice used in these different studies were derived from the same colony and were raised in the same institution. In historical cohorts, Thd and dUrd levels decreased slightly in younger, untreated mice, probably due to the lower cellular nucleoside requirements of the animals as their growth rate slowed and their metabolism became adapted accordingly. After this initial decrease, the levels of Thd and dUrd remained stable over long periods of time. 17
By contrast, in our LNP-treated mice nucleoside levels gradually declined. Our results constitute a proof of principle and are the first demonstration that a gene therapy approach based on gene editing may be of therapeutic benefit in MNGIE, as suggested in a murine model of the disease. Because the therapeutic effect is probably dose-dependent, whether higher doses will lead to further reductions in plasma nucleoside concentrations remains to be determined.
Further optimization of our protocol might be achieved using other combinations of LNP and rAAV2/8 vectors as well as different doses and administration schedules. LNP readministration followed by the close monitoring of nucleoside levels until the desired therapeutic effect is obtained may also be effective. Since the DNA template remains inside the nucleus over a long period of time, there is ample opportunity for LNP readministration (probably several weeks). In theory, this approach would be feasible, as LNPs are not immunogenic and their biosafety profile is good. However, Cas9 protein (and other bacterial nucleases) is potentially immunogenic, 53 –55 such that careful planning is needed to avoid inducing or exacerbating an immune response following sustained or repeated Cas9 expression in edited cells.
The fact that transgene expression is restricted to hepatocytes may, on the contrary, facilitate the induction of immune tolerance. 56 –58
One of the main advantages of gene-editing approaches such as the one presented here is a circumvention of the loss of transgene expression, which has been reported in some liver-targeted AAV-based conventional gene therapies. A previous study described the effective use of rAAV2/8 vectors targeting the liver in murine MNGIE, 14 but a long-term follow-up demonstrated a tendency of progressively lower transgene expression. 16 This reduction would be very relevant in humans, as AAV vectors induce humoral immune responses and can be administered only once. In a clinical trial, in which AAV5-based gene therapy was used to treat hemophilia A, FVIII expression decreased during the 3-year follow-up. 20 We believe that the biochemical correction obtained with gene editing will be permanent, as suggested in other reports, 59,60 but this remains to be confirmed by a longer follow-up.
Transgene insertion at the Alb locus has been used safely and successfully in preclinical models to express and secrete proteins of therapeutic value. 61,62 Since Alb exon 1 encodes a secretory signal peptide that is cleaved from the final product, the addition of a transgene cDNA containing a SA site into intron 1 allows the creation of a new protein that combines the secretory signal peptide and the protein of interest. Also, eventual indels due to NHEJ events at this intron are not expected to cause serious problems.
Correct editing at this locus precludes normal albumin expression, but the level of insertion is unlikely to reduce plasma albumin levels to a clinically relevant degree. Also, since albumin production is not essential for hepatocyte survival, its absence or reduction is not likely to confer a selective disadvantage. However, if the deficiency indeed turns out to be problematic, then strategies aimed at the coexpression of albumin and TP from the same transcript can be used. 63
Because TP is an intracellular enzyme, it was initially unclear whether a secreted form would be functional or very short lived. While human platelets exhibit strong TP activity, 6 murine blood cells and plasma have very low TP activity. 13 The secretion of TP by tumour cells and the presence of the enzyme in the plasma of cancer patients have been reported. 64,65 A study of the pharmacokinetics and tissue distribution of intravenously injected TP showed that the enzyme was stable, with a relatively long half-life in the circulation. 66 Although there is no evidence that TP is secreted physiologically, these findings suggest that secreted TP is active in the circulation and thus able to clear systemically accumulated nucleosides. Support for this hypothesis comes from previous experimental studies demonstrating correction of the metabolic defect in multiple tissues. 13 –18
The biochemical correction reported in those studies together with the therapeutic effect of the secreted enzyme observed in our experiments provide proof of concept for the treatment not only of MNGIE but also other hereditary, monogenic, and metabolic diseases as well. The low intracellular hepatic activity but very high plasma activity (>20-fold higher than in WT mice) of TP is consistent with efficient enzyme secretion.
Despite our demonstration that an Alb-hTP hybrid retains its activity in plasma, additional studies are needed to determine the protein's clinical potential. A positive result would support the use of editing at the Alb locus as a platform to treat not only noncell autonomous monogenic diseases, such as intracellular enzyme deficiencies that cause the accumulation and toxicity of diffusible metabolites (e.g., methylmalonic acidemia, propionic aciduria, or tyrosinemia), but also cell-dependent deficiencies, in which the affected cells take up the therapeutic protein or enzyme from the circulation (e.g., lysosomal storage diseases amenable to enzyme replacement therapy). 61
Unexpectedly, delivery of the templates alone was in some cases sufficient to allow efficient gene editing in vivo. In the respective mice, template insertion was confirmed and, in some of their liver samples, detectable TP mRNA expression as well, but without changes in plasma nucleoside levels. These results were especially surprising, as TP mRNA levels were higher in those mice than in mice treated additionally with the KH-ret PNP formulation carrying the CRISPR/Cas9 RNAs. The latter finding indicated that the PNP were inefficient or even deleterious under our experimental conditions.
However, much better results were obtained in mice treated with LNP and the rAAV2/8 template than in those treated with the templates alone. The insertion of DNA templates in the absence of nucleases has been previously described, 63,67 –71 but the efficiency of the insertion was variable. High levels of editing resulted in a therapeutic effect in a hemophilia B model but other studies found that such insertions can occur spontaneously, although with an efficiency several orders of magnitude below that obtained with nucleases. Indeed, successful applications of promoter-less gene editing have been reported in preclinical models. 63,69 Our results are consistent with those of other studies that also targeted the Alb locus, in which the additional presence of a nuclease clearly enhanced the therapeutic benefit. 34,42,43,61,72
Nevertheless, the advantages of nuclease-free gene editing include an avoidance of both the immunogenicity of the nuclease 53 –55 and the potential mutagenesis induced by the repair of DSBs in the DNA. 73 –75 Although the delivery of CRISPR/Cas9 in the form of RNA is probably much safer than delivery using other forms, the relatively efficient spontaneous insertion of template achieved under these conditions deserves further exploration.
Conclusion
In conclusion, this study demonstrated the feasibility of liver-targeted gene editing in a murine model of MNGIE. Transgene insertion at the Alb locus is likely to become an efficient platform for the expression of secreted proteins with therapeutic value, both in MNGIE and in other monogenic diseases.
Footnotes
Authors' Contributions
M.P. designed and conducted the experiments, analyzed the results, and wrote the article. C.F. and S.B. designed, synthesized, and characterized the PNP. S.F. and Y.T. synthesized and characterized the LNP. N.C. and F.V. performed the NGS analyses. F.V.-J. and R.M. assisted in determining nucleoside levels and TP activity. J.B. designed and supervised the study, analyzed the results, and wrote the article. All authors read and approved the final version of the article.
Acknowledgments
We thank Dr. X. Vidal (UAB) for statistical support, and Dr. M. Hirano (Columbia University) for creating and providing the dKO mice used in these experiments.
Author Disclosure
S.H.Y.F. and Y.K.T. are employees of Acuitas Therapeutics, a company focused on the development of lipid nanoparticle delivery systems for nucleic-acid-based drugs. R.M. was the recipient of personal fees associated with advisory tasks and of financial support from Modis Therapeutics for research outside the submitted work. R.M. also holds a patent “Deoxynucleoside therapy for diseases caused by unbalanced nucleotide pools, including mitochondrial DNA depletion syndromes” (PCT/US16/038110), with royalties paid to Modis Therapeutics.
Funding Information
This research was, in part, supported by the Instituto de Salud Carlos III/FEDER (Grants PI15/00172 and PI19/00295) and by MINECO/FEDER (Grant RTI2018-094734-B-C22). The support of the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) of the Generalitat de Catalunya, through grant SGR 2017 1559, is also acknowledged. Marta Parés was a recipient of a predoctoral fellowship from VHIR/Fundació La Caixa.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.