
Abstract
Oncolytic virus therapy is a promising novel immunotherapy. In this report, we engineered a novel oncolytic influenza virus (IV) carrying an antihuman programmed cell death 1 (PD-1) monoclonal antibody utilizing reverse genetics. A reassortant chimeric IV, named rFlu-huPD1, was synthesized as follows: the heavy chain of the PD-1 antibody was encoded on the PB1 fragment, and the light chain of the PD-1 antibody was encoded on the polymerase acid protein fragment. rFlu-huPD1 antibodies were produced in infected ovalantoic eggs and could replicate to high titers. Moreover, selective cytotoxicity of rFlu-huPD1 was upregulated in multiple hepatocellular carcinoma (HCC) cell lines compared with a control, as determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Furthermore, the activation of T cells in the spleen of tumor-bearing BALB/c mice treated with rFlu-huPD1 was observed, especially cytotoxic CD8+ T cell activation in vivo. In addition, in a patient-derived xenograft liver cancer mouse model, tumor growth was reduced and the overall survival of the mice was increased by intratumoral injections with rFlu-huPD1 compared with wild-type PR8 virus. Taken together, these findings provide evidence for the utility of a combination of oncolytic IVs expressing PD-1 inhibitors for use in HCC virotherapy.
INTRODUCTION
Oncolytic viruses (OVs)
Influenza virus (IV) is a segmented negative-sense single-stranded RNA virus and it has been considered a promising oncolytic agent. 7 –9 Previous studies showed that in a mouse melanoma model, a recombinant IV that expressed a single-chain antibody antagonizing cytotoxic T lymphocyte-associated antigen-4 (IAV-CTLA4) delayed tumor growth. 10 In a similar manner, the generation of recombinant IV encoding rflu4-CTLA4 has been reported in our previous studies. We found that in HepG2 allograft mouse models, rFlu-CTLA4 virus had selective cytotoxicity in hepatocellular carcinoma (HCC) cell lines and decreased tumor growth in vivo. 10
Programmed cell death-1 (PD-1), which is a checkpoint receptor, is expressed on T cells, B cells, and monocytes. PD-1 inhibits T cell activation when it binds to programmed cell death-ligand 1 (PD-L1) and programmed cell death-ligand 2 (PD-L2). 11 Anti-PD-1 immune checkpoint inhibitor 12,13 is a human IgG1 antibody that was approved by the U.S. Food and Drug Administration (FDA) for melanoma therapy in 2014. 14 In this study, we developed a chimeric IV that carries a single-chain antibody Fv segment against PD-1 using reverse genetics.
To investigate the oncolytic efficacy of rFlu-huPD1, in vitro studies and a patient-derived xenograft (PDX) liver cancer mouse model were applied. Through local injection of rFlu-huPD1, the PD-1 antibody can be delivered directly to the tumor site, thus enhancing its oncolytic efficacy accompanied by checkpoint blockade therapy. Our data provide support for its combination with checkpoint inhibition, and it represents a promising strategy in the clinical development of IV immunotherapy.
MATERIALS AND METHODS
Cell lines and viruses
HepG2, Huh7 (human HCC) and MIHA (normal liver cells) and H22 (murine liver cancer cells) cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). The MDCK cells were stored in our laboratory. These cells were maintained in dulbecco's modified eagle medium supplemented with 10% fetal bovine serum (FBS) and cultured at 37°C under 5% CO2. Ten-day-old specific pathogen-free (SPF) chicken embryos (Charles River Laboratories, Beijing, China) were used to culture IV A/PR/8/34 (PR8).
Generation of the chimeric IV expressing PD-1 antibody
A chimeric antibody-expressing IV rFlu-huPD1 was produced using IV reverse genetics technology. The coding sequences of the human PD-1 antibody were downloaded from GenBank. The full-length sequences of PB1 and PA were optimized and synthesized by Sangon Biotech (Shanghai). As shown in Fig. 1A, the heavy chain and light chain of the PD-1 antibody were cloned downstream of PB1 and PA, respectively, as previously described. 15,16 The bidirectional expression vector pHW2000 was used to clone the PB1-PD1 heavy chain and PB1-PD1 light chain.

Construction and characterization of recombinant oncolytic IV rFlu-huPD1.
After sequencing, two chimeric confirmed plasmids and the other six plasmids encoding influenza PR8 virus (pHW-NA, pHW-PB2, pHW-M, pHW-HA, pHW-NP, and pHW-NS) were cotransfected into MDCK cells (ratio 1:2) by Effectene Transfection Reagent (Qiagen, Hilden, Germany). The chimeric rFlu-huPD1 virus was harvested when a hemagglutination-positive result was observed with the hemagglutinin assay. The titers of the rFlu-huPD1 virus were determined with a 50% tissue culture infectious dose (TCID50) in MDCK cells.
Electron microscopy assay
The recombinant targeted oncolytic IV was concentrated by ultrafiltration in membrane encapsulation (100 kD) and centrifuged in a 30% and 60% sucrose density gradient. The purified virus RFLU-APD1 was obtained after deglycosylation. After negative staining, the morphology and size distribution of the recombinant virus RFLU-APD1 were observed by transmission electron microscopy.
Viral growth of recombinant virus rFlu-huPD1 in HepG2 cells
HepG2 cells were inoculated into 96-well plates at 2.0 × 104 to 2.1 × 104 per well. rFlu-huPD1 was added at a multiplicity of infection (MOI) of 0.1. The cells were cultured in an incubator at 37℃ and 5% CO2. Cell culture supernatants were harvested every 12 h until 96 h postinfection, and their viral titers were calculated as the logTCID50/mL. The virus titers of the recombinant oncolytic IV on HepG2 cells were measured and calculated by the Reed–Muchen method.
Flow cytometric assay
HepG2 cells were inoculated into six-well plates at 1 × 106 per well, and the chimeric oncolytic IV rFlu-huPD1 at an MOI of 3 was added. After culturing in a 37°C 5% CO2 incubator for 48 h, trypsin was added for digestion and harvest. Then, the cells were collected and washed twice with phosphate-buffered saline (PBS), and 5 μL Annexin-V and propidium iodide were added to each well and the cells were subjected to fluorescence activated cell sorter within 30 min.
rFlu-huPD1 expression of PD1 antibody by enzyme-linked immunosorbent assay
The PD1 protein was diluted with coating solution at a concentration of 2 μg/mL and applied at 50 μL per well, and the mixture was incubated at 4°C overnight. The supernatant was discarded and the wells were washed with phosphate buffer solution-tween-20 (PBST) five times and then incubated in blocking solution at 37℃ for 2 h. The PD1 polyclonal antibody was diluted in thiosulfate transferase for 10, 20, 40, 80, and 160 times and then added to two duplicate wells successively, at 50 μL for each well. The purified viral protein stock and 10-fold diluent were added and incubated at 37℃ for 1 h.
The plate was washed five times with PBST, and then the secondary antibody was added and incubated for 40 min at 37℃. The secondary antibody was discarded, and the cells were washed with PBST for five times. Chromogenic solution was used to develop the color. 17 The absorbance value at 450 nm was read. An absorbance >2.1 times that of the negative control was judged as positive, and GraphPad Prism was used to draw a titer graph.
Cell viability assays
The human hepatoma cell lines HepG2, Huh7, and MIHA were seeded in 96-well plates at 1.5 × 104 cells per well, and the rFlu-huPD1 virus at 0.1, 1, and 3 MOIs was added. Cell viability was determined with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at 48, 72, and 96 h postinfection.
Animal experiments
All animal experiments were performed in accordance with the institutional animal care and use committee and ethics committee of the Fifth Medical Center of Chinese PLA General Hospital. All facilities were approved by the animal care and ethics committee of the Fifth Medical Center of Chinese PLA General Hospital.
Murine liver cancer H22 cells were used to establish subcutaneous tumors in BALB/c mice and they served as immunocompetent mouse models. Tumor-bearing BALB/C mice were grouped to receive the following three treatments separately (n = 10 each group). When the volume of tumors reached ∼75 mm3, the mice were intratumorally injected with PBS, PR8, or rFlu-huPD1 seven times for 14 days at a dose of 3 × 106 TCID50 in a volume of 100 μL.
The PDX model was constructed by using NPIdKO mice (Beijing IDMO Co., Ltd). These mice are immunodeficient produced by knocking out the MHC I β2M (β2-microglobulin) gene and the MHC II IAβ gene on an NOD/SCID genetic background. Liver tumors (3 mm × 3 mm × 3 mm) were transplanted into the right back subcutaneous tissue of the NPIdKO mice. When the tumor volume was 80–150 mm3, the mice were randomly divided into three groups (8 per group). As shown in Fig. 5A, rFlu-huPD1 or PR8 virus was administered intratumorally for 7 consecutive days at a dose of 3 × 106 TCID50 in a volume of 100 μL.
PBS group was used as the control. The behavior and tumor volume of the mice were observed and recorded every 2 days until 46 days after the first injection. The vital signs, survival, and tumor volume of the mice were observed and recorded continuously. The mice were euthanized when the tumor volume reached 1,500 mm3.
T cell activation
To explore the immune cells in the spleen of tumor-bearing BALB/C mice, spleens were harvested from the mice and the infiltration degree of T lymphocytes was analyzed by flow cytometry at 21 days. Then, the spleen tissues were placed in PBS (pH 7.4) with 2% inactivated FBS and ground into single cells. The single cells were filtered through a 200 mesh cell sieve, centrifuged for 10 min at 200 g and washed with PBS. The cells were stained with CD45/CD3/CD4/CD8/CD69 antibody (APC antimouse CD45, Brilliant Violet 510™ antimouse CD3, FITC antimouse CD4, PerCP/Cyanine5.5 antimouse CD8a PE antimouse CD69, Biolegend, 1 μL) to separate the cytotoxic T lymphocytes (CD8+CD69+) and helper T cells (CD4+CD69+). Finally, flow cytometry analysis was performed according to the manufacturer's instructions.
Hematoxylin–eosin staining
The tissues harvested from the NPIdKO mice were fixed, embedded in paraffin, and sectioned. The sections were immersed in xylene I and II for 10 min and then passed through an alcohol gradient (100% [I, II], 90%, 80%, and 70% alcohol) for 5 min each to dewax and rehydrate the sections. Then, the sections were stained with hematoxylin for 6 min and rinsed with running water. A 1% solution of hydrochloric acid was used to differentiate the section, and the color became slightly lighter and blue after differentiation.
Subsequently, eosin was used to stain the sections for 6 min, and the sections were rinsed with running water. Finally, the sections were washed with 70%, 80%, 90%, and 100% alcohol for 10 s each to dehydrate them and they were placed in xylene for 1 min for clarification. Neutral gum was used to mount coverslips on the sections.
Statistical analysis
All statistical analyses were performed with GraphPad Prism v9.0.0 (GraphPad Software, CA, USA). Student's t-test was used to analyze the differences between two groups. Analysis of variance (ANOVA) was performed to compare multiple groups. Two-sided p < 0.05 was defined as statistically significant.
RESULTS
Generation and characteristics of the chimeric oncolytic IV rFlu-huPD1
To establish a chimeric IV that expresses antibodies during the virus life cycle, we explored fragments of oncolytic IV polymerase PB1 and PA as previously published. 18 The 2439 to 2716 nt chimeric PB1 segments were amplified to produce MU-PD1, and the 2312 to 2632 nt chimeric PA segments were amplified to produce MU-PD1. To translate PB1 and the heavy chain proteins, a sequence of porcine teschovirus 1 (PTV-1) 2A and a signal peptide sequence were inserted between PB1 and the heavy chain gene, respectively. The light-chain expression from the viral PA segment was designed on the same principle.
After optimizing the sequence, the heavy chain and light chain downstream of PB1 and PA were cloned, respectively, as shown in Fig. 1A. Furthermore, the chimeric oncolytic IV rFlu-huPD1 was observed by negative electron microscopy, and the viral particles examined by electron microscopy were >80% viral spherical structures 60–120 nm in size, with a characteristic lipid membrane bilayer on their outer surfaces (Fig. 1B). The chimeric virus rFlu-huPD1 was similar to the wild-type IV in both morphology and size.
Antibodies are produced during chimeric virus rFlu-huPD1 infection
The kinetics of antibody production were detected during rFlu-huPD1 infection. The virus rFlu-huPD1 was inoculated into eggs, and the antibody content in the allantoic fluid was measured. The results showed that the chimeric virus rflu-huD1 had an HA titer of 26 in the first generation of hemagglutination, an HA titer of 29 and a virus titer of 9.5 lgTCID50/mL after five successive generations of transmission through SPF chicken embryos (Fig. 2A, B). In addition, we examined whether the PD1 antibody expression was stable over time. The chimeric virus rFlu-huPD1 was subcultured in SPF eggs, passaging it five times, and its functional transgene expression was detected. We found that no loss of antibody production was observed (Fig. 2C), indicating that the PD1 antibody gene was stable in the rFlu-huPD1 genome.

Antibodies were produced during chimeric rFlu-huPD1 infection.
Furthermore, the pFlu-PD1-PB1 and pFlu-PD1-PA fragments from the second generation of chicken embryo allantoic fluid of the chimeric OV rflu-aPD1 were amplified by real time-PCR. pHW192-PB1 and pHW193-PA were used as positive controls, and the four amplified fragments were 2,948, 2,827, 2,341, and 2,233 bp, respectively. The amplified fragments of the positive control plasmids pHW197-M and pHW198-NS were 1,027 and 890 bp, respectively. The results showed that the gene fragment of the chimeric OV rFlu-huPD1 was of the expected size, indicating the successful rescue of the chimeric oncolytic IV (Fig. 2D).
The chimeric OV rFlu-huPD1 was diluted 10−3 and then inoculated into chicken embryos. Three eggs were inoculated at each time point. The allantoic fluid was collected on days 1, 2, 3, and 4. After purification, the antibody content was detected. In the control group, PBS and PR8 were added to the allantoic fluid after the virus was cultured for 3 days. The results showed that the chimeric OV rFlu-huPD1 could detect PD1 antibodies on the second, third, and fourth days. The antibody content was 0.42 ± 0.08 μg per chicken embryo, 0.58 ± 0.05 μg per chicken embryo, and 0.96 ± 0.11 μg per chicken embryo, but no antibody was detected in the PBS and PR8 control groups (Fig. 2E).
Effects of rFlu-huPD1 on cell viability and the apoptosis of different hepatoma cells
We hypothesized that a chimeric oncolytic IV expressing PD1 antibody would increase the oncolytic activity of oncolytic IV in treating liver cancer in vivo. First, it is necessary to clarify the infectious activity of rFlu-huPD1 on HCC cells. The recombinant oncolytic IV rFlu-huPD1 was used to infect the normal liver cell line MIHA and the liver cancer cell lines HepG2 and Huh7 at an MOI of 0.1 and 1 3, respectively. Cell viability was detected by MTS after 48, 72, and 96 h of infection. We found that there was no obvious killing effect induced by rFlu-huPD1 infection on the normal liver cell line MIHA (Fig. 3A). However, the viability of HepG2 and Huh7 hepatoma cells was significantly decreased in a time- and dose-dependent manner (Fig. 3B, C).
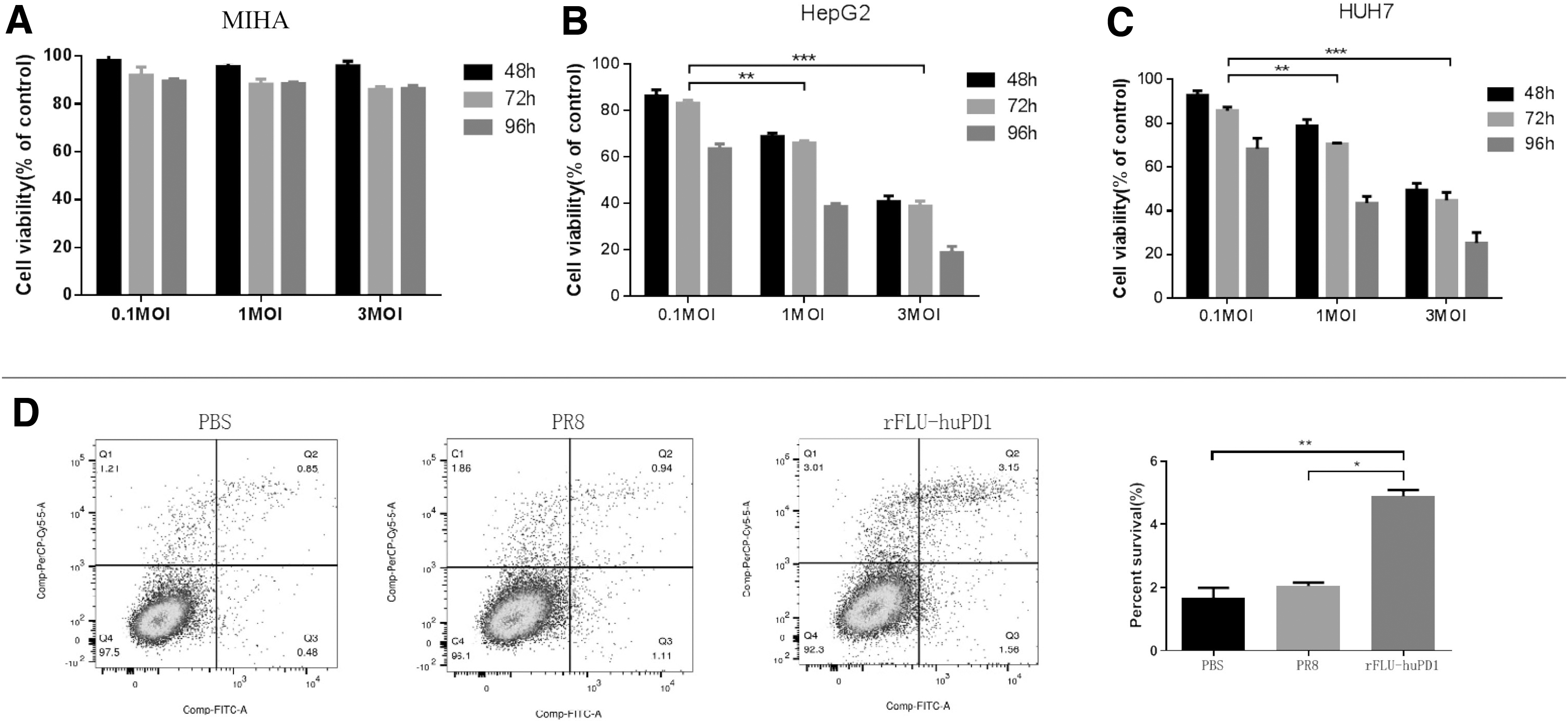
The infectivity of the recombinant OV rFlu-huPD1 was evaluated by MTT and flow cytometry in hepatoma cells and normal liver cells. MIHA
To detect whether rFlu-huPD1 exerts oncolytic effects by inducing tumor cell death, we infected HepG2 cells with the chimeric virus at a MOI of 3. After 48 h, flow cytometry was used to detect the apoptosis of HepG2 cells. The results showed that HepG2 cells treated with rFlu-huPD1 promoted cell apoptosis (Fig. 3D). These results suggest that rFlu-huPD1 selectively killed HCC cells in a time- and dose-dependent manner.
rFlu-huPD1 caused T cell activation in vivo
Murine liver cancer H22 cells were used to establish subcutaneous tumors in BALB/c mice to detect systemic antitumor immune responses. The involvement of CD8+ cytotoxic T lymphocytes (CTLs) and helper T lymphocytes (CD4+ T cells) is the most critical approach in tumor immunotherapy. CD8+ cytotoxic T cells can directly kill targeted cancer cells, whereas CD4+ T cells can maintain and strengthen the potential immune function. In addition, CD69 is the earliest inducible cell surface glycoprotein acquired during lymphoid activation and it is involved in early T cell events. The spleen, which plays an important role in immune regulation in vivo and contains a large number of lymphocytes, was used to detect T cell activation.
Hence, the activation of T cells in the spleen was further analyzed on day 21 after rFlu-huPD1 treatment. The percentage of CD8+ CD69+ T cells in the spleen was examined and analyzed by flow cytometry. As shown in Fig. 4A and B, both PBS injection and PR8 treatment failed to promote CD8+ CTL activation in the spleen.
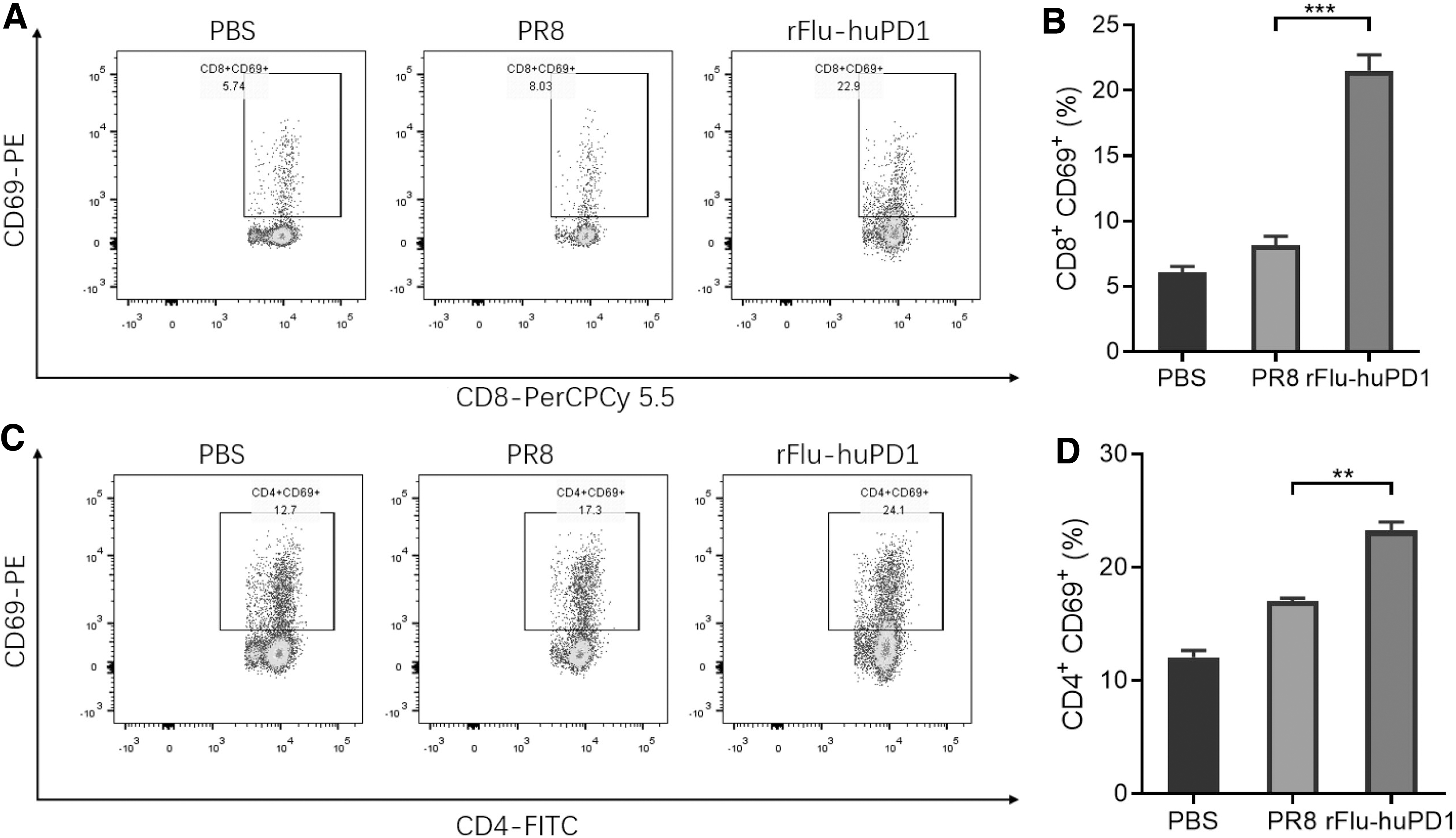
rFlu-huPD1-activated T lymphocytes in the H22 cell tumor-bearing model. Flow cytometric analysis of infiltrated CD8+ CD69+ T cells
In contrast, the percentage of CD8+ CD69+ T cells in the spleen of rFlu-huPD1-treated mice significantly increased to 22.9%, which was higher than that in the groups treated with PBS (5.74%) or PR8 (8.03%). Simultaneously, the percentage of CD4+ CD69+ T cells in the rFlu-huPD1 treatment group was effectively enhanced by rFlu-huPD1 treatment (Fig. 4C, D). These results demonstrated that after rFlu-huPD1 treatment, the infiltration of T cells within the spleen of mice was enhanced and activated.
Oncolytic effects of rFlu-huPD1 in the HCC PDX model
When the tumor volume reached 100–150 mm3, the PDX mice were treated with PBS, PR8, and rFlu-huPD1, the tumor volume was measured and recorded every 2 days, and the tumor volume curve of the mice was plotted. In the control group, the tumors grew rapidly, exceeding 700–800 mm3 on average within 30 days. However, the tumor volume of mice in the rFlu-huPD1- or PR8-treated group dramatically decreased, with mean volumes of 900 and 1,100 mm3 at 46 days after the first injection, respectively (Fig. 5A).
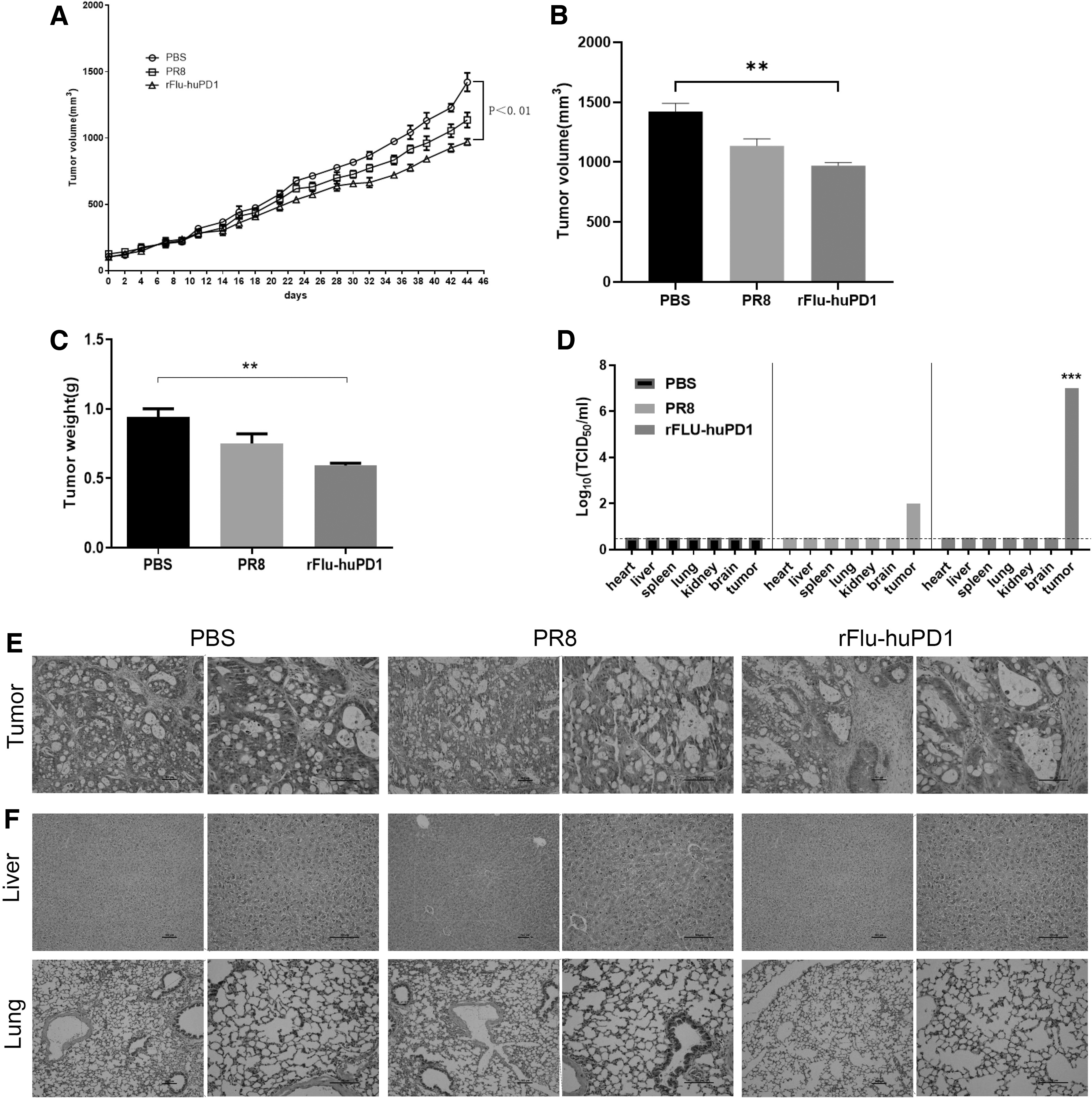
Recombinant OV rFlu-huPD1 in the treatment of liver cancer PDX mice.
In addition, compared with the PR8/PBS group, the tumor weight and tumor growth in the rFlu-huPD1 groups were significantly inhibited (Fig. 5B, C). As shown in Fig. 5D, the titers of TCID50/mL in the tumor tissue were markedly increased in the rFlu-huPD1 groups compared with the control group. These data showed that rFlu-huPD1 treatment reduced the tumor growth of a mouse hepatocarcinoma model in vivo.
Subsequently, hematoxylin-eosin (H&E) staining of mouse tumor tissues was used to confirm that tumor progression was suppressed by rFlu-huPD1. As shown in Fig. 5E, rFlu-huPD1 effectively decreased the cell viability in tumor regions, offering additional support for the antitumor efficacy of OVs in vivo. In addition, the in vivo safety of rFlu-huPD1 was evaluated with H&E staining of the liver and lung. Figure 5F shows that the livers and lungs of mice in the PBS group, PR8 group, and rFlu-huPD1 group had no obvious histological abnormalities.
DISCUSSION
Recently, cancer immunotherapies, including antibodies targeting immune checkpoint molecules, such as PD-1, PD-L1, and CTLA4, have emerged as an unprecedented breakthrough in cancer treatment. 19,20 In 2018, James Allison and Tasuku Honju shared the Nobel Physiology or Medicine Prize for their contribution to PD-1 therapy. 21,22 For this research, we described a novel OV, rFlu-huPD1, in which PR8 IV is designed to express PD-1 antibody in a combination immunotherapy strategy. Herein, we found that rFlu-huPD1-infected eggs produced and secreted hPD-1 antibody. The morphology and size of rFlu-huPD1 were confirmed by scanning electron microscopy.
Cell viability assays showed that rFlu-huPD1 could selectively kill cancer cells without damaging normal cells. Cytotoxic CD8+ T cells and CD4+ T cells in the spleen of tumor-bearing BALB/C mice treated with rFlu-huPD1 were activated in this study, suggesting that rFlu-huPD1 could trigger the mechanisms of immune system activation in vivo. In particular, CD8+ T cell activation, which could directly damage targeted cancer cells, may support rFlu-huPD1 as an effective antitumor agent. In a liver PDX mouse model, intratumoral administration of rFlu-huPD1 significantly reduced tumor growth and increased the viral loads in the tumor sites. Of course, the mechanism by which rFlu-huPD1 selectively kills liver cancer cells warrants further investigation in future studies.
To obtain a more potent antitumor response, several combined immune checkpoint blockades and various OVs strategies have been developed. PD-1, a negative regulator of pre-existing immune responses, is usually expressed on the surface of activated T cells. In this report, we generated for the first time a chimeric oncolytic IV carrying an antibody against human PD-1 using reverse genetics. However, our study has several limitations.
For example, there was no statistically significant difference in tumor volume between the rFlu-huPD1 and PR8 treatment groups. This may be related to the high degree of malignancy in the HCC model. Another limitation is the metabolism and secretion of antibodies against rFlu-huPD1, and possible adverse effects were not investigated in vivo. We will investigate these issues in our future studies.
In conclusion, we identified a chimeric IV expressing an anti-PD-1 antibody for use as an anticancer molecular reagent. Our study suggests that rFlu-huPD1 exerted enhanced oncolytic effects in preclinical HCC models. Our findings demonstrate that a chimeric rFlu-huPD1 virus may be a promising therapeutic agent for clinical applications.
Footnotes
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This study was funded by the Natural Science Foundation of Beijing, China (Grant No. 7202194).