
Abstract
Closing the gap in knowledge on the cause of neurodegenerative disorders is paving the way toward innovative treatment strategies, among which gene therapy has emerged as a top candidate. Both conventional gene therapy and genome editing approaches are being developed, and a great number of human clinical trials are ongoing. Already 2 years ago, the first gene therapy for a neurodegenerative disease, spinal muscular atrophy type 1 (SMA1), obtained market approval. To realize such innovative strategies, gene therapy delivery tools are key assets. Here, we focus on recombinant adeno-associated virus (AAV) vectors and report on strategies to improve first-generation vectors. Current efforts focus on the viral capsid to modify the host–vector interaction aiming at increasing the efficacy of target cell transduction, at simplifying vector administration, and at reducing the risk of vector dose-related side effects.
Introduction
The initial concept of gene therapy centered on curing a genetic disease by providing a functional gene copy or by counteracting the activity of a malfunctioning gene. 1 With the first market approval of a gene therapy in the Western World in 2012 for the treatment of lipoprotein lipase deficiency, this innovative treatment strategy has reached the clinic. 2,3 In the meantime, gene therapies for the treatment of the monogenetic diseases adenosine deaminase severe combined immunodeficiency, Leber congenital amaurosis, spinal muscular atrophy type 1 (SMA1), and beta-thalassemia as well as advanced therapy medicinal products for the treatment of cancer have followed. Given the large number of announced and ongoing human clinical trials and impressive activities in both fundamental and preclinical research, further approvals are expected, which are at least in the range of up to 20 new approvals per year from 2025 onward. 4 The focus in the area of genetic diseases is still set on rare diseases for which frequently no conventional therapy is available. The definition of what classifies as a rare disease is not consistent and thus the number of rare diseases vary; however, to give an estimation, we here refer to Haendel et al., who reported on more than 10,000 rare diseases that impact the human population. 5
Some of these diseases cause neurodegenerative disorders of the peripheral or central nervous system (CNS). Examples for the latter are Hungtinton's disease (HD), Parkinson's disease (PD), amyotrophic lateral sclerosis, lysosomal storage diseases (LSDs), Canavan's disease (CD), or SMA. 6 Current gene therapy approaches, which, except for the SMA1 therapy, are still in human clinical trial phases rely on adding new genetic information instead of exploring genome editing to repair the underlying defect. 6 However, independent of the actual gene therapy strategy, a key challenge in CNS-directed gene therapy is the difficulty of reaching the brain structure as physical barriers such as the blood brain barrier (BBB) have evolved to tightly control and to restrict access, thereby protecting the brain from damage. Depending on the underlying disease, either multiple or distinct cell populations need to be addressed and a wider distribution, rather than a locally restricted transgene expression, is desired. In addition, it might be advantageous to tightly control transgene expression.
Vectors based on the adeno-associated virus (AAV) are widely used in in vivo gene therapy and have become the delivery tool of choice for CNS-directed gene therapies. 6 Despite the unfortunate death of three boys in a human clinical trial on X-linked myotubular myopathy—a life-threating neuromuscular disease—in which extremely high vector doses were applied, 7,8 AAV vectors possess a remarkable safety record and, already with first-generation AAV vectors, a functional cure in patients is achieved. Indeed, three of the approved gene therapies mentioned earlier use AAV vectors as delivery tools and according to Brooks et al., AAV vectors have been explored in 250–300 human clinical trials so far. 9 In addition to AAV serotype 2 (AAV2) vectors—the prototype—vectors based on serotypes 1, 5, 8, 9 and rh10 are in use in CNS-directed gene therapies. 10 –13 These serotypes differ not only in epitopes recognized by the immune system, but also in their interaction with the host. Of interest in this regard is the ability of AAV9 and AAV.rh10 to cross the BBB, a finding that can be considered a game changer since it allowed the switch from intraparenchymal or cerebrospinal fluid (CSF)-based application routes to intravenous administration. 10 –13 Nevertheless, AAV vectors based on natural serotypes face limitations as the brain is by nature not a target tissue of AAV. Here, we report on strategies to optimize these first-generation vectors to become tailored vectors for CNS-directed gene therapy.
Brief Introduction to the AAV Vector System
AAV vectors are derived from natural AAVs that belong to the Parvovirus family and the newly formed genus Dependoparvoviridae, all known for their reliance on a co-infecting virus to replicate in the host cell. 14 –17 The first AAV was discovered in 1965 by Robert Atchinson and colleagues as a viral contaminant of simian adenovirus type 15 (SV15) preparations. 18,19 They noted that the particle was infectious but replicated poorly unless in the presence of adenovirus. 18,19 Since then, at least 13 human and non-human primate (NHP) serotypes and more than 100 natural variants have been identified from either tissue isolates or adenoviral preparations. 20,21
The AAVs are composed of an icosahedral capsid (20–25 nm in diameter) made up of 60 individual capsid proteins (Fig. 1A) and a single-stranded (ss) ∼4.7 kb DNA genome with distinct secondary structural hallmarks, namely the 145 bp Inverted Terminal Repeats or ITRs (Fig. 1B). 21,22 The ITRs are self-priming hairpins and are both origins of replication and key elements for packaging of the AAV genome into pre-assembled viral capsids. 21,22 The first vectorized AAV serotype was AAV2. The AAV2 genome contains a REP gene, which encodes for four non-structural multifunctional proteins (Rep78, Rep68, Rep52, and Rep40) that are important in genome replication and packaging. They are expressed through alternative splicing of the transcripts obtained from two different transcriptional start sites, p5 and p19 (Fig. 1B).

AAV2 monomeric capsid protein structure and AAV2 genome.
The AAV2 CAP gene encodes three viral capsid proteins—the 90 kDa viral protein (VP) 1, 72 kDa VP2, and 60 kDa VP3—the assembly-activating protein (AAP), and the recently discovered membrane-associated accessory protein (MAAP) (Fig. 1B). 23,24 Both MAAP and AAP coding sequences overlap with the capsid proteins at a +1 shifted reading frame, and although AAP has been characterized as an AAV protein key for—serotype-specific—AAV capsid assembly, the function of MAAP is not equally clear. 23,25 It is likely transcribed from a noncanonical start codon, CTG, and encodes a 15–16 kDa protein that associates with the host cell membrane during AAV production. 23
The capsid proteins are expressed from a single promoter, p40, and produced at a ratio of ∼1:1:10 (VP1:VP2:VP3) through an alternative splicing mechanism and an additional—unusual—translation start codon. 21 The capsid proteins of all characterized AAV serotypes contain a common “VP3 region,” which forms the actual capsid. 16,22 The additional sequences at the N-termini of VP1 and VP2 are buried within the capsid and only become exposed at lower pH values for endosomal escape and nuclear entry (see section “AAV infection biology”). 16,22
The common VP3 capsid protein structure shows a core eight-stranded antiparallel β-barrel motif (highly conserved among serotypes) running from strands βB to βI (Fig. 1A). There is a highly conserved α-helix between strands βC and βD (Fig. 1A). The surface-exposed loops between the β strands contain nine variable regions (VR), VR I-IX, which differ between AAV serotypes (Fig. 1A). The basic surface formation of the T = 1 icosahedral symmetry of AAV capsids includes depressions at the icosahedral twofold symmetry axes (termed twofold depressions), depressions around the fivefold symmetry axes, and three outward protrusions around the threefold symmetry axes. 26 The VRs mentioned earlier are primarily found at capsid protrusions, which represent sites of interactions between AAVs and the host cell and define receptor binding and immunogenicity. These variations have most likely evolved to escape host immune responses and at least partially explain the differences in antibody and receptor binding among the naturally occurring serotypes. The protrusions exposed at the capsid surface are formed by VR-IV, V, and VIII whereas VR-VI and VR-VII form their base. 27 Both VR-IV, at the highest peak, and VR-VIII, at the second highest peak, have evolved as popular sites for capsid engineering.
Besides differences in the amino acid sequence of the capsid proteins, serotypes differ in genome length, ITR sequence, and Rep proteins. Commonly, serotypes of human origin are more closely related to AAV2 than AAV serotypes from simian origin. AAV5, however, is an exception to this rule. 28 –30 AAV5, originally isolated from a human penile flat condylomatous lesion in 1984, exhibits only 54–56% sequence homology with AAV2, differs in promoter start sites (p7, p19, and p41), Rep proteins (Rep78, Rep52, and an Rep40-like), and genome integration profile, which is 99.7% distinct from that of wild-type (WT) AAV2 and was found to have a 39 bp integration motif that exhibits similar characteristics to AAV5's ITRs. 31,32
The AAV vectors are designed according to the gut-less concept, that is, all viral open reading frames are replaced by the transgene expression cassette or—in case of its use as a homology-directed repair template in genome editing—by the DNA sequence to be delivered (Fig. 2). The ITR sequence, commonly of AAV2, is the sole viral DNA sequence that remains (Fig. 2). AAV vector genomes (vg) can be packaged into various serotype capsids (pseudopackaging technology), which simplifies the switch between serotypes. 33 Vectors are produced either in mammalian cell lines or in insect cells. The most common strategy followed in research is transfecting HEK293/HEK293T cells with a vector plasmid providing the transgene to be packaged flanked by ITRs, an AAV helper plasmid encoding the REP and CAP genes, and an adenoviral helper plasmid delivering essential adenoviral genes for AAV progeny production not present in HEK293/HEK293 T cells. Also, combined AAV and adenoviral helper plasmids are frequently used. Due to the stability of particles, vector preparations can be purified by density gradient centrifugation and/or various affinity chromatography strategies. Stability is also an advantage regarding storage and transportation. (For comprehensive review on AAV production, we refer to Penaud-Budloo et al. 33 ).
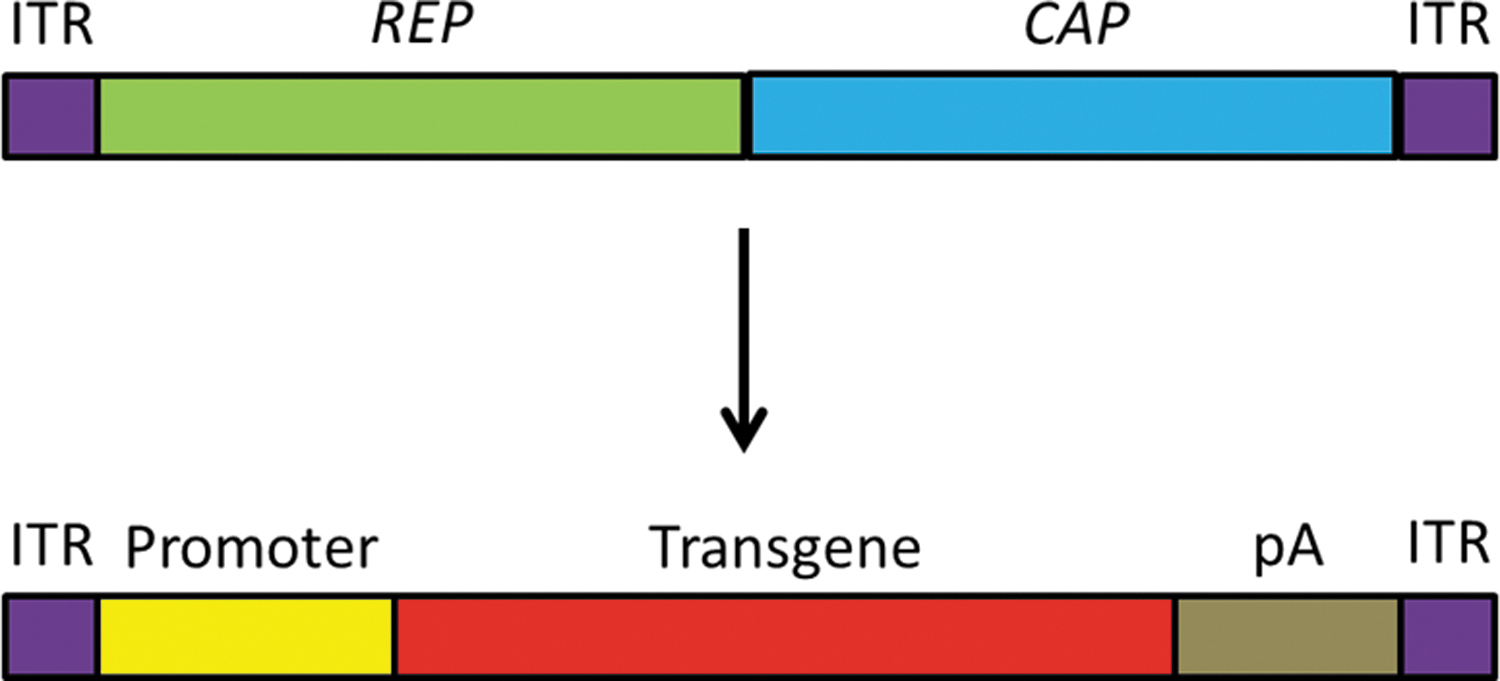
AAV as viral vector. Exchange of the REP and CAP genes of the wildtype AAV genome with a promoter, transgene, and pA sequence for use as AAV vector. Color images are available online.
The AAV vectors as gene therapy vectors have gained momentum over the past 10–20 years with increased understanding of AAV tropism, infection process, and gene expression, and the establishment of capsid-engineering techniques to optimize these activities (described in detail below).
AAV Infection Biology
The main component that defines the host–vector interaction is the AAV capsid. When applied intravenously, serum proteins interact with the capsid and might enhance or block transduction. 34 Human serum albumin, for example, increases transduction of AAV9 vectors in liver (but not into other tissues), whereas fibrinogen, fibronectin, alpha-1 acid glycoprotein, and plasminogen enhance global transduction most likely due to increasing vascular permeability. 34 On the contrary, the binding of neutralizing antibodies (NAbs) reduces vector efficacy. Also, when applied locally, it is expected that binding of host factors to the capsid will impact transduction efficacy and likely even immune responses.
Regarding cell transduction, AAV serotypes harness different types of glycans as primary or attachment receptors on the cell surface. 26 They include heparan sulfate proteoglycans (HSPGs) utilized by AAV2, 3, 6, and 13, O-linked 2,3-sialic acid for AAV4, N-linked sialic acid for AAV1, and 5 and 6 and N-linked galactose for AAV9. 35 –38 Binding to glycans is proposed to promote binding to the actual internalization receptors such as ανβ5 and α5β1 integrins in the case of AAV2. 39,40 This interaction, which also prepares the cell for vector entry, is stabilized by co-receptor binding involving, for example, fibroblast growth factor receptor 1 (FGFR-1), hepatocyte growth factor receptor (HGFR), or laminin receptor. 41 –43 Vector uptake is mediated by endocytosis through clathrin-coated pits, 44 although other routes have also been reported, including micropinocytosis. 36,45,46 Vector-containing endosomal vesicles are transported toward the nucleus via the cytoskeleton network. 47 Gradual acidification of the endosomal vesicles induces a conformational change of the AAV capsid, resulting in the exposure of the buried N-termini of VP2 and VP1. The latter contains a phospholipase A2 (PLA2) domain and nuclear localization signals within the unique VP1 region and the shared VP1/VP2 sequences. 48,49 The catalytic activity of the PLA2 domain mediates particle escape from the endosomal compartment by lipolytic pore formation. 50 This acidification step also primes the capsid for release of the vector genome. 16 Particles that are released into the cytoplasm are prone to ubiquitination and, thus, proteosomal-mediated degradation is a possible outcome. 51 Although the process of nuclear translocation is still not entirely understood, the transport of particles through the nuclear pore complex (NPC) might occur. 44,52
Besides receptors, distinct host factors are also required for successful transduction. A prominent example is the adeno-associated virus receptor (AAVR), a type I transmembrane protein that is mostly associated with the trans-Golgi network in steady state cells. 53 However, similar to other trans-Golgi network proteins, AAVR recycles from the plasma membrane to the trans-Golgi network via the endosomal pathway. 53 AAVR is an essential factor for AAV1, 2, 3b, 5, 6, 8 and AAV9-mediated cell transduction, and it associates with the AAV capsid via one of its five polycystic kidney disease repeat domains. 54 –56 This interaction appears to be required for proper intracellular trafficking of AAV, and it has been observed that AAVR accompanies the particles on their route toward the trans-Golgi network. Another example of host factors required for AAV transduction is GPR108, a member of the G-coupled receptor protein family present in late endosomes and the trans-Golgi network. 57 For many AAV serotypes, except for AAV5, it acts downstream of AAVR and binds to the N-terminus of VP1, perhaps supporting endosomal escape and nuclear delivery. 57
Before transgene expression, vector genomes need to be released from the capsid followed by conversion of their ssDNA conformation into a transcriptionally functional double-stranded DNA (dsDNA) template. Second-strand synthesis is known to restrict transduction and can be circumvented by using a self-annealing vector genome conformation, called self-complementary (sc). 58,59 sc-AAV genomes fold onto themselves upon uncoating to generate a double-stranded vector genome that increases vector transduction efficiency and transgene expression speed. 60,61
The AAV vectors lack an active genomic integrase activity but can become integrated at a very low frequency in a non-specific way as part of DNA repair events. 62 Commonly, however, AAV vector genomes form episomes that remain extrachromosomal.
Of interest for the capsid-engineering endeavor is the recent report of a capsid-vector genome interaction. An AAV9 capsid, for example, was reported to contribute to cell-selective gene expression in the CNS when combined with distinct promoters. The chicken β-actin (CBA) promoter used in AAV9 drove gene expression exclusively in the neurons of the rat striatum, whereas a truncated CBA hybrid promoter shifted expression to preferentially striatal oligodendrocytes. 63 This difference could not be observed with AAV2, regardless of the constitutive promoter used. 63,64 Interestingly, 6 years earlier, Salganik et al. postulated that AAV capsids may play a role in second-strand synthesis via the pH quartet, a group of four interacting amino acids found at the intersection of the twofold, threefold, and fivefold interfaces, which undergo structural changes when subjected to an acidic environment as found in the endosome. 16
With further regards to this capsid–genome interaction, AAV2 variants with mutations at the twofold interface and carrying enhanced green fluorescent protein (GFP) encoding transgene expression cassettes were observed to enter HeLa cells, travel to the nucleus, release vector genomes, and perform second-strand synthesis but then showed a significantly reduced ability to express the transgene as indicated by the extremely low accumulation of GFP transcripts. 65 Changing glutamic acid (E) 564 to alanine (A) resulted in an almost 50% increased release of vector genomes but this mutant then showed reduced GFP messenger RNA (mRNA) levels of 5-log. Two further mutants, D529A and K692A, presented with ∼260- and 226-fold-lower levels of mRNA per released vector genome, respectively, even with almost identical levels of uncoating compared with the unmodified AAV2 vector. 65
Together, physical barriers, lack of receptor expression, cellular uptake, vesicular trafficking, endosomal escape, nuclear entry, viral uncoating, and conversion of the ssDNA genome into a double-stranded genome and/or transcription of the AAV vector coding sequence all affect tropism and transduction efficiency and might reduce efficacy of the AAV vector system. 16,21,22
Advantages and Challenges of the AAV Vector System
The number of human clinical trials using AAV vectors already indicates that this delivery system possesses advantageous features. In contrast to other virus-based vectors in frequent use in gene therapy, AAV vectors are derived from a virus that is not associated with a disease in humans. 66 The risk of vector mobilization is low, as vector production depends on the presence of factors provided by non-related viruses and thus vector mobilization in patients requires the simultaneous presence of an AAV, adenovirus or herpes virus, and AAV vectors in the same cell. The AAV vectors are very stable and sustain shifts in pH and/or temperature. Innate immune responses elicited by the vectors are dose-dependent, transient, and commonly mild, although sufficient to prime de novo vector-specific adaptive immune responses. 67 By nature, a portfolio of serotypes and variants are available, differing in their host–vector interaction. Use of this toolbox in addition to the constantly growing number of capsid-engineered vectors is customer-friendly, as the mentioned pseudopackaging technology is applicable to all these types of AAV. 33 Natural AAV, and thereof derived vectors, use distinct but broadly expressed attachment and internalization receptors. 36,37 Consequently, a broad set of different cell types can already naturally be transduced.
However, this so-called broad tropism comes at the expense of gene delivery into off-target cell types, that is, gene delivery into cell types not defined as targets for a given application. In addition, interactions with factors in the blood or prolonged binding to cell surface glycans might induce unspecific uptake or engulfment. Also, in the case of local instead of intravenous administration, binding to cell surface glycans interferes with the spread of vector particles, which restricts the area that is transduced. To overcome these challenges as well as to mediate transduction of cells that are refractory to natural serotypes, the vector–receptor interaction is modified by capsid engineering using rational design-based as well as library-based approaches, which will be presented here in the context of tailoring AAV vectors for CNS-directed gene therapy (for a comprehensive reading of capsid engineering we refer to Vandenberghe et al., 68 Wang et al., 66 Büning and Srivastava, 69 and Li and Samulski 70 ).
Preexisting immunity represents a further challenge. Many AAV serotypes are endemic to humans and, consequently, the seroprevalence is high with approximately 30–60% of the human population showing NAbs against AAV, although this does vary by region and is AAV serotype-specific. 71 Since NAbs reduce the effectiveness of gene therapy, patients are tested for the presence of preexisting antibodies and then possibly excluded from clinical trials. Consequently, several strategies are in development to overcome this challenge. Lee et al. reported, for example, on a 2.3-fold enhanced evasion from NAbs without significantly affecting transduction efficiency by shielding parts of the capsid via conjugated biocompatible polyethylene glycol (PEG) molecules, a technique termed PEGylation. 72 –75 Another NAb evasion technique is the use of AAV serotypes with lower preference in the human population such as AAV5 or AAV8. 72 NAb against AAV8 serotype, isolated from rhesus monkeys, has immunity in only 6% of humans, whereas for AAV5 an NAb prevalence of only 3.2% is reported. 76,77 The low prevalence fostered their use in hemophilia B clinical trials. 76,77
Also, capsid engineering strategies hold promise as major epitopes recognized by preexisting antibodies are changed. 78 These strategies include site-directed mutagenesis of the known NAb epitopes on the AAV capsid and the use of AAV library-based techniques such as error-prone polymerase chain reaction (PCR) and capsid shuffling. AAV2 NAb epitopes were originally mapped by using gene fragment phage display libraries and peptide competition experiments, which localized many of the epitopes, including those of the monoclonal antibodies (mAbs) A20, A1, A69, and B1, which are frequently applied tools in AAV research. 79 A20 epitopes were mapped to three different sites on the linear VP sequence, including sites on VR I, II, and V; A1 epitopes were mapped to the C-terminus of VP1; A69 epitopes were mapped to a sequence along VP2; and B1 epitopes were mapped to the C-terminus of VP3. 79
Later work showed that peptide insertion in surface-exposed capsid positions reduces the affinity of anti-AAV antibodies. Specifically, insertion of the L14 peptide (QAGTFALRGDNPQG) inserted at positions 534 and 573, respectively, of the AAV2 capsid proteins reduced affinity of antibodies to these AAV2 mutants by up to 70%. 80 In addition, capsids containing the L14 peptide at amino acid position 587 were further tested and seen to transduce HeLa cells to a significantly greater degree than the unmodified ( = WT) AAV2 after incubation with NAbs. 80
A decade later, cryo-electron microscopy imaging identified a region in VR VIII of AAV8 to be a major epitope for the neutralizing mAb ADK8. 49,81 Gurda et al. then examined the NAb epitopes of AAV serotypes 1, 2, and 5 in parallel and determined that they were also located mostly at threefold protrusions and overlapped with key receptor binding sites. These studies help to provide useful data for rational design-based capsid engineering strategies to generate AAV vectors for gene therapy with immune escape phenotypes.
Of the library-based approaches, capsid shuffling showed promise as it resulted in variants with improved transduction efficiency and an immune escape phenotype. This system involves the random shuffling of the CAP sequences from different serotypes to generate chimeric AAV capsids (Fig. 3). 66 This strategy results in the disruption and elimination of the NAb epitopes due to the recombination of capsid sequences and structures that originally defined the epitope. 66,82 For example, Pei et al. isolated the capsid shuffled mutant, AAV LP2–10, with enhanced NAb evasion characteristics after screens in mice with circulating human intravenous immunoglobulin (IVIG). This vector required 300 times more IVIG than its parental serotypes (AAV2, AAV6, AAV8, and AAV9) to be neutralized. 83 Another chimeric AAV, cA4, holding high capsid sequence homology with AAV2, was found to only be neutralized at a 10-fold higher concentration of IVIG compared with AAV2 and the chimeric AAV cB4 was even 400 times more resistant to neutralization by IVIG than AAV2. 84
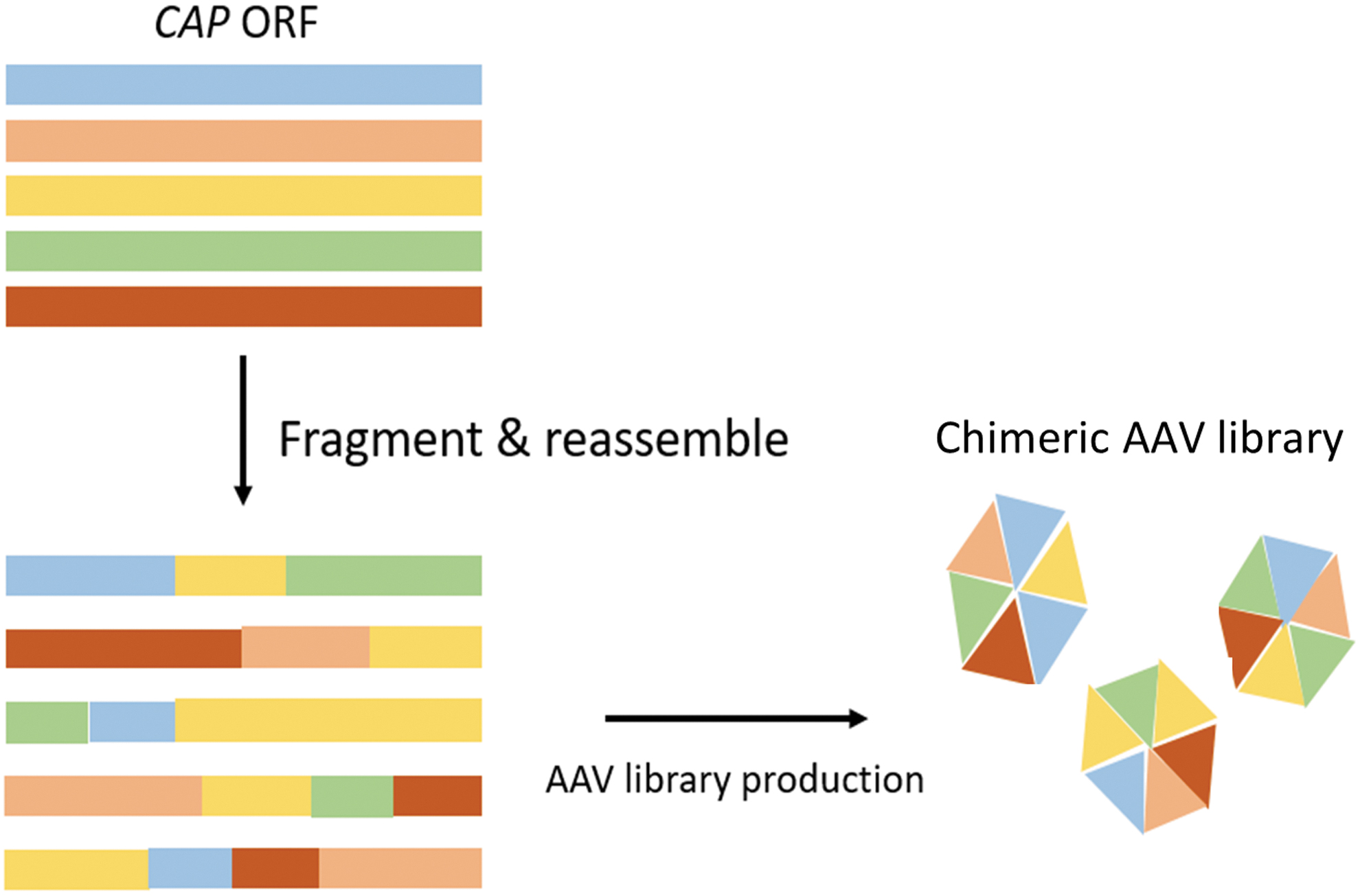
Schematic representation of AAV capsid shuffled library production. This is a directed evolution technique that involves recombining the CAP sequences of different AAV serotypes to generate a mosaic capsid. Color images are available online.
Recent work by Leborgne and colleagues revealed the potency of a totally different strategy. They confirmed that the presence of a circulating streptococcal cysteine endopeptidase called Imlifidase (or IdeS) enhances AAV cell transduction in both mice and NHPs even with preexisting antibodies due to its ability to cleave immunoglobulin G (IgG) molecules in vivo. 85
The removal of antibodies by plasmapheresis is another option to reduce an AAV-triggered immune response. This technique is well established and widely accessible. 86 A study in 2011 by Monteilhet et al. tested this procedure to reduce NAb titers in AAV1, 2, 6, and 8 in 10 seropositive patients by performing 5 plasmaphereses with 1–5-day intervals between each. NAb titers before plasmapheresis were 1:20–1:800 (AAV1), 1:5–1:12,800 (AAV2), 1:5–1:200 (AAV6), and 1:5–1:400 (AAV8). 87 After 5 plasmaphereses, NAb titers became almost undetectable for some patients and for those with initial high NAb titers (>1:800) they could be significantly reduced to 1:20–1:200. 87 However, even low NAb titers of 1:5–1:10 are still able to completely inhibit AAV transduction efficiency. 88,89 More recent work confirmed plasmapheresis to be able to reduce anti-AAV antibody plasma concentrations through an immune absorption column alongside the use of empty AAV particles to deplete even higher concentrations of anti-AAV antibodies from the plasma. 86 Further analysis in a C57BL/6 mouse model, which had been treated with an AAV8 vector encoding coagulation factor IX (AAV8-hF.IX), showed a significant decrease from a 1:1,000 NAb titer before plasmapheresis to an NAb titer of <1:1 after plasmapheresis by using this technique. 86 In general, a more targeted approach as just described will likely be preferred in the future to avoid coincidental depletion of non-AAV specific immunoglobulins.
A further drawback of AAV is the limited packaging capacity. Comprehensive analysis on the quality of vector preparations revealed that vector genomes, including the ITR sequences, should not exceed 5 kb. 33 Thus, when designing AAV vectors with a natural genome conformation (ssAAV vectors), transgene expression cassettes ought not exceed the size of 4.5 kb, whereas half of this size is recommended for scAAV vectors. Many genes or combinations of tissue-specific promoters and transgenes exceed this size, forcing the development of artificial promoters and shorter versions of the proteins. An alternative strategy is co-delivery of two or more AAV vectors each carrying parts of the transgene or of the genome editing machinery. Recombination of vector genomes occurs by nature, resulting in “repair” of oversized vector genomes. This is possible, as sense and anti-sense versions of the genomes are packaged with equal efficiency and the actual length of the oversized genome that is packaged within a single preparation is varying.
However, to improve efficacy, rational design-based approaches have been developed (reviewed in Tornabene et al. 90 ). The easiest strategy relies on the use of two or more AAV vectors carrying parts of the transgene expression cassette flanked by ITRs that recombine in the transduced cell to form the full-length coding sequence. 91 This system of overlapping vectors has shown some success in animals in the delivery of large genes such as dysferlin in muscle cells of dysferlin-deficient mice. 91,92
In addition, trans-splicing or hybrid strategies have been developed. 90,93 –95 In trans-splicing, two AAV vectors are supplied, one with a splice site donor sequence and one with a splice site acceptor sequence; the splice sites recombine and are spliced out, leaving behind the full-length transgene. 91 The hybrid system combines both identical overlapping regions of the transgene for recombination and a trans-splicing system. 91
More recently, the split intein system was introduced. The split intein system uses genetic elements—called inteins—originally identified in unicellular organisms, which can posttranslationally self-excise themselves out of the surrounding polypeptide. 90 The C- and N-termini of the inteins are inserted at the C- and N-termini of two separately expressed transgene halves delivered through two different AAV vectors. On translation, the inteins associate and cause their own trans-splicing, which results in the formation of the full-length transgenic polypeptide. 90
A further issue, broadly discussed among AAV experts and beyond, was a report by Nault et al. on fragments of WT AAV2 detected in hepatocellular carcinoma (HCC) samples. 96 –98 HCC had also been reported in two other studies after intravenous AAV administration in neonatal mice. 96,99 However, further larger studies using 695 mice and 132 mice, respectively, did not observe this phenomenon. 100,101 Nevertheless, the study in 2015 by Chandler et al. revealed that HCC development occurred in mice where the AAV genome had integrated at the RNA imprinted and accumulated in nucleus (Rian) locus on chromosome 12. This integration appeared to change the expression of neighboring regulatory RNAs such as microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), and long intergenic noncoding RNAs (lincRNAs). 96 However, it is important to note that this locus is not actually present in larger mammals such as NHPs and humans. 102 Further, the mice used in these studies were administered with very high vector doses (1 × 1014 genome copies [GC] per kg) and mice were in the neonatal period where considerable development occurs. 96,102 Interestingly, the study also observed that HCC development only occurred when using CBA or thyroxine-binding globulin (TBG) promoters and not when a human α-1 antitrypsin (hAAT) promoter was used, although in all cases the integrations were still detected. 96 This observation suggests that the enhancer-promoter choice may influence the likelihood of tumorigenesis. Although this specific integration pattern is unlikely to occur in humans, these studies can provide us with information on promoter choices and possible sites in the human genome that might be considered as hot spots and should be carefully guarded. 103
A major challenge for current in vivo approaches using intravenous administrations are the high vector doses (up to 1.5 × 1013–2 × 1014 vg/kg body weight [kgbw]) required to achieve therapeutic efficacy. 104,105 High vector doses might result in complement activation, as discussed, as a cause for adverse events (transient renal impairment) reported from two recent Duchene muscular dystrophy clinical studies. 67 Earlier studies on hemophilia B revealed that vector dose also correlates positively with cytotoxic T cell responses directed against vector-transduced hepatocytes and that it is likely caused by a memory response. 88,96,105 –111 These responses induce a transient transaminitis, loss of the vector-transduced cells, and, thus, reduction of therapeutic efficacy. 106 Similarly, high vector doses showed adverse events in the context of CNS-related diseases such as increased transaminase levels in 4 out of the 15 patients treated with either a low dose (6.7 × 1013 vg/kgbw) or a high dose (2.0 × 1014 vg/kgbw) of the AAV9-SMN vectors. 104 Even more recently, as already mentioned earlier, a phase II AAV gene therapy clinical trial for X-linked myotubular myopathy had to be suspended after three deaths due to progressive liver toxicity in response to the high AAV vector doses. 7,112 It is assumed that preexisting antibodies as well as preexisting hepatobiliary disease might have played a role. 67
Strategies to lower the vector dose include usage of scAAV, which allows higher transgene expression levels and earlier onset of expression. 105 However, scAAV have significant packaging size constraints, lower production yields, and increased innate immune activation potential, which are considerable drawbacks. 70,113 As immune activation has been correlated with toll-like receptor (TLR) activation, particularly TLR9 recognizing unmethylated CpG motifs in the transgene expression cassette, 113 direct incorporation of short DNA oligonucleotides into the vector genome that antagonizes TLR9 activation has been assayed. This study, indeed, showed substantially reduced innate immune and T cell responses while improving transgene expression in mice and pigs. 114 Also, the depletion of residues prone to target the AAV capsid for proteasomal degradation (and subsequent presentation by the major histocompatibility complexes) resulted in improved transgene expression and reduced immune responses. 115,116 In addition, or alternatively, restricting vector tropism to the target cell in conjunction with improved intracellular processing will allow the reduction of vector doses and an increase in vector safety. Although strategies for this are developed, transient immunosuppression can be applied. However, optimal timing of immunosuppression is the key as regulatory T cells need to be activated to induce peripheral tolerance against the transgene product. 117 –119
Altering AAV Tropism, Genetic Versus Non-Genetic Techniques
With our current knowledge on what defines AAV tropism and which structures and amino acid residues of the capsid are involved, strategies applied to modify vector tropism (as well as immunogenicity) are becoming more and more sophisticated. 70 Both non-genetic and genetic strategies are followed.
Genetic strategies encompass those that change the amino acid composition of the capsid. Examples are addition of unnatural (or designer) amino acids (UAAs) to the capsid protein sequence (Fig. 4A) or antibody binding domains to function as acceptor sites (Fig. 4B), insertion of design ankyrin repeat proteins (DARPins) or receptor-targeting ligands to the N-terminus of capsid proteins, or insertion of peptide ligands or nanobodies at the tip of capsid protrusions. 70
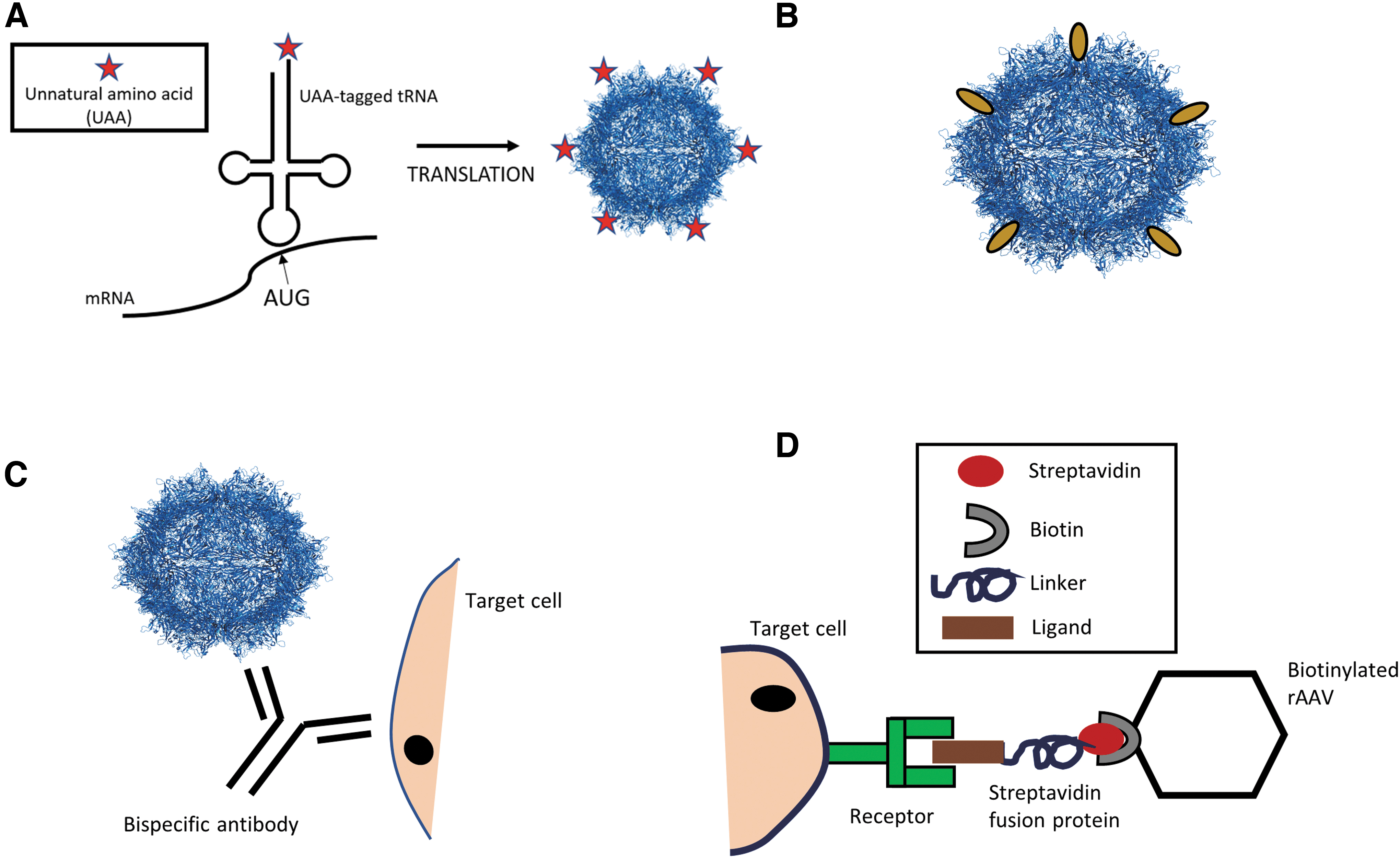
Genetic and non-genetic techniques for altering AAV tropism.
Specifically, UAA incorporation involves the use of an engineered specificity-altered transfer RNA (tRNA) synthetase/tRNA pair within the host cells that incorporate the UAA at a site called the blank codon (commonly TAG) within the target mRNA (Fig. 4A). 120 There is the potential for an almost infinite number of UAAs—defined as chemically synthesized variations of natural amino acids—and many of these allow bio-orthogonal chemical conjugation between target biological molecules. 120 A good example of the use of this technique is the study by Kelemen et al., where azide-UAAs were incorporated at specific sites on the AAV2 capsid to redirect the tropism of AAV2 vectors to αvβ3 integrins, which are found at high levels on tumor vasculature. The azide-UAA-altered AAV2 capsid were retargeted through chemical attachment of a cyclic peptide containing an αvβ3 integrin-binding motif, and it was, indeed, observed that the engineered vectors exhibited dramatically enhanced transduction of the target cell line. 121 Advantages of this technique include an almost unlimited selection of chemical molecules that can be attached to the capsid surface not limited by the genetic code, and the ability to use a variety of artificial linkers to enhance presentation of the binding motif of interest on the AAV capsid.
The N′-terminus of VP2 proteins allows for fusion of not only peptides but also proteins (Fig. 4B). Although the initial attempt of fusing an antibody fragment into the AAV capsid resulted in vector preparations with poor titers, 122 later approaches were successful in labeling the capsid for infection or biodistribution studies of the WT capsid or in re-directing vector tropism. 123 –126
Indeed, the first example of true AAV vector retargeting was obtained by using the N′-terminus of VP2. Specifically, AAV2 capsids blinded for binding to HSPG by substituting in each of the 60 capsid proteins' arginine (R) 585 and R588 to alanine (A) were equipped with DARPins recognizing human Her2/neu fused to the N′-terminus of VP2. 127 DARPins are engineered high-affinity binding ligands derived from ankyrin repeat protein motifs found prevalently at protein–protein interfaces in nature. 128 The available DARPin libraries were developed by the Plüchthun lab in 2013 and can be screened by using ribosome display to identify target-specific DARPins. 129 The initial study by Münch et al. (2013), as well as the follow-up study, reported that fusion of DARPins to the VP2 protein of AAV2 did not impair capsid assembly and were well tolerated. 126,127 However, to achieve off-target-free on-target in vivo transduction, vector preparations had to be depleted of AAV particles that did not incorporate the DARPin-VP2 fusion protein. 127 Further, exchange of Her2/neu directed DARPins for DARPins with other specificities can switch AAV tropism, such as in the studies by Münch et al. and Hartmann et al., who successfully retargeted AAV to human CD4, epithelial cell adhesion molecules (EpCAM), glutamate receptor subunit GluA4 expressing cells, endothelial surface marker CD105 expressing cells, and the natural killer cell-specific marker NKp46, respectively. 127,130
When using the N-terminus of VP2, up to five targeting ligands can be incorporated. To increase the number of targeting ligands displayed on the capsid and to avoid the need of separating VP2-fusion containing from targeting ligand-deficient particles, receptor-binding ligands are inserted at capsid protrusions. The most common position for peptide insertion into AAV capsids of different serotypes is the tip of the second highest protrusion, that is, I-587 or I-588 in the case of AAV269 (I = insertion site). The insertion of peptides in I-587 of AAV2 also simultaneously impacts AAV2's natural binding to HSPG, as the R585 and R588 become separated (Fig. 5). 69,131 In addition, the tip of the highest peak accepts peptide insertions that mediate target receptor binding, although an extra modification—so as to destroy the primary receptor binding motif—is required to re-direct vector tropism. 132 The highest peak also accepts nanobodies, which are the single immunoglobulin variable domains from heavy-chain antibodies naturally found in camelids. 133 The number of nanobodies, however, was restricted by modifying only the VP1 capsid subunits. Eichhoff et al. showed that insertion of either CD38-targeting, ARTC2.2-targeting, or P2X7-targeting nanobody sequences resulted in highly specific transduction of HEK293 cells engineered to express the respective membrane protein (CD38, ARTC2.2, or P2X7) and that the same strategy can be used to re-target other AAV serotypes.

Highest and second highest peaks of the AAV2 capsid proteins.
Many of the initially tested peptide ligands for AAV vector retargeting were derived from phage display library screens. Phage display libraries were first described by George P. Smith in 1985, 134 who discovered that a foreign peptide sequence can be inserted into the pIII coat protein of the filamentous bacteriophage M13, generating so-called “fusion phages”. 134,135 The peptide:pIII coat protein fusion is presented on the surface of the bacteriophage and does not affect pIII's function or general structure but can be used to identify peptides that bind target antigens, as shown in Fig. 6. 135,136
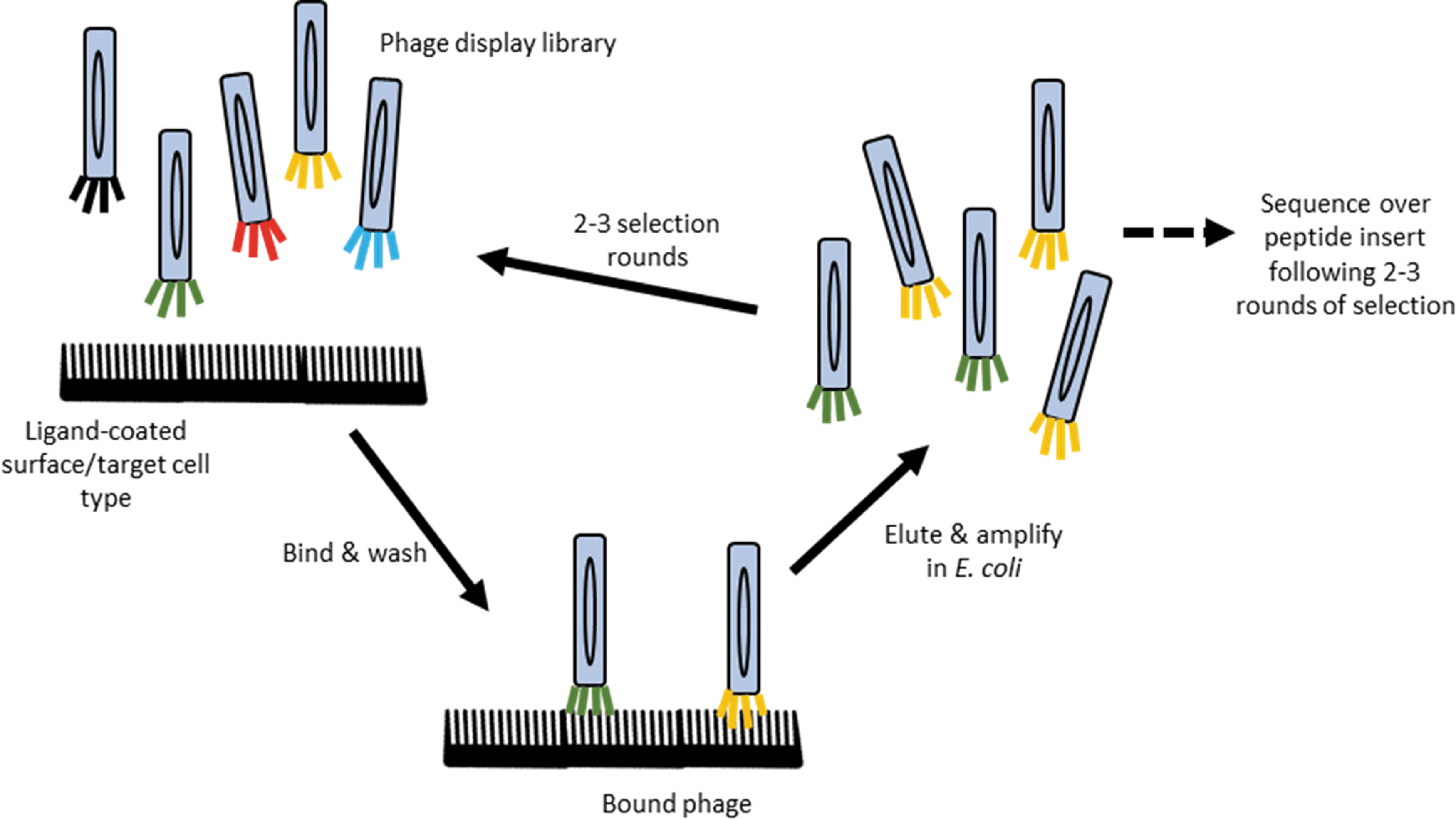
Phage display libraries. Panning and in vitro/in vivo selection process of peptide library displayed on pIII coat protein of M13 bacteriophages (termed phage and depicted as long colored ovals) to identify peptides with high affinity for target antigen. Color images are available online.
Phage display library screens have been used to a greater extent for the identification and generation of mAbs against specific antigens. 137 Among them are mAbs for cancer therapies—with affinities for antigens such as PD-L1 to increase activity of T cells, 137,138 CD22 for enhanced cancer cell apoptosis, 137,139 and the epidermal growth factor receptor (EGFR) for inhibition of cancer cell growth. 137,140 Further, mAbs have also been developed with this technique for the treatment of optical, infectious, and immunological disorders. 137
Similarly, peptide sequences displayed on the phage pIII coat protein and screened in an in vitro or in vivo system for high target cell affinity can be used to identify peptides that might re-direct AAV vectors. 69,135 Specifically, AAV vector capsids engineered to present phage display library-derived peptides have been successfully used for the targeting of vascular endothelium cells, 141 –143 atherosclerosis plaques, 144 smooth muscle cells to impair neointimal formation, 145 and skeletal muscle aiming at treating muscular dystrophies and heart failure. 146
More specifically regarding neurodegenerative disease, a human single-chain variable fragment (scFv) antibody phage display library was used to isolate a fragment specific for amyloid-beta (Aβ), a major driving force behind the progression of Alzheimer's disease (AD). 147 This fragment, when delivered as a transgene via intrahippocampal infusions within an AAV1 vector to a triple transgenic mouse model of AD, resulted in significant reductions in Aβ depositions and hyperphosphorylated tau in the mouse brain and the mice showed improved cognitive ability by 12 months of age. 147 Almost a decade later, Elmer et al. delivered a silent scFv-IgG (a single-chain antibody fused to part of an Fc domain to allow enhanced antigen clearance) via AAV1 into the brains of a different Aβ mouse model (APP mouse); scFv-IgG transgene expression resulted in improved clearance of Aβ plaques in the hippocampus and cortex of the treated mice. 148
However, although phage display libraries exhibit some beneficial characteristics for the isolation of peptides with preferences for binding distinct cell surface or intracellular molecules, there are also some significant drawbacks. Fusion phages are generated in Escherichia coli, which do not perform the same post-translational modifications as mammalian cells, and this could alter peptide affinities, biodistribution, and pharmacokinetics of the peptide when transferred to a mammalian system. 137 Further, phage display libraries isolate peptides based only on receptor or antigen-binding affinities, which does not necessarily confer good internalization and processing features when used as a targeting ligand for AAV vectors. Further, the peptide sequence might interfere with capsid assembly, or its affinity might be lost when presented as part of a loop structure at capsid protrusions.
In response to these challenges, two laboratories developed in parallel, but independently, the AAV peptide display technology with first reports published in 2003 for AAV2. 149,150 In the meantime, AAV peptide display libraries based on various serotypes have been developed and screens have moved from ex vivo to in vivo. All libraries have in common that they consist of millions of randomly created AAV capsid variants that differ in the peptide they display at either the highest or second highest capsid peaks (Fig. 7). They are screened for variants with tropism for the desired target cells—if possible—in conditions that mimic the latter application as closely as possible. Such screens not only ensure that the peptide insert is recognizing the target antigen in the context of the AAV capsid, but they also allow identification of variants that overcome post-entry, in addition to pre-entry, barriers. Further, screens can be performed in the presence of NAbs for variants with immune escape features.

Peptide display AAV library generation. The red bands represent the inserted oligonucleotide/peptide sequences within the CAP ORF, and the different coloured AAV variants each represent a variant with a different and unique peptide insert. ITR, inverted terminal repeat; ORF, open reading frame. Color images are available online.
The selection process itself involves administering the library to the target cell line in vitro or to an animal model in vivo. The AAV sequences are then amplified from the target cell population; on the one hand, they are re-cloned to produce a sub-library and on the other hand, they are analyzed by next-generation sequencing. The sub-library is then subjected to two to three further rounds of selection. From the AAV variants that accumulate, candidates are chosen based on criteria such as overall charge of peptides, specificity, and fold of accumulation. Candidates are produced as AAV vectors followed by studies to confirm target cell transduction efficiency and to assay transgene expression levels.
Nowadays, 7-mer insertions are the favored insertion length as this sequence is long enough to generate high diversity in a peptide display library but not too long that it would cause severe structural or stability changes in the capsid. 150 Anything shorter than this may not provide enough diversity to the library, and anything longer than this will not significantly increase the library diversity. 151 Hence, 7-mer insertions have become the most optimal insertion length. Successful AAV peptide display library screens include, for example, the identification of an AAV9 peptide display variant, AAVMYO, showing enhanced transduction of musculature, including skeletal muscle, heart, and diaphragm 152 ; an AAV2-based vector, AAV-VEC, with superior targeting of human macrovascular endothelial cells alongside enhanced transgene expression 153 ; and a family of AAV peptide display variants that was found to exhibit a transduction efficiency of up to 38% in dendritic cells normally non-permissive to AAV vectors. 154
Further recent work resulted in identification of AAV peptide display variants—designated AAV2.GL and AAV2.NN—which have a strong tropism for photoreceptors on ex vivo explants of the human retina and allow widespread and efficient retinal transduction of photoreceptor cells after intravitreal administration in mice, dogs, and NHPs. 155 Analysis of the AAV2.GL variant also showed success of this vector in restoring Cnga3 protein expression in the cones of Cnga3−/− achromatopsia mice after intravitreal administration. 155 These are only very few examples that are, nevertheless, sufficient to reveal the great potential and width of application of the AAV peptide display technology for improving the vector system for clinical application (for more examples, we refer to Büning and Srivastava, 69 and Büning et al., 22 ).
The AAV peptide display libraries were the first of the directed evolution techniques, which now also includes error-prone PCR and capsid-shuffled AAV libraries. 22,66,156 Of these, particularly peptide display libraries and capsid-shuffled libraries have added a huge number of AAV capsid variants with improved transduction efficiencies to the AAV toolbox. 22,84
One of the first studies testing the applicability of capsid shuffled libraries was reported in 2008 by Grimm et al. They recombined the CAP genes from eight AAV serotypes—AAV2, AAV4, AAV5, AAV8, AAV9, avian AAV, bovine AAV, and goat AAV—and screened their library on human hepatocytes after incubation with IVIG or mouse sera. They thereby identified the variant AAV-DJ, which contained a mixture of the serotype 2, 8, and 9 CAP sequences and that exceeded AAV2 in its liver targeting ability in vivo in IVIG-immunized mice. 157
Another study in the same year was performed in R.J. Samulski's lab where they isolated a chimeric AAV—chimera-1829 (a combination of AAV1, AAV2, AAV8, and AAV9)—with high transduction efficiency of melanoma cells commonly only weakly permissive to AAV. 115 In 2014, Lisowski et al. then used the capsid shuffling technique on AAV1, 2, 3, 4, 5, 6, 8, 9, avian AAV, and bovine AAV CAP genes. After four rigorous selection rounds in humanized murine chimeric liver models followed by in vitro analyses of prime candidates on primary human hepatocytes, they identified the variant AAV-LK03, which could transduce human hepatocytes 67-fold better than AAV8, 6.5 times more than AAV3B (which shares the majority of sequence homology with AAV-LK03), and 3-fold more than the AAV-DJ variant. 158 AAV-LK03 shares 98.9% of its amino acid sequence with AAV3B, in spite of also having sequence homology with AAV2, AAV4, AAV8, and AAV9 mainly at the N-terminus of VP1. 158,159 Importantly, it appeared to show species-specific targeting of human liver cells 158 and this success resulted in its use in human clinical trials to improve efficacy of hemophilia A gene therapy (NCT03003533).
In contrast to genetic strategies, non-genetic strategies include those approaches that do not change the viral amino acid sequence, that is, the WT capsid is used as backbone. Examples of non-genetic strategies are the use of bispecific antibodies (Fig. 4C), biotin–avidin interactions to cross-link specific fusion proteins with biotinylated AAV vectors (Fig. 4D), or other variations of chemical coupling approaches using drugs and conjugation moieties. 72,121,160
Indeed, the first non-genetic strategy to alter AAV tropism was the use of bispecific antibodies that allow retargeting of AAV vectors without any knowledge of the capsid structure or function. One part of the bispecific antibody has a high affinity for the AAV2 capsid, whereas the second part recognizes the target cell surface receptor (Fig. 4C), and so the antibody acts as an adapter to direct AAV to cells expressing the target receptor. 161
The high affinity binding of biotin and avidin has also been used to redirect AAV tropism through attachment of a target ligand expressed as a fusion protein with streptavidin to biotinylated AAV capsids (Fig. 4D). 162 Based on this strategy, a chemical coupling technique was established by Mével et al., where they linked N-acetylgalactosamine (GalNAc) molecules containing a conjugation moiety to the surface of AAV2 vectors, which further enhanced the transduction of hepatocytes via interaction with the highly expressed and hepatocyte-specific asialoglycoprotein receptor (ASGP-R). Intriguingly, these chemically modified capsids also resulted in significantly reduced NAb formation in vivo in mice, which was also confirmed to occur when AAV8 and AAV3b were used as backbone. 163 Thus, it appears that covering the capsid surface with targeting moieties not only influences AAV tropism but may also hinders a host immune response to the AAV capsid.
Strategies for CNS-Directed Gene Therapy
The CNS has been a challenging target for intravenously administered AAV vectors, because most serotypes are unable to cross the BBB. 11,104,164,165 Thus, AAV gene delivery to the brain has focused on intraparenchymal injections directly into brain regions that are affected by the disease. 166 –170 Although this type of local injection exhibits good transduction efficiency, it results in transduction of cells only around the local site of injection, especially in higher order mammals (yet many neurological diseases are spread throughout different sites in the CNS 6,11,166 ). Other administration strategies include CSF-based gene delivery, intracerebroventricular (ICV) or cisternal or lumbar intrathecal (IT) administration. However, these approaches require high amounts of vectors, particularly when compared with intraparenchymal injections. There is also a risk of leaking into the blood circulation, causing off-target transduction and undesirable immune responses (reviewed in Hocquemiller et al. 6 ). Further, administering intracranial injections in humans is more invasive and thus more stressful compared with an intravenous administration.
Common AAV serotypes for gene delivery into the CNS via intraparenchymal administration include AAV1, AAV2, AAV4, AAV5, AAV7, AAV8, AAV9, and AAVrh10. 171 Direct injection into the brain preferentially transduces neurons, although serotypes such as AAV5, AAV9, and AAVrh.43 show stronger astrocyte transduction 171 –173 and AAV4 preferentially targets ependymal cells. 174 Minor changes to the capsid were shown to target AAV vectors to more refractory cell types such as microglial cells. By introducing distinct amino acid substitutions into the AAV6 capsid such as tyrosine to phenylalanine (Y731F+Y705F) as well as threonine to valine (T492V), Rosario et al., for example, achieved transductions of microglia in vitro and in vivo. 175 Of importance in this regard is also the study of Gilkes et al. assessing two intracranially administered AAV8 vectors with double-(Y444 + 733F) and triple-mutations (Y444 + 733F+T494V) in neonatal mice for the treatment of Mucopolysaccharidosis type IIIB (MPS IIIB), an autosomal recessive lysosomal disease characterized by severe CNS degradation. 176 The AAV8Y444+733F+T494V was superior to WT AAV8 in vector distribution and transgene expression in the CNS. 176 On the same line, an AAV8 capsid variant with Y447F and Y733F substitutions showed ∼17-fold higher transduction efficiencies of rat striatal neurons in vivo compared with the WT AAV8 vectors and should thus be used to optimize, for example, scAAV8-small interfering RNA (siRNA) vector-mediated pyruvate dehydrogenase (PDH) knockdown approaches when studying PDH deficiency. 177
The AAV vector administration route has, however, been observed to affect AAV biodistribution in the brain. For example, intrastriatal injection of AAV5 vectors in rats resulted in vectors targeting the striatum, cortex, thalamus, and hippocampal areas whereas IT administration showed transduction in the cortical-, striatal-, thalamic-, hippocampal-, cerebellar area, and brainstem including detection of vectors in the spinal cord. 173 The ICV administration was then seen to exhibit a more evenly distributed vector transduction pattern throughout the rat brain and spinal cord. 173 Considering that many neurological diseases affect different cell types and areas of the brain (such as dopaminergic cells in the substantia nigra in PD, cells in the basal ganglia in HD, and cells of the hippocampus in AD), the administration route needs to be carefully considered to increase efficacy of the AAV vector-based gene therapy.
A huge step forward in CNS-related gene therapy was the seminal observation that AAV9 crosses the BBB in neonatal mice as well as adult mice and can robustly target motor neurons in the spinal cord, astrocytes throughout the spinal cord and brain, and also shows widespread and strong transduction of many cell types in all brain regions. 164 Since then, AAV9 vectors have been successfully established as a gene delivery tool to treat SMA1, through intravenous administration of vectors encoding survival motor neuron (SMN) protein as revealed by its market authorization as Zolgensma. 104 AAV9 vectors also show promise in treating rare LSDs, 178 –180 CD, 181 type II GM1 Gangliosidosis (NCT03952637), PD (NCT04127578), and dementia (NCT04408625).
An AAV9 vector encoding the Niemann-Pick disease type C1 (NPC1) gene for example was shown to increase the lifespan, delay the weight loss and decline in motor function when intravenously delivered to Npc−/− mice, which are the murine model of human NPC1 disease. 180,182,183 NPC1 in humans is an LSD causing neurodegeneration (specifically of cerebral Purkinje cells), ataxia, weight loss, and early death. The rescue of the disease phenotype in the Npc−/− mice and maintenance of the Purkinje cells after AAV9-NPC1 administration argues that gene therapy is a likely option to treat NPC1 in humans.
However, AAV9 does not work equally well across species. 6,184,185 On intravenous administration of an scAAV9 vector encoding for GFP, young adult NHPs showed strongest GFP expression predominantly in glial cells in the CNS whereas most transduction in mice occurred in the neurons and astrocytes. 184,185 Moreover, using a comparable dose (9–9.5 × 1012 vg/kg), NHPs showed significantly lower peripheral organ targeting than mice. 184,185 Further IT and intracisternal AAV9-GFP administrations in 5-day-old pigs—in contrast to mice—showed widespread motor neuron and dorsal root ganglion transduction alongside transduction of cerebellar Purkinje cells, nerve fibers and nuclei within the medulla. 185 Of note, the IT and intracisternal injections ensured almost no transduction of peripheral organs, 185 which would overcome some significant barriers for human gene therapy.
A study in 2012 also made an important observation, namely that molecular changes of tissues caused by a disease can impact transduction efficiency of AAV vectors. 186 In a mouse model of the LSD, mucopolysaccharidosis (MPS) VII, levels of sialic acid in the CNS are elevated. 186 This appears to hamper AAV9 vector-mediated transduction in the CNS but an AAV2 variant—AAV-PFG, originally created through insertion of a brain vascular endothelia-targeting peptide from a phage display library 187 —was able to both transduce the brain vasculature and alleviate symptoms in this LSD mouse model. 186
Besides AAV9, also AAVrh.10, AAVrh.39, AAVrh.43, AAV7, and AAV8 were shown to cross the BBB after an intravenous injection in 10-week-old mice and transduce cells to varying degrees throughout the CNS, where this had only earlier been shown in neonates. 11 Top candidates included AAVrh.8 and AAVrh.10, which exhibited the strongest transduction of CNS cells in adult mice and showed reduced peripheral organ targeting. 10,11,165 AAVrh.10 is now being tested in clinical trials for its safety and efficacy in children with Late Infantile Neuronal Ceroid Lipofuscinosis (NCT01414985) and children with Metachromatic Leukodystrophy (NCT01801709). In addition, AAVrh.8 encoding the β-hexosaminidase A enzyme subunits (HEXA and HEXB) is studied for the treatment of children with neurodegenerative GM2 Gangliosidosis (NCT04669535).
The discovery of the BBB-crossing AAV serotypes is considered a game changer, although one must acknowledge that these serotypes transduce cells in peripheral organs to a significantly higher degree when administered intravenously. 188 Vectors must, therefore, be used at very high doses—for example, 2 × 1014 vg/kgbw in the gene therapy of SMA1—to obtain sufficiently high particle numbers in the CNS. 104 The downside of high vector doses is an increased risk of side effects, including host immune responses (as described in detail earlier).
Deciphering the mechanism of the abovementioned AAV serotypes' trafficking across the BBB is an active field of research. An in vitro study by Merkel et al. showed that AAV9 undergoes transcytosis through brain microvascular endothelial cells (BMVECs) of the BBB without compromising the BBB itself, where AAV2 vectors show only endocytosis into the BMVECs and an inability to cross the BBB. 12,189 Albright et al. later identified several key residues for BBB-trafficking in the BBB-crossing serotype AAVrh.10. They randomly exchanged capsid amino acid residues of AAV1—a serotype not able to traverse the BBB—by the corresponding ones from AAVrh.10. When amino acid residues N262, G263, T264, S265, G267, S268, T269, and T273 were substituted, the AAV1 chimera could cross the BBB and transduce neurons in the motor cortex and cortical neurons across the entire cortex. 13 These amino acids are conserved in AAVrh.8 and AAVrh.39. Further work by Mietzsch et al., which analyzed the structural differences between the neurotropic serotypes AAVrh.10 and AAVrh.39, and AAV8, which does not traverse the BBB, showed that only four amino acids—S269 (VR I) and N472, A475 and A468 (VR IV)—appear as key residues for BBB crossing. 190 These residues are also conserved in the AAV9 capsid.
As natural AAV serotypes (except for the BBB crossing serotypes) must either be administered intracranially (which is not so efficient for widespread CNS cell transduction) or administered intravenously at very high doses (which often triggers an immune reaction and off-target transductions), research has focused on strategies to improve transport via the BBB or on engineering the AAV capsid to re-direct tropism toward the CNS.
Strategy 1: BBB shuttle peptides
The BBB is a highly selective barrier of endothelial and glial cells, which restricts the entry of molecules from the circulating plasma into the brain cells. The earlier mentioned BMVECs that make up the BBB have unique properties compared with peripheral endothelial cells (ECs), including highly restrictive tight junctions connecting the ECs, and the significantly lowered rates of transcytosis, which reduces vesicle-mediated transcellular movement of molecules into the CNS. 191 This barrier is vital in protecting the CNS from infection, harmful toxins, and injury; however, it is a major obstacle in medicine.
As an alternative to the use of the BBB crossing serotypes mentioned earlier, or to enhance the BBB crossing of AAV9 and other vectors, vectors can be applied together with BBB shuttle peptides. 188,192 The BBB shuttle peptides are short peptide sequences that can bind to biological molecules, including DNA, proteins, and small drugs. This then allows enhanced transport across cell membranes and even through tissue. The exact mechanism of this transport is not fully understood but the overall basic properties of the shuttle peptides allow for electrostatic interactions with the negatively charged proteoglycans and phospholipids on the host cell membranes that trigger cell entry. 193
The first shuttle peptide was discovered in 1988–1989 by two separate groups who found that the Trans-Activator of Transcription (Tat) protein of human immunodeficiency virus (HIV) could enter cultured cells and trans-activate viral gene expression from a host viral promoter. 194,195 Further mapping of the domains determined an 11 amino acid peptide sequence, which became the first shuttle peptide. 194,196 A further study in 1999 by Schwarze et al. confirmed that fusion of the Tat peptide with a 120 kDa beta-galactosidase protein allowed delivery of the beta-galactosidase into all tissues in mice, including the brain, after an intraperitoneal administration. 197
A more recent study by Meng et al. tested the effect on cell transduction of AAV9 after incubation with three different shuttle peptides—LAH4, LEPTIN30, and APOE. 188 They found that incubation with any of these significantly enhanced transduction of HEK293 cells (indicated by increases in GFP expression to 540%, 230%, and 170% compared with AAV9 not incubated with the respective shuttle peptide), ECs (to 360%, 200%, and 170%, respectively), and human astrocytes (to 750%, 590%, and 410%, respectively) by AAV9 and that the LAH4 peptide consistently had the strongest effect. 188 They further showed that this occurred in a dose- and temperature-dependent manner, with 20–40 μM of the shuttle peptide and 37°C being the optimal parameters. 188
An in vitro BBB model comprising monolayers of BMVECs similarly showed that AAV9-LAH4 exhibited enhanced crossing of the monolayer compared with AAV9 alone. In line with this, in vivo tests in mice showed increases in GFP expression of 250% in the hippocampus, 160% in the cerebellum, and 140% in the cortex after intravenous administration of AAV9-LAH4 vectors containing a GFP transgene. 188 However, the liver also showed a 400% increase in GFP expression with AAV9-LAH4 administration compared with AAV9 vectors alone. Further, immunohistochemistry revealed that use of the shuttle peptide with AAV9 vectors enhanced transduction of neurons, astrocytes, and ependymal cells. 188 Interestingly, use of AAV9-LAH4 also reduced the levels of interleukin-1β (IL-1β) and tumor necrosis factor α (TNF-α) in the brain to 55% and 60%, respectively, compared with solely AAV9. 188
This and other recent studies testing the applicability of BBB shuttle peptides 192,198 show enhanced BBB crossing, CNS cell transduction, and even a lowered immune response when BBB shuttle peptides and AAV vectors are combined. However, as revealed by the earlier mentioned study, off-target transductions were also found to be enhanced.
Strategy 2: AAV peptide display library screens
Since the discovery that AAV9 crosses the BBB, 164 AAV9 peptide display libraries have been screened to further enhance CNS tropism aiming at lowering vector doses, reducing off-target transductions, and attaining higher transgene expression levels in the target cells. One such AAV9 peptide display library was created by Nonnenmacher and Weber. 36 This TRACER (Tropism Redirection of AAV by Cell-type-specific Expression of RNA) library is unique, as it selects for variants expressing the modified CAP gene via a tissue-specific promoter. The team used the common strategy of inserting random 7-mer peptides between residues 588 and 589 of the VR-VIII loop of the capsid protein but replaced the REP gene to make space for the promoter sequence such as human synapsin 1 (SYN) or a gfaABC1D (GFAP) promoter to ensure specific expression and variant isolation from only the target cells. They identified 10 top candidates (namely 9P03, 9P08, 9P09, 9P13, 9P16, 9P31, 9P32, 9P33, 9P36, and 9P39) from the isolated pool of 330 brain-enriched variants after two rounds of in vivo selection in mice. 199 These top candidates exhibited up to 400-fold increased CNS transduction and up to 1,000-fold higher spinal cord transduction in C57BL/6 mice compared with the parental AAV9 after intravenous injection and showed almost 100-fold reduced targeting toward liver compared with AAV9. 199
Another study aimed at identifying an AAV9 variant to efficiently knock down the Huntingtin gene (HTT) in the CNS of a mouse model of HD using a miRNA specific for Htt (miHTT). 165 This time, random peptides were inserted at the N-terminus of the VP2 capsid protein of an AAV9.47 backbone (this variant is a quadruple mutant of AAV9 with the changes S414N, G453D, K557E, and T582I, which has been shown to have an alike CNS tropism to AAV9 with decreased liver tropism 165 ). Surprisingly, the control vector with an insertion of 19 alanines—named AAV-AS—showed a 6- and 15-fold increased transduction of the spinal cord and cerebrum compared with the parental AAV. 165 This enhanced CNS transduction was also confirmed in a cat model and was observed to knock down Htt expression by 33–50% in the HD mouse model. 165 In line with the earlier discussion of how the AAV capsid proteins might play a role in controlling expression from the AAV vector genome, the surprising feature of AAV-AS might become explainable. 65,200
Besides AAV9, other AAV serotypes have also been used as the backbone of peptide display libraries for the identification of BBB-crossing and CNS-transducing AAV variants. Körbelin et al., 201 for example, screened an AAV2 peptide display library in the FVB/N mouse model. They performed four rounds of selection utilizing intravenous injections of the viral library. They thereby identified AAV2-BR1, a variant containing the NRGTEWD peptide at capsid amino acid position I-588, which outperformed the parental serotype, exhibiting 65-fold higher transgene expression levels in the mouse brain measured by luciferase activity in tissue lysates. Interestingly, the AAV2-BR1 variant also exhibited negligible off-target tissue transduction. 201 This peptide display variant showed extremely high levels of targeting to the BMVECs, which may allow CNS entry via transcytosis. 201 The specific targeting of the brain ECs by AAV2-BR1 also holds great potential for treating neurological diseases such as stroke, multiple sclerosis, and epilepsy, which involve a strong vascular component.
Also, the CREATE (Cre Recombinase-based Targeted Evolution) peptide display library platform was applied successfully. 202 The AAV capsid variants in CREATE deliver a Cre-inducible fluorescent protein to detect and quantify AAV peptide display variants that express the transgene in the target cells of respective transgenic animals. 202 The AAV9-based library contained 7-mer random peptide insertions and was administered intravenously into GFAP-Cre mice. 202 In a single round of selections, Deverman et al. identified an AAV-PHP.B capsid containing the peptide insertion TLAVPFK. This variant showed a >40-fold increase in widespread CNS transduction compared with AAV9 when injected intravenously, particularly transducing the majority of astrocytes and neurons across multiple CNS regions while demonstrating similar tropism to peripheral organs as AAV9. In a separate trial in GFAP-Cre mice, the variant AAV-PHP.A (with peptide insert YTLSQGW) was identified after two rounds of selection. Even though this variant showed even higher efficiency and selective CNS astrocyte transduction as well as reduced off-target tropism, for example, for liver in mice compared with PHP.B, only PHP.B could efficiently transduce human glial fibrillary acidic protein (GFAP)-expressing astrocytes and MAP2-expressing cortical neurons when compared with AAV9 or PHP.A. However, further studies have shown that the transduction profile of AAV-PHP.B could not be reproduced in all mouse strains commonly used in research as well as NHPs. 203 –205 The difference in permissiveness is explained by the difference in receptor expression. 206 Specifically, the stem cell antigen-1 (SCA-1), also known as LY6A, was independently identified by both the Deverman and Wilson labs as the receptor used by PHP.B to gain access to the CNS. 203,207,208 Even though this variant is likely not clinically relevant for human gene therapy, this work demonstrates the complexity of CNS targeting by AAV and provides seminal insight on the mechanism of vector-mediated BBB crossing.
A further advancement of Deverman's capsid selection strategy is the multiplexed CREATE (M-CREATE) platform, which accelerates data processing and analysis. 209 In addition, based on CREATE, Hanlon et al. introduced the iTranduce library selection strategy, which does not rely on the use of Cre transgenic mice but instead uses engineered AAVs that encode capsids with peptide insertions along with a Cre expression cassette. 210 When administered intravenously into mice with a Cre-sensitive fluorescent reporter, the identified candidate AAV-F, containing the FVVGQSY peptide insert, showed 65-fold higher astrocyte and 171-fold greater neuronal transduction with significantly higher expression levels in the spinal cord compared with the parental AAV9. 210 The rate of liver transduction for AAV-F remained identical to that of AAV9. 210 Although PHP.B showed a similar transduction profile in C57BL/6 compared with AAV-F, greatly enhanced transduction of the CNS of the popular mouse strain BALB/c was shown for AAV-F.
An alternative strategy, the barcoded rational AAV vector evolution (BRAVE) screening approach, was developed by Davidsson et al. 211 BRAVE allows for large-scale selection of peptide display libraries by only requiring a single round of in vivo screening. 211 By combining the strategies of rational design and directed evolution, each virus particle was equipped with a peptide, derived from proteins known to interact with the human CNS, and a unique molecular barcode sequence of 20 nucleotides within the packaged genome, which later also contained the virus expressing RNA. With the aim of identifying AAV capsid variants capable of retrograde axonal transport in vivo, the Davidsson lab screened their AAV2-based library in the forebrain of adult rats. Five of the initially 25 capsid variants identified as candidates by this screen were characterized in more detail. MNM004 (containing the Herpes Simplex Virus-derived peptide sequence VMSVLLVDTDATQQ in I-587) and MNM008 (containing the Canine Adenovirus-2-derived peptide sequence SFTSPLHKNENTVS), for example, showed impressive retrograde transport features. 211 When MNM004 was compared with a newly identified AAV2 vector displaying the Retro peptide LADQDYTKTA, 212 the new variant showed equal or even better transport efficacy to projection neurons. 212 MNM017 containing the Tau-protein derived peptide sequence SIPGFPAEGSIPLP is of interest, as this variant transduces GFAP-positive glial cells with high efficiency. With ∼10,000 different peptide sequences, the BRAVE library shows a relatively low diversity, which is maybe compensated by rational design-based decision on the candidate proteins from which the peptides are chosen. It should also be mentioned that this approach shows great promise in identifying AAV capsid variants that are able to target more refractory cell types relevant in CNS-related disorders, such as microglial cells.
Overall, peptide display libraries are a versatile and promising technique to tailor AAV vectors for gene therapy, specifically for the targeting of challenging organs such as the brain where little is known about the pre- and post-entry barriers.
Strategy 3: AAV capsid shuffled libraries
Capsid shuffling has shown promise in generating AAV variants with new features. One prominent advantage is that these variants are commonly not recognized by antibodies against natural serotypes due to the new combinations of capsid amino acid sequences. 72,157
To improve quality and production of capsid shuffled libraries, a computationally designed DNA shuffling strategy, called SCHEMA, 213 has been developed. SCHEMA can identify ideal crossover points for the generation of stable mosaic AAV capsid libraries. 214 A SCHEMA-derived AAV capsid shuffled library with the CAP sequences flanked by loxP sites was screened in transgenic mice expressing Cre with a neural stem cell (NSC)-specific promoter to identify variants specific for NSCs. 215 On AAV infection of cells expressing Cre, the floxed CAP becomes inverted, allowing the recovery of AAV variants with inverted CAP gene sequences (through use of a set of primers that amplifies only the inverted CAP orientation) and that indicate successful targeting and release of viral genomes within the nuclei of NSCs. 215 The capsid-shuffled variant, SCH9 (composed of a mixture of capsid sequences from AAV2, 6, 8, and 9), was isolated, which transduced 60% of adult mouse NSCs after ICV administration and reached a 24-fold higher transgene expression level than AAV9. 215 Further, 2- to 10-fold higher NAb concentrations were required to neutralize SCH9 compared with its parental serotypes. 215
With further regards to the CNS, a study in 2010 isolated a variant that can cross the BBB after kanaic-acid induced seizure in rats to provide potential treatment for intractable epilepsy. 216 The library—composed of AAV variants with chimeric capsids made from AAV1–6, 8, 9 and an AAV8 with a E531K mutation—was applied intravenously via the rat tail vein 24 h after kanaic-acid induced seizure. 216 Three rounds of in vivo selection led to clones 32 and 83, which showed increased transduction of cells in the piriform cortex (the CNS compartment most affected by seizures) with CAP gene sequences from AAV1, 8, and 9. 216 The shuffling process appeared to combine and enhance the abilities of AAV8 to cross the seizure-compromised BBB and target the seizure-affected regions of the brain, AAV9 to cross the BBB and transduce cells in the CNS and AAV1 to remove tropism of peripheral organs such as the liver. 216 Interestingly, although clones 32 and 83 showed a de-targeting from peripheral organs (with virtually no liver and heart targeting) in both treated and non-treated animals, they were not able to cross the BBB in non-compromised animals. 216 This reveals a further example of how the change of the microenvironment impacts vector tropism, an important parameter to be considered when tailoring vectors for gene therapy.
In 2016, Choudhury et al. isolated AAV-B1 from a capsid shuffled library of AAV1, 2, 4, 5, 6, 8, 9, rh8, rh10, rh39, and AAVrh43, which outperformed AAV9 regarding transduction efficiency in the neonatal mouse brain, including motor neurons in the spinal cord, and demonstrated a 3.6 times lower liver targeting. 166 A similar transduction profile was seen in juvenile cats and, further, an IVIG neutralization assay showed that AAV-B1 was—in comparison to AAV9—less sensitive to NAbs. 166 AAV-B1 possesses high sequence homology with AAV8; however, many of the surface-exposed VR loop regions in the capsid proteins contained amino acid sequences from AAVrh.10, which might account for AAV-B1's ability to cross the BBB and transduce cells in the CNS. 11,166
More recently, the capsid shuffled variant AAV/Olig001—a chimera composed of capsid sequences from AAV1, 2, 6, 8, and 9—was identified after the capsid shuffling of AAV1, AAV2, AAV2i8, AAV2.5, AAV6, AAV8, AAV9, AAV9.47, AAVrh10, and other capsids from unrelated projects and screening in 6-hydroxy dopamine (6-OHDA)-treated rats for targeting of oligodendrocytes. 217 The 6-OHDA treatment causes degeneration of dopamine-containing nerve terminals and infiltration of oligodendrocyte precursor cells, and the AAV/Olig001 variant was identified to show strong oligodendrocyte targeting after both intravenous and intrastriatal administration. 217 Further, compared with AAV8, the most highly represented parental serotype in AAV/Olig001, AAV/Olig001 showed de-targeting from peripheral organs. 217
This variant was then later tested in the nur7 mouse model of Canavan disease. 218 The nur7 mouse model is based on a point mutation in the aspartoacylase (Aspa) gene, which results in accumulation of N-acetylaspartate (NAA) in the CNS, which is similarly seen in human cases of Canavan disease. An AAV/Olig001-GFP reporter vector determined ICV administration as the most optimal delivery route for targeting of oligodendrocytes exhibiting the characteristic NAA accumulation. 218 With this result, ICV administration was employed to deliver the ASPA coding sequence by AAV/Olig001 vectors, which resulted in rescue of the nur7 mouse motor function as well as significant reductions in vacuolation in the brain. 218 Compared with AAV9-ASPA, the AAV/Olig001-ASPA vector demonstrated improved rescue 218 and again illustrates how engineering of AAV capsids holds potential for optimizing AAV vectors. Based on these promising results, a Phase I/II clinical trial is underway for testing the applicability of intracerebroventricularly administered rAAV-Olig001-ASPA in children afflicted with CD (NCT04833907).
Gene Therapy for Inner Ear Disease
The inner ear is one of the links between the sensory system of the body and the CNS. Most hearing loss cases are categorized as sensorineural hearing loss, which affects the cells of the inner ear, auditory nerves, and central auditory pathway. As many of these cases also have an underlying genetic cause, they present as potential gene therapy targets. Current treatments and medical help for sensorineural hearing loss are presently limited to hearing aids and cochlear implants, which are invasive and do not restore natural hearing. Development of a gene therapy treatment is much needed in this area, and many research groups are now focusing on engineering AAV capsids to target and treat inner ear disorders.
The mechanism of hearing involves the transfer of sound vibrations from the tympanic membrane to the auditory ossicles (the three middle ear bones named malleus, incus, and stapes). The movement of the ossicles in reaction to vibrations of the tympanic membrane causes the stapes bone to tap against the oval window and this movement is transferred as fluid-filled vibrations into the inner ear.
The inner ear comprises the semi-circular canals and the spiral-shaped cochlea, which is filled with endolymph (containing ∼154 mM potassium ions and 0.91 mM sodium ions) and perilymph (containing ∼6.9 mM potassium ions and 138 mM sodium ions). Vibrations traveling within the perilymph of the cochlea will move a key membrane of the organ of Corti called the basilar membrane. Sitting on the basilar membrane are the supporting cells (SCs), the inner hair cells (IHCs), and the outer hair cells (OHCs), which then also move in response to the underlying membrane. The endolymph is forced to rush between the hair cells (HCs) and the tectorial membrane, which lies just above the HCs, causing the HCs to beat. This beating opens channels along the stereocilia, allowing an influx of potassium ions into the HCs, triggering their depolarization. 219 This depolarization prompts fusion of glutamate-filled vesicles with the plasma membrane at the base of the HCs and release of their contents into the synaptic cleft between the HC and the spiral ganglion neuron (SGN). Binding of the glutamate to receptors on the SGN will cause a further depolarization of the SGN, which then continues to propagate the signal along the auditory nerves and the central auditory pathway to the auditory cortex of the brain.
Common cases of genetic deafness often result in degeneration of HCs and SCs—most notably with GJB2 mutations (the most common hereditary deafness gene) 220 and MYO7 mutations—but degeneration of many other cell types of the inner ear is also seen, and so it is vital to find a way to prevent the death of these cell types. A promising avenue is the delivery of a therapeutic transgene to these cells via an AAV vector.
One of the more recent developments that resulted in successful transduction of the inner ear restoring hearing in a mouse model of genetic deafness is the Anc80L65 vector. 221 –223 This vector was designed in 2015 after an in silico ancestral sequence reconstruction of various AAV serotypes and the Anc80L65 variant was identified as the likely ancestor of AAV1, 2, 8, and 9. 224 Landegger et al. showed that the Anc80L65 variant encoding GFP from the cytomegalovirus (CMV) immediate-early promoter transduced 60% and 100% of IHCs and OHCs, respectively, on cochlea explants of P4 C57BL/6 and CBA/CaJ mice. This strong GFP expression in IHCs and OHCs was noted throughout the cochlea at both the apex and the base, and it was also seen very strongly in all other cell types within the cochlea. 221 The AAV serotypes tested alongside this (AAV1, 2, 6, 8, and 9) exhibited much weaker GFP expression in these cells. They then performed an in vivo comparison where they administered all the AAV vectors encoding GFP directly into the inner ear of P1 mice and harvested the cochlea at P10. 221 They observed that the Anc80L65 variant had transduced the IHCs and OHCs at both the apex and the base, 100% and 90%, respectively, and that this strong GFP expression was seen at a 20-fold and 3-fold lower dose (1.7 × 1012 vg/mL) than with AAV1 or AAV2, respectively. 221 These results suggest that the Anc80L65 vector may be a promising gene delivery system to cells in the inner ear—studies in mice so far have shown a restoration of hearing to almost WT levels after using this vector for therapeutic gene delivery in the Ush1c c.216G>A knock-in mouse model of Usher syndrome. 222
A separate study by Tan et al. identified the AAV variant AAV-ie to strongly transduce cochlear SCs and to induce differentiation of the SCs into HCs through AAV-ie delivery of the Atoh1 gene. 220 This finding may be key in the eventual treatment of many types of hearing loss; not only for the treatment of hereditary hearing loss but also for non-inherited sensorineural hearing loss caused by noise damage, aging, or ototoxic drugs. 220 As HCs become damaged and have very low regeneration potential, a therapy in which new HCs can be constructed is vital to restore hearing in these individuals. AAV-ie was created through the insertion of a short peptide (DGTLAVPFK) into the VP1 capsid protein of the AAV-DJ variant. 157,220,225 Ex vivo analyses on mice cochlea explants showed that the AAV-ie vector could transduce 90% of SCs, and the in vivo analyses in mice provided a similar result. 220,225
Further, previous work identified the key transcription factor ATOH1 for the development of HCs in the inner ear of mice, 226 and so Tan et al. performed several experiments to see whether the delivery of Atoh1 via AAV-ie to SCs would induce differentiation of SCs into HCs in vivo within the cochlea of mice. The results showed that, indeed, new HC-like cells appeared in the mice cochlea after 14 days following AAV-ie-Atoh1 administration—as much as 82 new HCs per 100 μm—but some of them still expressed SOX2 (a SC-specific marker) and they were only minimally excitatory. 220 It can, thus, be assumed that ATOH1 expression in SCs results in the generation of a transition state between SCs and fully developed HCs, and that more work needs to be done to identify other transcription factors and signals required for HC generation. 220 The delivery of Atoh1 via an adenovirus gene therapy vector has shown moderate restoration of hearing in a guinea pig model of genetic deafness, 227 and so delivery of this gene to the inner ear shows promise.
Another direction from which to treat hearing loss would be to downregulate the mutant protein that is causing the disorder. Single dominant missense mutations are a common occurrence in many genetic forms of deafness, and a study in 2016 by Shibata et al. used an AAV9 vector-based RNA interference (RNAi) to suppress the expression of the mutant transmembrane protein TMC1 in Tmc1Bth/+ mice. 228 The Tmc1Bth/+ mice are typically profoundly deaf by 35 weeks and model an autosomal dominant non-syndromic hearing loss observed in humans, DFNA36. 229 However, after administration of the mutant TMC1 miRNA delivered by AAV9 vectors directly into the cochlea, hearing loss was significantly slowed. 228
Further work on gene delivery by AAV vectors to rescue genetic deafness include a study in 2012 where mice lacking the vesicular glutamate transporter-3 (VGLUT3) protein, which show congenital deafness (the mouse model for human autosomal dominant DFNA25 deafness), regained their hearing after AAV1 cochlear delivery of VGLUT3. 230 Likewise, a study in 2019 showed revival of exocytosis and replenishment of the key glutamate vesicles of IHCs in Otof−/− mice after administration of a dual AAV2/6 vector where the large OTOF gene was divided into one AAV2 vector and one AAV6 vector to overcome the limited packaging size of AAV. 219 The OTOF gene encodes the otoferlin protein, a large multi-C2-domain protein expressed mainly in IHCs, which, when mutated, causes profound deafness in mice and in humans due to severely disrupted IHC vesicle exocytosis, replenishment, reformation, and endocytosis. 219 This study was able to show a recovery of hearing in the Otof−/− mice, 219 demonstrating that a dual AAV vector system also holds potential to treat genetic forms of deafness where the mutated gene may be too large to be delivered within one AAV vector.
Conclusion
The AAV vectors have emerged as the delivery tool of choice for CNS-related gene therapy. Of utmost importance was the discovery that some natural AAV serotypes cross the BBB, fostering the move from local to intravenous administration. To tackle current limitations of the AAV vector system, capsid engineering is performed. Employing this technique, a substantial number of novel AAV variants have been added to the toolbox and we here presented details on the variants developed to improve the use of AAV vectors for the CNS. However, more work still needs to be done, including the need to identify parameters conferring BBB crossing abilities, the impact of disease-related changes of the microenvironment, the interaction of AAV with the immune system, and characterization of host factors, including cell surface receptors that might affect AAV-CNS transduction patterns. The potential of capsid engineering in improving the efficacy and safety of AAV vectors in CNS-related gene therapy approaches has been clearly demonstrated and will remain a critical determinant for success.
Footnotes
Authors' Contributions
Jo.M., Je.M., and H.B. performed the literature search and wrote the review article. Jo.M. designed and drew the figures. H.B. drafted and edited the review article.
Acknowledgments
The authors apologize to the investigators whose work was not cited owing to limited space.
Author Disclosure
No competing financial interests exist.
Funding Information
The authors thank the BMBF and MWK Lower Saxony-funded Professorinnenprogramm Niedersachsen, the DFG-funded cluster of excellence REBIRTH, and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 819531) for support. In addition, they would also like to express their sincere appreciation to the Hannover Biomedical Research School (HBRS) for financial support.