
Abstract
Gene therapy is a relatively novel field that amounts to around four decades of continuous growth with its good and bad moments. Currently, the field has entered the clinical arena with the ambition to fulfil its promises for a permanent fix of incurable genetic disorders. Hemoglobinopathies as target diseases and hematopoietic stem cells (HSCs) as target cells of genetic interventions had a major share in the research effort toward efficiently implementing gene therapy. Dissection of HSC biology and improvements in gene transfer and gene expression technologies evolved in an almost synchronous manner to a point where the two fields seem to be functionally intercalated. In this review, we focus specifically on the development of gene therapy for hemoglobin disorders and look at both gene addition and gene correction strategies that may dominate the field of HSC-directed gene therapy in the near future and transform the therapeutic landscape for genetic diseases.
Hematopoietic Stem Cell Gene Therapy for Monogenic Diseases: Lessons Learned and Promises Fulfilled
The field of gene therapy has a history that covers ∼40 years; if we try to set a point in time that certain concepts were materialized in a living animal, we may have to travel back to the 1980s. In a seminal paper published in Nature by Dzierzak et al., a gamma-retroviral vector (gRVV) was used to transfer a complete human beta hemoglobin transgene, into murine hematopoietic stem cells (HSCs); expression was documented over a period of 4.5 months on average although at levels, which was quite low to be translated into clinical applications. 1 For the record, the level of human beta globin mRNA was 0.4–4% of the endogenous mouse beta globin levels and this expression was achieved with very vector low copy numbers per cell (0.07–0.2). It was early on evident in the scientific community that for a meaningful gene transfer approach, improvement of vector technology was a sine qua non.
The transition from gene transfer to gene therapy can be viewed as a scientific quest to tackle emerging complexities; when gene transfer was achieved, gene expression had to be improved. When gene expression was achieved, gene silencing had to be avoided. When persistence was achieved, genotoxicity emerged as a serious adverse event. All of what has been cited set the wheels of vector improvement in motion, resulting in the current Phase-III trials of gene therapy products, not to mention the chimeric antigen receptor T-cells (CAR-T) revolution of genetically engineered T cells.
A major milestone of the field was the introduction of lentiviral vectors (LVVs) as gene transfer vehicles. 2,3 It is worth mentioning that in the 1995 paper of Miyoshi et al., retroviral vectors were used as negative controls in the transduction of CD34+ cells, indicating the potential of lentivectors in the improvement of gene transfer technology. Another retroviral vector genus that presented favorable characteristics for HSC gene transfer were the foamy virus (FV) derived vectors. 4,5
Both vector systems use a split plasmid approach where all the essential genes are provided in trans, whereas the transgene carries the gene of interest along with the necessary cis-acting elements for vector packaging. In addition, a safety feature that has been incorporated in the vectors is the self-inactivating (SIN) design; in brief, a deletion of the U3 region in the 3′ long terminal repeat (3′LTR) is transcribed in the 5′LTR, in effect de-activating the endogenous retroviral promoter and leaving the transgene promoter as the sole functional unit. At our current times, LVV have dominated the gene therapy field since they have a number of advantages such as high titers, storage feasibility, and long-term expression.
Be that as it may, one should mention the success stories that gRVV vectors have produced over the years in diseases such as the adenosine deaminase (ADA) deficiency and X-linked severe combined immunodeficiency (X-SCID) immune deficiencies caused by the lack of adenosine deaminase and common gamma chain of the IL2 receptor. 6,7 Over the ensuing years, after gene transfer of the correct gene in the patients' HSC, gene-mediated therapy seemed to be a realistic prospect with no major adverse events till the announcement of five cases of clonal T cell proliferation in patients who received the retroviral vector for X-SCID. 8 Similar problems emerged with the gene therapy trials in patients with chronic granulomatous disease (CGD) and Wiskott-Aldrich syndrome (WAS). 9,10
The problem was associated with the actual process; retroviral integration into the HSC genome caused the uncontrolled activation of nearby genes (LMO2, CCN2, MECOM) that transformed them into real oncogenes driving the patients' blood cell proliferative disorder. This was a turning point that set in motion the quest for understanding the integration profile of the vectors in the HSC host cell genome. This was largely facilitated by high-throughput technologies such as linear - amplification mediated PCR and next generation sequencing.
What we learned from this research effort was that the three retroviral vectors have different preferences when the moment of genome integration arrives. gRVVs show a strong preference for integrating near proto-oncogenes and transcription start sites (TSS), a feature that can explain the genotoxic potential of these vectors in the gene therapy setting. 11 In contrast, the LVVs seem to prefer actively transcribed genes whereas FV vectors show no preference for transcribing genes and only a weak preference for TSS. 12,13
A final introductory note should be addressed to the HSC per se; these cells have the unique potential of unlimited self-renewal and differentiation, ensuring that any implemented genetic modifications will be passed onto their progeny. This property, along with the relatively simple means for their isolation, has made them an attractive target for gene therapy. These valuable cells, for the purpose of gene therapy, are manipulated ex vivo, a process that dampens their stem cell potential. Optimal vectors are those that can deliver their payload to HSC with minimal ex vivo manipulations.
Of all retroviral vectors, gRVVs are fully dependent on nuclear membrane break down for entering the nucleus; this requires active cell divisions that reduce the stem cell potential of HSCs. 14 The FV vectors also require cell division but can form a stable intracytoplasmic pre-integration complex and deliver their cargo on cell division; this feature allows for minimal overnight manipulations of HSCs that are suitable for clinical applications. 15 Finally, LVVs have the unique capacity to integrate into quiescent cells, setting the gold standard in optimal HSC gene transfer conditions.
Over the past 30 years, gene transfer has evolved into gene therapy over a tumultuous path. In Fig. 1, some of the major milestones are depicted in a chronological order; gene addition for monogenic disorders will gradually evolve to gene editing whereas for the foreseeable future, gene addition will be the dominant technology for CAR T cell production. In the ensuing chapters, a detailed account of the major milestones will be presented, placing emphasis on hemoglobinopathies.
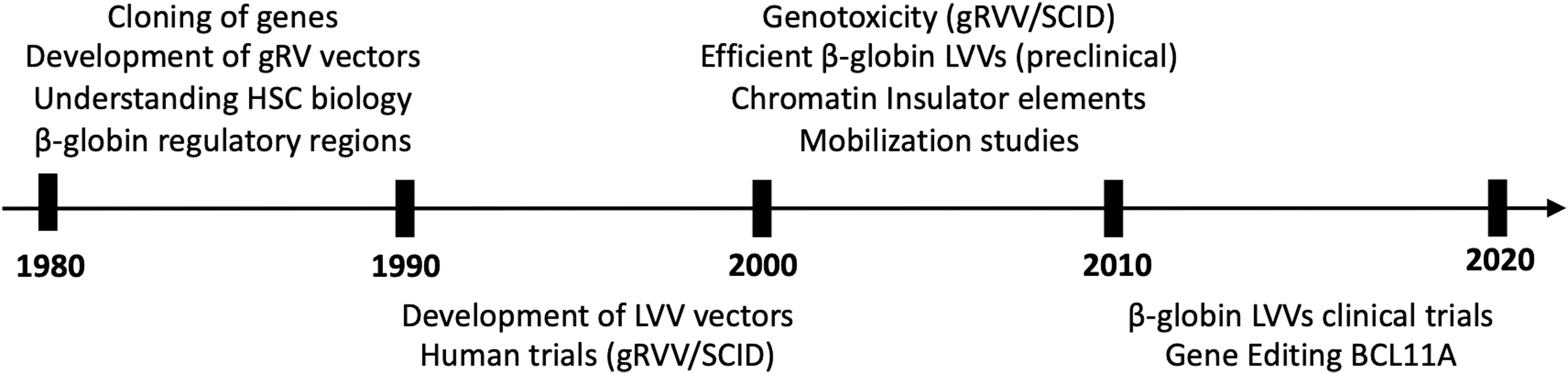
The major milestones in gene therapy vector development for hemoglobinopathies and in the clinical application of the technology are shown in chronological order.
Long Standing Challenges and Expectations: Hemoglobinopathies as Model Disease
The clinical success of HSC gene therapy for primary immunodeficiencies in the early 2000s was later extended to metabolic conditions such as leukodystrophies and recently to red blood disorders. Beta-hemoglobinopathies, such as thalassemia and sickle cell disease (SCD), have been among the very first targets of gene therapy due to the early elucidation of their genetic basis; however, they only recently reached the clinic.
Up to date and for six decades, allogeneic HSC transplantation (allo-HSCT) used to be the only curative therapy, limited however, from donor availability, immunological complications, and patient age. 16 For those patients not fulfilling the criteria to proceed with allo-HSCT, conventional treatment, consisting of red blood cell transfusions in combination with iron chelation (thalassemia) or Hydroxyurea along with supportive care of painful crises (SCD), represents the mainstay of treatment. Such a lifetime treatment affects the quality of life, leading to treatment non-compliance and shortened life expectancy.
A form of one-time therapy that would be curative, largely safe, broadly applicable and ensuring a good quality of life has been an unmet medical need in hemoglobinopathies for decades. Although gene therapy has recently provided clinical evidence as a one-time curative autologous treatment for hemoglobinopathies, for years it faced a plethora of formidable challenges, including (1) the need for high and sustained levels of engraftment and globin gene expression (20% to 30% protein levels) for a robust therapeutic effect, (2) the traditionally suboptimal efficiency of globin vectors, (3) the demanding disease background, and (4) the requirement for increased safety. 17,18
The discovery of the human β-globin gene locus control region (LCR) and the incorporation of micro-LCR cassettes (∼3 kb) in globin vectors as well as the switch from gRVVs to SIN-LV globin vectors greatly improved the safety and efficacy of gene therapy for hemoglobinopathies and facilitated the clinical translation. 19 In addition, both the high numbers of HSCs collected after granulocyte-colony stimulating factor (G-CSF) and Plerixafor (thalassemia) or Plerixafor-only (SCD) mobilization and the use of myeloablative conditioning generated favorable conditions for sufficient engraftment of genetically modified HSCs in a non-competitive manner, overcoming the lack of a natural selective advantage (at the level of corrected HSCs), as opposed to other diseases. 20 –22
All these hurdles in treating patients chronically suffering from a non-malignant disease with gene therapy, in combination with the demand for a clinical benefit that would fully justify the procedure risk, have significantly delayed the transition of gene therapy from bench to bedside. An outline of genetic interventions targeting the HSC is presented in Fig. 2.
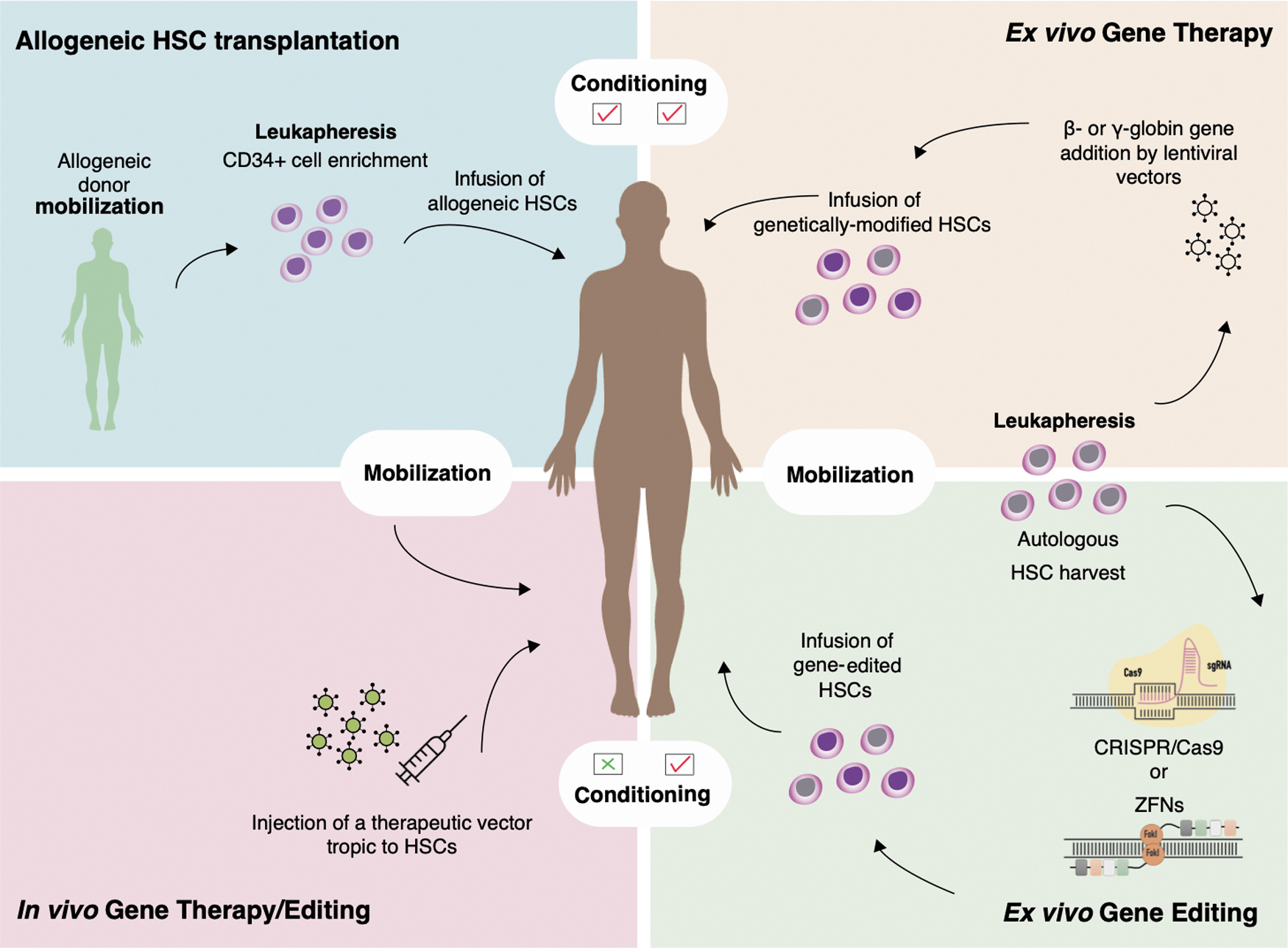
The basic four procedures for HSC-oriented correction of a monogenic disorder starting (clockwise from top left) with allo-HCT, ex vivo gene therapy with gene addition, ex vivo gene editing, and in vivo gene therapy/editing without the need for host conditioning. allo-HCT. allogeneic hematopoietic cell transplantation; HSC, hematopoietic stem cell. Color images are available online.
Gene therapy by gene addition: ex vivo gene transfer
The rationale of gene therapy for β-thalassemia is based on the restoration of the α/β-globin chain balance, whereas for SCD it is based on the inhibition of hemoglobin S (HbS) polymerization. Both can be accomplished either by the insertion of a normal copy of the β-globin gene (HBB) or by correction of the mutated β-globin gene or by the transfer or induction of the γ-globin gene, using several approaches.
The ex vivo β- or γ-gene addition has been the most well-studied gene therapy approach for the correction of globin chain imbalance or inhibition of HbS polymerization in hemoglobinopathies and involves the ex vivo insertion of a normal copy of the β- or γ-globin gene, along with its regulatory elements (beta-globin LCR), into autologous HSCs, using a gRVV or an LVV as delivery vehicles. After the emergence of serious adverse events caused by insertional mutagenesis in clinical trials using gRVVs, those were largely replaced by the SIN LVVs of increased safety. 10,22,23 The incorporation of longer genomic fractions of the beta-globin LCR in SIN-LVVs, having the capacity to transduce non-diving cells and a SIN machinery along with a safer integration profile over SIN-gRVVs, made them ideal vehicles for globin gene addition. 24
The first studies using LVVs (HPV403, HPV436, and TNS9 vectors) published by the groups of Leboulch and Sadelain demonstrated correction of murine beta-thalassemia or SCD. 25,26 Since then, a number of preclinical studies using similar LVVs verified the feasibility of this approach in major hemoglobinopathies. 27 –29 Nevertheless, those pioneering studies also brought to light the susceptibility of the elements controlling the globin gene expression to chromosomal position effects that may result in transgene silencing or variegated, position-dependent expression. 28
As a means of minimizing vector-genome interactions by protecting globin-LVVs against the encroachment of adjacent heterochromatin at the site of integration (barrier activity) but also by shielding the genome from the activation of cellular genes by vector enhancers (enhancer-blocking activity), short genetic regulatory elements called chromatin insulators (CIs) have been incorporated within LVV backbones to flank the transcriptional unit. 30 However, the prototypic cHS4 vertebrate insulator derived from the Dnase I hypersensitive site (DHS)-4 of the chicken β-globin LCR has been associated with a significant reduction in vector titers and a more efficient, although still incomplete, insulation in mouse over human HSCs. 31,32
The need for more potent, small-sized human insulators has been recently met by the discovery of 27 high-affinity CCCTC-binding factor (CTCF)-binding sites, functionally characterized as robust enhancer-blocking insulators, which displayed in their majority, superior enhancer-blocking activity than the prototypic cHS4. 33 By screening 15 of the 27 novel enhancer-blocking CIs for potential barrier activity, we identified two (B4 and C1) as ideal, dual-functioning CI having the capacity to prevent both the unwanted events of genotoxicity and loss of gene expression. 34
Gene therapy clinical trials
Ex vivo gene transfer/addition: thalassemia
The first gene therapy trial for transfusion dependent (TDT) β-thalassemia started in 2007 in France; a βE/β0-thalassemia major patient was transplanted with HSCs transduced with an SIN-LVV (HPV569 vector-bluebird bio) flanked with the cHS4 insulator and encoding an HBB gene with a βT87Q mutation, following myeloablative busulfan conditioning. The βT87Q globin gene serves to provide an anti-sickling effect and also to distinguish, by high-performance liquid chromatography, the vector-derived Hb from the endogenous or transfused HbA. The patient became transfusion independent after 1 year; however, the total Hb consisted of only one-third of the vector-derived HbAT87Q (the rest two thirds were fetal hemoglobin [HbF] and endogenous HbE). 35 Importantly, the HbAT87Q fraction was derived by a benign, although dominant, clone at an integration site close to the HMGA2 gene. This clone persisted for almost 9 years, before eventually subsiding, fortunately without causing oncogenesis (Table 1). 35
Clinical trials of gene therapy for β-hemoglobinopathies
CIRM, California Institute for Regenerative Medicine; CRISPR, clustered regularly interspaced short palindromic repeats; DSMB, Data and Safety Monitoring Board; HbF, fetal hemoglobin; HSCs, hematopoietic stem cells; HSPCs, hematopoietic stem and progenitor cells; LVV, lentiviral vector; SCD, sickle cell disease; SIN, self-inactivating; TDT, transfusion-dependent β-thalassemia; ZFN, zinc finger nuclease.
Subsequently, bluebird bio after vector optimization (named BB305) initiated multinational phase I/II studies for β-thalassaemia (HGB-204, NCT01745120), SCD (HGB-206, still active), or both (HGB-205, NCT02151526) (Table 1). At a 5-year follow-up, the majority of the non-β0/β0-thalassemia individuals in HGB-204 and HGB-205 studies (11/14, 78%) became transfusion-independent (TI), but only 38% (3/8) patients with β0/β0 genotypes reached the definition of TI, as a weighted average Hb ≥9 g/dL without red blood cell (RBC) transfusions for ≥12 months (Lal, ASH 2019, updated results of Ref. 36).
Toward improving the clinical outcomes, especially in β0 patients, a refinement in the manufacturing process was applied early after the initiation of phase III trials for TDT; HGB-207 trial (NorthStar-2/NCT02906202) enrolled all but β0/β0 genotypes, and HGB-212 trial (NorthStar-3/NCT03207009) enrolled double or compound β0 or IVSI-110 heterozygotes. Impressively, after manufacturing refinement, 86% (25/29) of patients across both Phase 3 studies, who completed the 2-year follow-up, achieved the primary endpoint of TI. 37
In another gene addition phase I/II trial for β-thalassemia recently conducted in Italy, three adults and six children with TDT were intraosseously treated with beta-globin SIN-LVV (GLOBE)-transduced cells following a myeloablative, although reduced toxicity, conditioning (treosulfan–thiotepa) (NCT02453477, Table 1). 38 In this study, the majority of the pediatric population (3/4 reported) achieved TI, whereas all of the three reported young adults demonstrated, at best, reductions in transfusion volumes or frequency. Although the difference in the outcome between pediatric and young adult patients could be reasonably attributed to the “younger” stem cell niche of kids, the high rates of TI in adult patients in the bluebird bio's trials do not support the notion of an impaired, “aged” bone barrow (BM) niche that may inhibit the engraftment of gene-modified HSCs and affect the overall clinical outcome.
The striking difference between GLOBE trial and all other HSC gene therapy trials has been the intra-bone marrow delivery of transduced HSCs. Although this route of administration was selected to avoid trapping of the cells and bypass filter organs, it may also generate technical difficulties and impracticalities affecting cell delivery and limiting broad applicability. In addition, the faster platelet engraftment observed in this trial in comparison to other running clinical trials that was initially thought to be derived from the direct access of gene-modified cells to the bone marrow could also be attributed to the higher CD34+ cells (≥2-fold) infused in the GLOBE trial.
One of the first clinical trials for thalassemia was initiated in 2012 at Memorial Sloan Kettering Cancer Center (MSKCC), New York (NCT01639690) (Table 1). None of the three β-thalassemia subjects who received the TNS9.3.55 β-globin vector-transduced HSCs became transfusion independent. One patient with a severe genotype (β0/IVSI-110) who received partial myeloablation had no response. From the other two patients who experienced sustained (5–8 years post gene therapy) reduction in blood transfusions, one with a milder β+ genotype received non-myeloablative conditioning and the other with a severe β0/IVSI-110 genotype received a myeloablative conditioning. 39,40 The important message from this trial in association with trials using a myeloablative conditioning is that myeloablation, at least for now, is a conditio sine qua non for a substantial clinical benefit.
Ex vivo gene transfer/addition: SCD
In parallel with bluebird bio's HGB-trials for major thalassemia, the HGB-206 trial (NCT02140554) is running for SCD using the same Lentiglobin vector. The transition during the trial, from bone marrow harvest to plerixafor mobilization, 41 and the refinement in the manufacturing process (HGB-206, group C) greatly improved outcomes, as represented by <50% HbS and an average proportion of RBCs containing βA-T87Q ∼90% by month 24 as well as complete resolution of vaso-occlusive crisis (VOC) post-treatment. 42 A phase III trial for SCD (HGB-210, NCT04293185) is also ongoing (Table 1).
On February 2021, the U.S. Food and Drug Administration (FDA) had placed the ongoing Bluebird bio's trials on a temporary suspension, due to a serious adverse event of myeloid malignancies that developed in two patients in the HGB-206 trial post-gene therapy. Similar to a previous event of malignancy (myelodysplastic syndrome [MDS] progressing to acute myeloblastic leukemia [AML]) in the HGB-206 trial, the investigation demonstrated that the events were very unlikely related to gene therapy and led to lifting the FDA clinical hold for all suspended trials. 43 Plausible explanations for the genotoxic events are discussed in the last section of the review.
In an ongoing phase I trial initiated at 2014, Kohn's group is also evaluating the safety and evidence for efficacy of autologous HSCs transduced with an LVV containing a β-globin transgene with three anti-sickling mutations (βAS3-FB, NCT02247843, Table 1) in SCD adults, while results are awaited.
Drebaglobe, a gene therapy product of autologous CD34+ cells, which have been transduced ex vivo with the GLOBE vector, expressing an anti-sickling β-globin protein (AS3) and containing three amino acid substitutions in the wild-type β-globin gene, is currently being tested in SCD patients after intravenous administration of transduced HSCs (NCT03964792, Table 1). Using an LVV encoding a modified γ-globinG16D gene (ARU-1801) and a reduced-intensity conditioning in an ongoing phase I/II study (NCT02186418, Table 1), Malik's group have recently announced stable expression of 22–41% total anti-sickling globin and remarkable improvement in clinical outcomes of three treated βS/β0 SCD patients, up to 2 years post-treatment. 44 In particular, patients experienced 85–100% reductions in the number of VOCs, translating into an average 93.8% reduction in cumulative hospitalization days.
Alternative approaches for viral vector gene therapy
RNA interference (RNAi)
A pioneering approach for gene therapy of hemoglobinopathies has been applied at Boston's Hospital, using the RNAi technology to target the γ-globin suppressor BCL11A with a microRNA (miRNA)-adapted short hairpin RNA (shRNAmir) under the transcriptional control of β-globin locus regulatory elements being transferred to HSCs with an SIN-LVV. This approach has been tested preclinically in SCD patient-derived CD34+cells showing potent induction of HbF with phenotype reversal and is currently being investigated clinically (NCT03282656, Table 1). Results were reported for six patients with an at least 6 months follow-up post-gene therapy. All engrafted successfully with a safety profile consistent with the preparative chemotherapy. The patients achieved post-infusion, robust and stable HbF induction (percentage HbF/[F+S] 20.4 to 41.3%), at levels of HbF per F cell up to 18.6 pg/cell (within the range of sickle cell trait), resulting in resolution of VOCs and acute chest syndrome. 45
In vivo gene transfer
Ex vivo gene therapy is a highly sophisticated and expensive approach, requiring expertise and specialized facilities for HSCs' harvesting and transplantation. To overcome barriers associated with the ex vivo correction of HSCs, including also the the extensive culture conditions compromising the cell “stemness” and the requirement for myeloablative conditioning to facilitate the engraftment of gene-modified cells, the in vivo gene therapy, namely the direct injection of the therapeutic vector into the patient in the absence of conditioning, has been recently pursued.
Dr Lieber's group has developed a platform for in vivo HSC gene therapy based on mobilization of HSCs from the bone marrow and their transduction while in circulation, by an injected HSC-tropic, helper-dependent adenovirus HDAd5/35++ γ-globin gene transfer vector system armed with the integration-promoting machinery of Sleeping Beauty. 46 A murine thalassemia intermedia model showed a near to complete phenotypic correction maintained after serial transplants, by in vivo γ-globin gene transfer in combination with minimal doses of an in vivo hematopoietic stem and progenitor cell (HSPC) selection regimen (mgmtP140K gene/low-dose O6-benzylguanine). 47 To overcome the serum inactivation of the commonly used vesicular stomatitis virus glycoprotein envelope of LVVs or the potent immune responses to ADV vectors, Dr Kiem's group used FV and pseudotyped LVV with the cocal envelope to in vivo correct X-SCID in a large animal model. 48
In vivo gene transfer approaches hold great promise for the treatment of hemoglobinopathies and may simplify the gene therapy procedure by avoiding leukapheresis, ex vivo HSC manipulation with subsequent loss of engraftment potential, conditioning, and transplantation. This approach will be further discussed in the last section of the review.
HSC Gene Editing for Monogenic Diseases: The Evolution of Gene Therapy
Among different gene editing platforms, including zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), the discovery of the prokaryotic system of the clustered regularly interspaced short palindromic repeats (CRISPR) represents a unique, efficient, and versatile genetic tool for the manipulation of the genome via gene editing approaches, and therefore holds great promise for the long-term rational and radical treatment of monogenic diseases originating from defects of the HSC. 49
The concept and the tools
In contrast to the gene addition approaches of the conventional gene therapy technology, where the therapeutic transgene is primarily transferred into the target cells via modified viral vectors which in turn integrate randomly into the host genome, the gene editing technological platform can directly modify the target genomic area of the defective sequences and either restore them with the normal incoming sequences or modify other distant regulatory sequences, which can eventually exert a permanent beneficial therapeutic effect. Therefore, from the genetic therapeutic point of view, gene editing represents a significant step of evolution of human gene therapy. Further, due to its versatility, additional properties of the system are continuously being developed, optimizing fidelity and safety, and improving genetic manipulations, such as single base editing, transposition, recombination, and epigenetic regulation. 50
The system is based on a simple principle mediated by three individual components, as shown in Fig. 3. Specifically, it consists of (1) a CRISPR-associated (Cas) effector nuclease; (2) a crRNA (CRISPR RNA) molecule, which consists of the guide RNA (gRNA), (followed by a 12 nt stretch of repeat sequences, highlighted in orange, and a GAAA tetraloop), which contains the complementary sequences for the one strand (3′ to 5′) of the target DNA, and thus locates and recognizes with precision the correct target sequence of the host DNA to be cleaved by Cas nuclease; and (3) a tracrRNA (trans-activating CRISPR RNA) that binds via base pairing in a hairpin loop to crRNA, leading to the formation of an active complex. Thus, the resulting crRNA-tracrRNA complex guides CAS protein to the crRNA-specified sites in the genome with high fidelity. It should be noted that a combined RNA consisting of a tracrRNA and at least one crRNA is often designated as single guide RNA (sgRNA). This formulation is usually applied in cases when multiple crRNAs are packaged together to form an sgRNA. This approach can be further combined with the gene encoding the Cas nuclease in the same plasmid, which can then be used for the transfection of the target cells.
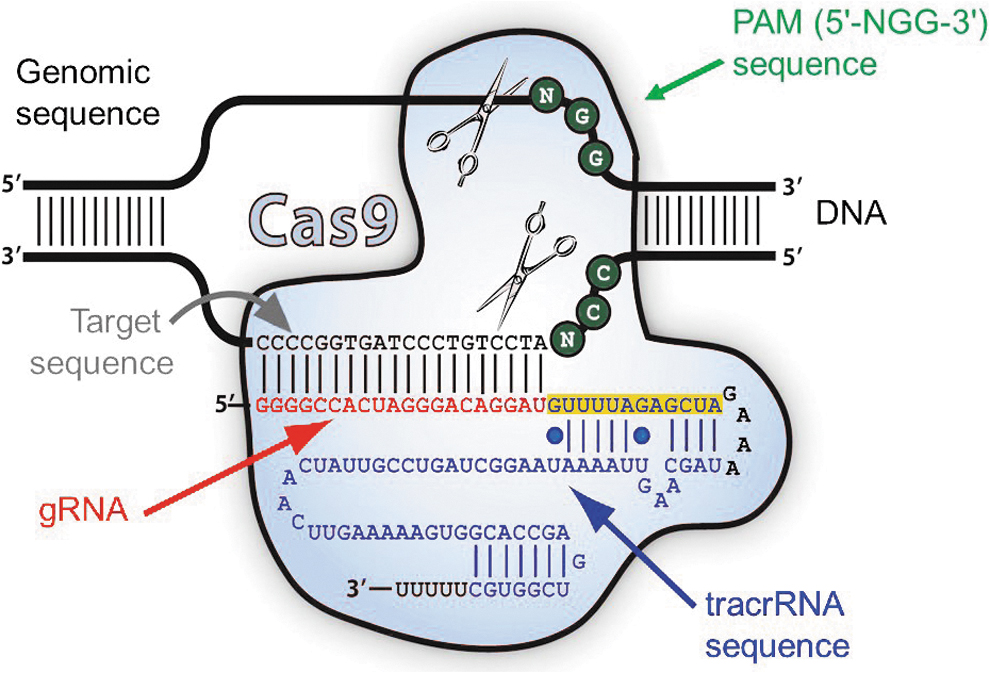
The CRISPR/Cas9 system includes (1) a CRISPR-associated (Cas) effector nuclease (light blue), (2) a crRNA (CRISPR RNA) molecule, which consists of the gRNA shown in red (followed by a 12 nt stretch of repeat sequences, highlighted in orange, and a GAAA tetraloop, shown in black), which contains the complementary sequences for the one strand (3′ to 5′) of the target DNA, and (3) a tracrRNA (trans-activating CRISPR RNA) shown in dark blue, which binds via base pairing in a hairpin loop to crRNA, leading to the formation of an active complex. The PAM sequence, located immediately adjacent to the target sequence, is shown as green circles (Modified by permission from System Biosciences LLC). CRISPR, clustered regularly interspaced short palindromic repeats; gRNA, guide RNA; PAM, protospacer adjacent motif. Color images are available online.
The most studied and widely used system for gene editing, so far, is the CRISPR-Cas9 nuclease derived from Streptococcus pyogenes, which contains the SpCas9 effector nuclease and the appropriate sgRNA (Fig. 3). The two RNAs of the system, that is, the crRNA and the tracrRNA, can be readily engineered together as a sgRNA, containing a spacer sequence for target sequence hybridization and scaffold for SpCas9 binding.
The high degree of fidelity of the CRISPR-Cas system depends on two major factors for its specificity: (1) the nature of the target sequence and (2) the protospacer adjacent motif (PAM) sequence, located immediately adjacent to the target sequence (Fig. 3). For the SpCas9 nuclease in particular, the required PAM sequence is 5′-NGG-3′, which fortuitously occurs roughly every 8 to 12 bp in the human genome, underscoring the flexibility of the system to select convenient target sites. For the gRNA, the target sequence is ∼20 nucleotides long as a part of each CRISPR locus in the crRNA array. A typical crRNA array has multiple unique target sequences.
Since the gRNA sequences are not part of the SpCas9 protein and thus customizable, they can be independently synthesized. After the binding of gRNA to SpCas9, the nuclease molecule undergoes a conformational change into an active DNA-binding form. Based on the requirements mentioned earlier, the sgRNA has the capacity to recognize and direct the SpCas9 nuclease to the target DNA site, via hybridization with the target DNA sequence and the simultaneous recognition of the PAM motif. Importantly, the spacer region of the gRNA remains free to interact with target DNA. SpCas9 will only cleave a given locus if the gRNA spacer sequence shares sufficient homology with the target DNA. Once the SpCas9-gRNA complex binds a putative DNA target, the seed sequence of 8–10 bases at the 3′ end of the gRNA targeting sequence will start to anneal to the target DNA, and provided that the seed and target DNA sequences do actually match, the gRNA will continue to anneal to the target DNA in a 3′ to 5′ direction.
On target binding, spCas9 undergoes a second conformational change that positions the two nuclease catalytic domains called RuvC and HNH, to cleave the opposite strands of the target DNA, as shown in Fig. 3. The end result of SpCas9-mediated DNA cleavage is a double-strand break (DSB) within the target DNA and ∼3–4 nucleotides upstream of the PAM sequence. The resulting DSB can then be repaired by one of two general repair pathways, that is, either via the efficient but error-prone non-homologous end joining (NHEJ) pathway, or via the less efficient but high-fidelity homology directed repair (HDR) pathway. The NHEJ pathway usually leads to small deletions or insertions (indels) at the site of the joining ends. The fact that this repair mechanism is random, it can lead to a diverse array of such types of mutations among a population of transfected target cells.
Pros and cons and further optimization of the system
Due to its versatility, the CRISPR/Cas system is undergoing continuous development to overcome current limitations that have been observed during the early period of its application as a potential gene editing tool. These obstacles include off-target effects, narrow targeting scope, and issues associated with the delivery of CRISPR components for genome engineering and therapeutic approaches. 50 Improvements of its specificity, employing structure-guided, and complementary evolution-based engineering of SpCas9 nuclease have led to increased PAM plasticity by the SpCas9 variants. Further, efforts toward increased targeting specificity, coupled with reduced frequency of off-target effects employing structure-guided mutagenesis, have generated novel variants exhibiting high fidelity, by combining reduced off-target effects, while maintaining on-target specificity. 50 Additional progress has also been documented in engineering Cas9 orthologs, with a scope of increasing their editing efficiency, by introducing mutations, and recognizing a series of different PAM motifs.
A significant step for minimizing the off-target effects was recently accomplished, employing a new alternative technological approach in a gRNA-dependent fashion, designated as base editing. This is accomplished by fusing a deaminase enzyme to an inactive form (dCas9) of the nuclease to avoid cleavage. This approach significantly extends the spectrum of editing implemented by the CRISPR/Cas9 system.
Currently, two general types of base editors and their improved variants for targeted base mutagenesis are used: the cytidine (CBE) and the adenine base editor (ABE), using different modes of action, exhibiting increased editing efficiency and reduced off-target effects. For example, the CBE form converts a targeted C-G bp to a T-A bp, via an intermediate deaminated pair of U-G, which is then converted to T, by the cell's DNA mismatch repair mechanism. More recently, combined dual CBE-ABE and glycosylase base editors have been developed, whereas a new tool of prime editors, generated by fusing a reverse transcriptase molecule to spCas9 with a mutated domain called nickase (nCas9), can introduce a single-strand cleavage with the same specificity, and thus generate precise modifications while avoiding the HDR pathway, and this is under study. 50
For the eventual large-scale application of the CRISPR/Cas system in the clinical setting, the serious caveats of off-target effects should be efficiently addressed by further modifying the individual components as discussed earlier, and by suppressing as much as possible the resulting tolerance of mismatches between the sgRNA and target DNA sequences. This can be greatly facilitated by the use of several bioinformatic tools for the initial design of the appropriate sgRNAs. Finally, the issue of immunogenicity of the CRISPR components should be considered and addressed systematically. 51
Preclinical and clinical development
Regardless of the reservations and limitations cited earlier, several preclinical studies involving the gene editing systems (excluding hemoglobinopathies, which are discussed separately later) have been conducted with variable outcome in HSCs of specific monogenic disorders, such as SCID, WAS, Fabry disease, Hurler syndrome, Wolman disease, CGD, Hyper IgM syndrome, Fanconi anemia, Hemophilia A and B, Amegakaryocytic thrombocytopenia, and AIDS. 52,53 Further, in the clinical setting, a total of five industry-sponsored clinical trials, all employing in vivo strategy, either via ZFN or CRISPR/Cas9 technology, have been initiated involving Mucopolysaccharidosis I and II, Hemophilia B, and AIDS. 52,53
These trials are expected to provide significant insights regarding the degree of efficiency of the approach and underscore the technical challenges for future clinical protocols, which currently include issues of the appropriate sources and yield of the HSCs, efficiency of the HSC delivery of the gene editing system by increasing the transduction rate utilizing CD34+-specific viral envelope glycoproteins, optimizing the precision repair mechanism by HDR-enhancing conditions without affecting HSC stemness, maintenance of long-term stability and harnessing the immunogenicity risk, along with the need for international collaborative studies for further definition of novel genetic biomarkers predicting the outcomes of patients with HSC monogenic disorders. 54 –56
HSC Gene Editing: Hemoglobinopathies as Model Disease
Mutated gene correction
All genome editing tools (ZFNs, TALENs CRIPSR/Cas9) have been extensively assessed as therapeutic modalities in β-globin disorders. The most widely pursued gene editing methods include the reactivation of the endogenous, developmentally silenced HbF, the decrease of the excess alpha globin production, and the in situ correction of disease mutation.
Reversing the causative mutation by genome editing seemed, in theory, a straightforward procedure; however, it has met several challenges. The primary obstacle has been that correction requires activation of the HDR pathway and suppression of the NHEJ mechanism, which has not been very prolific in human adult HSCs. However, a small number of important preclinical breakthroughs on that front have occurred for both SCD and β-thalassemia. Either an AAV6 cassette or a single-stranded oligonucleotide has been used so far as the template vehicle and both methods have been partially successful in patient cells in vitro or in SCD animal models. 57,58 In β-thalassemia, in particular, an alternative method not requiring HDR has been tried by two independent groups in which correction of IVSI-110(G>A) and/or IVS2–654C>T β-thalassemia was achieved by a simple allelic disruption. 59,60
Template- and HDR-independent, base or prime editing could represent a quantum leap for gene correction of hemoglobinopathies. A group of hemoglobinopathy experts led by David R. Liu have successfully applied an ABE that converted the SCD β-globin (HBBS) to the non-pathogenic Makassar β-globin s (HBBG) in patient-derived CD34+ cells. 61
Reactivating HbF by downregulating γ-globin silencers
Hereditary persistence of fetal hemoglobin (HPFH) is a benign genetic condition that has been linked with ameliorated clinical manifestation of hemoglobinopathies. 62 Specifically, γ-globin presence in β-thalassemia can reduce the levels of excess alpha chains and restore the globin chain balance; however, in SCD, the antisickling properties of gamma globin inhibit polymerization of βS hemoglobin, prevent sickling, reduce hemolysis, and decrease microvessel occlusion. Therefore, reactivating HbF expression represents a universal strategy that can be effective—independently of the specific mutation-, for both SCD and β-thalassemia patients, allowing the development of single therapeutic products for treating both diseases.
The fetal-to-adult globin switching occurs soon after birth and is completed by the second year of postnatal life. The regulation of this switching mechanism is governed by several cis and trans regulatory elements. Perturbation of these elements driving a sustained HbF production in adult life has long been considered a desirable approach to treat β-globin disorder.
A major breakthrough toward this perspective was the identification of BCL11A as a critical transcription factor regulating the suppression of γ-globin expression. 63 Early studies targeting the coding region of BCL11A successfully increased fetal globin in mouse models and human erythroid cells. However, BCL11A's involvement in HSC proliferation and survival as well as differentiation of lymphocytes rendered the approach not clinically applicable. The obstacle was addressed by the discovery of an erythroid-specific enhancer of BCL11A, composed of three DHSs, +55, +58, and +62. 64 Two independent studies achieved deletion of a GATA footprint in +58 DHS, using ZFNs and CRIPSR/Cas9, respectively, which significantly decreased BCL11a expression and increased γ-globin levels. 65,66 Since these two initial studies, a number of groups have pursued disruption or precision editing of the erythroid enhancer in normal and patient samples and evaluated the effect of the mutation in small and large animal models. 67 –70 Overall, the results of these studies converge toward high BCL11A editing rates ex vivo, irrespective of the nuclease used; however, after transplantation, a substantial loss of the edited cells occurs in vivo. This result, as suggested by Demirci et al., 70 can be potentially avoided by increasing the number of infused cells.
Recently, as a result of this universal scientific effort, BCL11A-enhancer knock-out is being applied in patients with β-thalassemia and SCD, with promising outcomes. The first clinical trial initiated by Sangamo Therapeutics and Sanofi is based on electroporation with an mRNA-coding ZFN targeting the BCL11A-enhancer in autologous CD34+ cells from thalassemic and SCD patients (NCT03432364 and NCT03653247, respectively, Table 1). Results from the first two enrolled thalassemic patients presented in late 2019 showed rapid hematologic reconstitution, HbF elevation, and persistence of indels, 6 months post-infusion. Further, the first three SCD patients receiving ZFN-modified autologous CD34+ cells remain symptom-free at 52 and 13 weeks, and 29 days post-infusion. 71
Another clinical trial based on CRISPR/Cas9 editing of the same site is being carried out by a CRISPR Therapeutics-Vertex partnership for TDT and SCD. 72 Recently, results from the follow-up of 10 thalassemic and 4 SCD patients at 24 and 19 months post-CTX001 infusion, respectively, were presented (trials NCT03655678 and NCT03745287, respectively, Table 1). 73 CTX001 infusion increased and retained at high levels the total unsupported Hb and HbF with F cell pancellularity in both TDT and SCD. The TDT patients were transfusion-free within 2 months post-CTX001 infusion, and SCD patients were VOC symptom-free immediately after CTX001 infusion throughout the follow-up. Similar to gene therapy by gene addition, the CTX001 safety profile was shown to be generally consistent with myeloablative conditioning and autologous HSC transplantation.
Among other HbF regulators (LRF, ZNF410, HRI), which have recently emerged through high-throughput genome editing assays, only the ZNF410 could serve as a potential therapeutic target without a major effect in erythropoiesis and hematopoiesis. 74
Reactivating HbF by HPFH induction
In addition to silencer deletion, genome editing strategies mimicking HPFH mutations have been explored. HPFH is caused either by large deletions in the β-globin gene cluster (deletional HPFH) or by mutations in the promoter of the γ-globin gene (non-deletional HPFH). A 13.6 kb deletional-HPFH induction in wild-type and SCD-patient-derived HSCs led to a significant increase in γ-globin production coupled with a partial phenotype correction of sickle cell-edited erythroblasts. 75
Exact replication of non-deletional mutations would require activation of the HDR pathway and rather not unexpectedly, efforts have moved toward the deletion of the BCL11A- and LRF-binding sites within the HBG promoter. 76 Targeting the BCL11A-binding distal TGACCA motif at HBG-115 has been performed by using several genome editing modalities and proven to be as efficient as the BCL11a-erythroid enhancer knock-out in elevating endogenous HbF. 67,77 Further, this approach resulted in long-term HbF reactivation in a non-human primate model; however, genome editing levels abruptly dropped shortly after transplantation. 78
In addition to the BCL11a-binding site, perturbation of the LRF-binding site at around HBG-200 also results in high expression; however, intensity varies in relation to the nuclease-binding site. 79,80 Recently, multiplex mutagenesis combining HBG-promoter and BCL11A-enhancer mutations was performed ex vivo and in vivo in thalassemic-xenografts, further increasing the HbF yield. 80
All aforementioned methods of editing the γ-globin promoter have a seemingly safe profile; however, there is the theoretical possibility of genomic translocations due to the presence of two almost identical promoters, leading to excision of a large genomic region. An alternative method based on in vivo base editing was recently reported, where even monoallelic −113A>G substitution increased γ-globin production in erythroid clones and a significant elevation was observed in a mouse model on selection of the edited cells. 81
HSC Gene Therapy for Hemoglobinopathies: New Challenges Ahead
Due to the complexity of globin gene expression and regulation, the clinical translation of gene therapy for hemoglobinopathies was significantly delayed, despite this family of genetic diseases being among the very first targets of gene therapy. During the past three decades of strenuous research in the field, several lessons were gained, eventually being translated to an unequivocal clinical success. Notwithstanding success, gene therapy for hemoglobinopathies still faces limitations or/and new challenges.
Long-term safety and sustainability of clinical benefit
Any new therapeutic intervention for patients suffering from a chronic disease such as thalassemia major and having a close to normal life expectancy, if adequately compliant to conventional therapy, should provide a robust, long-term benefit that will fully justify the procedure risk.
In all HSC gene transfer trials, patients are monitored for 2 years by integration site analysis at regular time-points, for emerging clonal expansion post-gene therapy. According to the requirements of regulatory authorities, patients with hemoglobinopathies treated with gene therapy by either gene addition or gene editing should undergo a prospective evaluation for a minimum of 15 years after the completion of 2-year follow up of the parental studies. These important long-term follow-up studies will allow firm conclusions to be drawn on both the sustainability of the clinical benefit and the long-term safety.
Recently, disclosed data from the long-term follow-up LTF-303 trial (Table 1) 37 enrolling patients with hemoglobinopathies who have completed the 2-year follow-up of the parental PhI/II (HGB-204 and 205) studies and the PhIII (HGB-207 and 212) studies with the Lenti-globin for TDT expressing the βA-T87Qglobin gene (51 patients enrolled) demonstrated that TI for those who reached TI (PhI/II: 68.2%/PhIII: 86.2%) has been durable and maintained for a maximum of 7 years. Importantly, no insertional mutagenesis, malignancies, vector-derived replication competent lentivirus, or deaths have occurred.
As opposed to thalassemia gene therapy, SCD gene therapy (HGB-206 trial) has been related to oncogenesis. Three patients developed myeloid malignancies after gene therapy, at 3 and 5.5 years (MDS progressing to AML and de novo AML, respectively) or 6 months (MDS). In all cases, however, the investigation showed that these side effects were not gene therapy-related. In the first case of MDS that subsequently progressed to acute leukemia, the investigation demonstrated that the event was unrelated to the integrating BB305 vector with no evidence of vector integration in the CD34+ blast cells. 43 In the recent case of AML, the vector integrated within the vesicular-associated membrane protein 4 (VAMP4) gene, having no known association with oncogenesis whereas no considerable expression changes were found within a 10 MB region around that gene. In this patient, typical de novo AML-related mutations and chromosomal abnormalities were detected. The initial diagnosis of an early onset post-gene therapy MDS event based on the detection of trisomy 8, an MDS-related cytogenetic abnormality, was later revised to transfusion-dependent anemia due to the absence of blast cells or cytomorphological changes in the BM.
These cases of suspected or confirmed leukemogenesis in SCD (in contrast to thalassemia gene therapy) raise the question of potential vulnerability of SCD patients to oncogenic events. Indeed, an increased risk for myeloid malignancies in SCD has been reported; 82 plausible host factors that may create a permissive environment for the development of malignancies are the chronic hypoxia and inflammation, reduced apoptosis, erytrhopoietic stress, increased cell turnover, and clonal hematopoiesis of indeterminate potential (CHIP)-related mutations. 83 The SCD patients undergoing gene therapy may have additional risk factors for oncogenesis derived from the conditioning regimen (myeloablative or non-myeloablative) or/and a low engraftment of gene-corrected HSCs, allowing for clonal proliferation of the endogenous cells. The chronic use of Hydroxyurea in sickle cell patients has not been directly associated with an increased risk of mutagenic effects, although case reports of myeloid neoplasms and other malignancies have been described. 84,85 Overall, the leukemogenesis risk in SCD patients could be driven by consecutive accumulation of mutations in the context of the natural history of the disease and its treatment.
True genotoxic events with SIN-LVVs remained, until very recently, only a theoretical safety concern pertaining to vectors using powerful enhancers (i.e., globin vectors). 86 Unfortunately, 1 month after the European market approval of Skysona, a gene therapy product of BlueBirdBio for early adrenoleukodystrophy (ALD), a participant in a now on-hold trial, developed MDS 1 year after treatment while another two treated patients present abnormalities in their bone marrow cells. Unlike the genotoxic events in SCD patients, the ALD case was developed by LVV integration at a site in the genome linked to MDS. 87 The vector used in that study carried a very strong promoter of broad activity to ensure that brain cells could produce high levels of the ALD protein to reach a clinically meaningful result, at the expense however of risking to turn on nearby oncogenes.
Chromatin insulators flanking viral vectors have been proposed as a means to minimize the risk of insertional oncogenesis by protecting the environment adjacent to the insertion sites from the activating power of strong enhancers as described earlier in this review. 88 CIs have been brought to the forefront recently, due to the discovery of novel, human CTCF elements with considerable advantages over all previous insulators, shown to provide powerful enhancer-blocking capacity as well as protection from chromosomal position effects. 33,34
Genome editing may eliminate the risk of near random viral vector integration and insertional mutagenesis, of gene addition by viral vectors, but it faces the risk of unintended “off target” mutagenesis that could generate nonspecific and undesirable genetic modifications with unexpected consequences. Various tools have been developed for in silico detection of off-target mutations along with improved on-target efficiency to ameliorate the off-target effects. In addition, novel engineered Cas9 variants and new gene-targeting techniques replacing the DSB-based editing have been developed, which can mitigate the off-target effects, although they are still subject to substantial optimization. 52
Myeloablative conditioning: a major current limitation
Early toxicity
Whether full or partial myeloablation should precede the infusion of gene-corrected HSCs used to be an issue of debate for the design of the first gene therapy clinical trials for thalassemia. Full myeloablation would create adequate space for the gene-corrected HSCs to engraft and allow for maximum genetically modified HSC chimerism, at the expense however of increased peri-transplant or later toxicity. Partial myeloablation would ensure a safer procedure but also generate the challenge for the transduced HSCs to compete for a niche over the endogenous BM cells, which might lead to low engraftment.
The lesson gained from those early studies was that the highly demanding disease background in hemoglobinopathies and the lack of survival advantage of the genetically corrected HSCs, makes the use of myeloablative conditioning rather mandatory for a clinically meaningful outcome. Indeed, full myeloablation allowed patients to achieve transfusion independence in several trials 36,38 whereas in the NY trial, a non-myeloablative conditioning resulted, at best, in reduced RBC transfusion volume or frequency. 89
Peri-transplant hematopoietic and non-hematopoietic toxicity in thalassemia NorthStar-2 and NorthStar-3 trials was overall well tolerated and consistent with the Busulphan-associated known toxicity, most commonly including stomatitis, febrile neutropenia, and epistaxis. Liver veno occlusive disese (VOD) was a suspected and rather common, although reversible with defibrotide treatment, adverse or serious adverse event in these trials (2/22 and 4/38 patients, respectively), before anti-VOD prophylaxis was introduced in all patients. Engraftment was successful, although relatively delayed, especially for platelets. 36 A conditioning protocol with reduced toxicity but sufficient myelosuppression has been applied in the Italian GLOBE-trial; this has been accomplished with the use of treosulfan and thiotepa. 38
Late toxicity
Apart from early toxicity, chemotherapy-based conditioning is also associated with a known late side effect: secondary malignancies. However, the previously held belief that secondary leukemogenesis is driven by certain cytotoxic drugs causing DNA damage has been challenged by the currently effective concept that cytotoxic chemotherapy selects for, or generates conditions enhancing pre-existing, pre-leukemic cells harboring DNA lesions associated with chemotherapy-related mutations, that is, TP53-mutations, which develop into acute leukemia in a niche where other normal cells have been eliminated by the treatment. 90 Having excluded the vector-driven leukemogenesis, the myeloid malignancies in patients with SCD treated with gene therapy were interpreted in the aforementioned context. Infertility is another well-known risk associated with traditional conditioning, which becomes of higher importance when kids and adolescents with chronic (and non-malignant) disease are treated.
Non-chemotherapy-based conditioning
Genotoxic conditioning remains a substantial obstacle of HSC transplantation or gene therapy. A highly appealing alternative is the targeted host conditioning, devoid of DNA-damaging agents, that specifically depletes HSCS and other hematopoietic cells from the bone marrow niche while sparing non-hematopoietic cells and maintaining intact marrow cellularity. Anti-c-kit monoclonal antibodies were shown to allow donor HSC engraftment in immunodeficient mice and when combined with CD47-blockade to extend anti-c-kit effector activity to fully immunocompetent mice. 91 Similarly, an internalizing immunotoxin targeting the hematopoietic-cell-restricted CD45 receptor (CD45-saporin [SAP]) effectively conditioned immunocompetent mice, enabling >90% donor chimerism without aplasia or long immunosuppresion and full correction of a murine sickle-cell phenotype. 92
Biological agent-only, non-genotoxic, host conditioning could improve the safety and allow broader applicability of HSC transplantation or gene therapy in the context of non-malignant blood diseases, thus transforming the current practices of HSC cellular therapies.
No host conditioning: in vivo HSC gene therapy
As already briefly described, an interesting alternative platform to ex vivo modification that could bypass the need for conditioning in the genetic correction of HSC has been recently introduced. This in vivo approach involves the mobilization of HSCs from the bone marrow, their transduction in the periphery, and then their return to the bone marrow. 46 Dr. Lieber's group has developed a platform for in vivo g-globin gene delivery, in which G-CSF+plerixafor-mobilized HSCs, while in circulation, are being transduced by a system of a helper-dependent adenoviral vector targeting CD46 (a uniformly expressed receptor on HSPCs) and a hyperactive Sleeping Beauty transposase (SB100X). Targeted HSCs later home in their bone marrow niche and stably express the transgene. 46 This platform has been successfully tested in mouse models of hemoglobinopathies for random or targeted γ-globin gene addition, single or multiplex CRISPR/Cas9 gene editing, base editing, and combined γ-globin gene addition with gene editing; the system is currently being investigated in proof-of-concept studies in non-human primates. 47,80,81,93 –96
Such a simplified, cost-efficient, and overall “portable” gene therapy approach, without the need of leukapheresis, conditioning, and transplantation expertise, would potentially allow broader clinical applicability of gene therapy for hemoglobinopathies even in resource-poor regions where the greatest demand for a β-thalassemia therapy lies.
Beyond hemoglobinopathies, other genetic diseases could also be targets for in vivo gene therapy. Dr. Kiem's group has recently shown clinically relevant results in a canine model of X-SCID with direct i.v. cocal-enveloped LVV infusion in the absence of conditioning. 48 Moreover, in Fanconi anemia, in which the genotoxicity risk by conditioning is higher and the HSPC manipulation could be deleterious for the “fragile” and scarce HSCs, in vivo gene therapy may offer a viable alternative.
Nevertheless, compared with ex vivo gene therapy, in vivo gene therapy is still at an infancy period, requiring optimization to ensure therapeutically meaningful globin gene expression and overcome the hurdle of inadvertent immune response.
Marketing authorization and new challenges ahead
The long-awaited gene therapy for the global community of hemoglobinopathies has ultimately reached clinical practice. The bluebird bio gene therapy product, the autologous CD34+ cells encoding the β A-T87Q-globin gene, received conditional approval, in June 2019, for the treatment of transfusion-dependent β+ thalassaemia in patients 12 years and older, who are eligible for stem cell transplantation but do not have a matched related donor [betibeglogene autotemcel(beti-cel); Zynteglo; LentiGlobin for β-thalassemia].
In recent years, a number of advanced therapy medicinal products have received marketing authorization based on their curative potential for difficult to cure or incurable diseases, including ADA-SCID deficiency, congenital blindness, spinal muscular atrophy, B cell malignancies, and, recently, thalassemia. This new era of personalized therapies has not only drastically changed the conventional pharmaceutical model of standardized and bulk drug production but has also given prominence to new challenges affecting the 3-A pillars of health; Availability, Accessibility, Affordability.
In particular, the new curative treatments for β-type hemoglobinopathies are expected to financially challenge the health system payers—even in-high income countries—due to burdensome price tags (e.g., €1.57 million for Zynteglo). Even though the life-time costs for treatment of chronic diseases such as thalassemia, including also the indirect costs of patient follow-up, hospitalization needs, reduced patient productivity, and individual and family psychological suffering, surpass in total the price of one-time gene therapy treatment, the demand for such high prices to be paid ahead and at once, challenges the national economies and complicates patient access to such expensive genetic treatments. This is likely to happen even in developed or rich countries, but it is expected to be seen universally in low- to middle-income countries. Such high costs have also generated new models of payment; an outcome-based model of payment has been proposed for Zynteglo and Kymriah by bluebird bio and Novartis (CAR-T cell therapy for B cell malignancies), respectively, where full reimbursement requires a full response to treatment.
The long-term follow-up of patients with hemoglobinopathies treated with gene therapy will establish the durability of the clinical benefit and the long-lasting safety; the question is whether, indeed, such a one-time expense will be cost-effective overall.
Additional challenges include the inability of such sophisticated therapies to reach all patients in need in a timely manner, not only because of the exorbitant costs but also because of the complexity of the process requiring specialized infrastructure and transplantation expertise. Such challenges may deepen inequality between patients and either make impossible or critically delay the access to curative treatments for those living in poor-income countries. Alternative and less complex procedures such as the in vivo gene therapy may facilitate the availability, accessibility, and affordability of genetic interventions for β-type hemoglobinopathies in developing countries.
The urgent need for changes in market access policies to enable gene therapies to be made available globally is reflected on the Bluebird bio's decision to wind down their operations in Europe and focus on the U.S. market. 97 In any case, all stakeholders involved, including the pharmaceutical industry, policy makers, patients, and health care providers, need to closely collaborate to create solutions that are acceptable to payers and companies and make gene therapy for hemoglobinopathies broadly available and accessible to patients.
Footnotes
Author Disclosure
E.Y provided advisory board and speaker's bureau services to BlueBirdBio and CripsR Therapeutics/Vertex - received clinical trial support, paid to
Funding Information
This work was funded by the Hellenic Society of Gene Therapy and Regenerative Medicine.