
Abstract
Repeat dosing poses a major hurdle for the development of an adeno-associated virus (AAV)-based gene therapy for cystic fibrosis, in part because of the potential for development of an immune reaction to the AAV1 capsid proteins. Here, to dampen the immune response to AAV1, we treated Rhesus monkeys with methylprednisolone before and after the instillation of two doses of AAV1Δ27-264-CFTR into their airways at 0 and 30 days, followed by a single dose of AAV1-GFP on day 60. Animals were euthanized on day 90, except for one monkey that was sacrificed at 1 year. No adverse events occurred, indicating that the two AAV1 vectors are safe. rAAV1-CFTR and AAV1-GFP vector genomes and mRNA transcripts were detectable in all lung sections and in the liver and pancreas at day 90 and after 1 year at levels comparable with animals necropsied at 90 days. The numbers of vector genomes for cystic fibrosis transmembrane regulator (CFTR) and green fluorescent protein (GFP) detected here were higher than those found in the monkeys infected without methylprednisolone treatment that we tested previously.1 Also, lung surface and keratin 5-positive basal cells showed higher CFTR and GFP staining than did the cells from the uninfected monkey control. Positive immunostaining, also detected in the liver and pancreas, remained stable for at least a year. All animals seroconverted for anticapsid antibodies by 90 days post-treatment. The neutralizing antibody titer declined in the animal necropsied at 1 year. Conclusion: AAV1 safely and effectively transduces monkey airway and basal cells. Both the presence of vector genomes and transduction from AAV1-CFTR and AAV1-GFP virus seen in the monkeys 4 months to 1 year after the first instillation suggest that repeat dosing with AAV1-based vectors is achievable, particularly after methylprednisolone treatment.
INTRODUCTION
Beginning with the first regulatory approval after many years of research, 2 and coupled with an explosion of vaccines based on viral and mRNA approaches, 3 gene therapies (Approved Cellular and Gene Therapy Products | FDA) (see Braendstrup et al. 4 ) are finally having an impact on human health. Despite these advances, however, genetic therapies that could benefit patients with a variety of genetic disorders are still unavailable. One such disorder is cystic fibrosis (CF), an autosomal disorder caused by a defective gene encoding the cystic fibrosis transmembrane regulator (CFTR). 5 Despite years of intense research, 6 there is still no gene therapy for CF. There are more than 1,700 mutations in CFTR, with the folding mutation F508-del being the most prevalent. 7
Recently, there has been enormous progress in the development of a triple combination of two correctors, VX-661+VX-445, and a potentiator, VX-770, that can rescue and activate F508-del CFTR trafficking and function, respectively. 8 However, even if successful in the F508-del context, this combination cannot treat patients with other mutations, such as splice variants, large deletions, or premature stops. 9 In addition, there are trafficking mutants such as N1303K that are notoriously refractory to rescue. 10 Thus, there is still a critical unmet need to develop alternative strategies such as gene therapy.
AAV2 gene transfer has been tested in humans. 11 The major findings in clinical studies were that AAV2 administration by itself does not lead to any detectable clinical sequela but does not show a sustained clinical effect 12 in patients with CF. One reason is that AAV2 is not very efficient in transducing lung cells. 13 Second, there is not enough protein expression from the weak promoter containing within the vector to be effective. 14
Since these early studies, much progress has been made in overcoming these hurdles. Newer viral vectors based on the AAV1 serotype have been shown to be more efficient than AAV2 in targeting the airway. 15 More powerful promoters such as chicken β-actin (CBAA) have been identified, which increase the expression of CFTR from adeno-associated virus (AAV) vectors. 16 These advances have moved the field significantly farther forward.
AAV serotypes for CF gene therapy
Since the first clinical trials with AAV, many more serotypes of AAV have been discovered making it possible to boost gene transfer, particularly in airway epithelia using naturally occurring serotypes. 17 One of these is AAV9, which has a higher efficiency of gene transfer and higher ability and to transduce lung cells, particularly alveolar cells. 18 We have studied AAV519 containing a pseudotyped virus with the ITRs from AAV2 and the capsid proteins from AAV5. We found that this construct transduces Rhesus macaque lungs to levels much higher than those achieved with AAV2. The increase in airway transduction of AAV5 over AAV2, estimated from recombinant gene expression, is ∼100-fold.
To take this one step forward, we and our collaborators conducted a nonlethal study in chimpanzees. We chose chimpanzees because they are the closest genetic relative to Homo sapiens. 20 We cloned the cDNA for firefly and Renilla luciferase 21 into AAV1 and AAV5, respectively, and used a dual reporter assay to assess gene transduction. 21 The results of the analysis performed on bronchial brushings showed much greater transduction with AAV1 than with AAV5 (as measured by luciferase activity). The same dual reporter assay was used on primary human airway cells grown in tissue culture; in these cells, AAV1 was ∼100-fold more effective in transduction than was AAV5.
Several investigators have shown that AAV6 is tropic for the airways 22 –24 and can penetrate airway mucus, a potential benefit for patients with CF. 25 For example, Halbert et al. 22 showed that AAV6 was highly effective in transducing large and small airways, estimating that AAV6 is between 15 and 74 times more efficient than AAV2. This result is comparable with the result we observed when we compared AAV5 with AAV219 but was less than what we observed for AAV1. 21 Thus, although AAV1 and AAV6 belong to the same clade (A) 17 for CF, AAV1 may be a better choice than AAV5 or AAV6.
Truncating CFTR to package AAV
AAV2 has a 4.7-kb packaging capacity, 26 which was just large enough to package the original tgAAV2-CFTR vector. 27 However, to improve the expression of CFTR the addition of new, more efficient promoters necessitated trimming of some of the CFTR coding sequences to allow it to be packaged within the size limit of AAV. To accomplish this, Sirninger et al. 28 designed a new AAV vector, Δ264 CFTR, missing the first four transmembrane segments. In subsequent studies, a rAAV2-CBA-Δ264 CFTR construct was packaged into an AAV5 capsid and delivered into the lung of a Rhesus monkey by bronchoscopy. 19
In this study, both robust gene transfer and transduction were detected. One notable observation was enhanced expression of endogenous wild-type (wt) CFTR in the monkey airways after infection with rAAV5-CBAA-Δ264 CFTR. 19 This enhanced expression of the endogenous CFTR was caused by transcomplementation, which occurred by the binding of certain truncation mutants such as Δ264 CFTR to both F508-del and wt CFTR, thereby increasing their trafficking and function. 28 We created a similar truncated version of AAV1Δ27-264-CFTR used in this study, which behaves through a similar mechanism. 29
Repeat dosing
In a previous study, 1 we tested the effectiveness of repeat dosing of our AAV1-based vector by spraying two doses of AAV1Δ27-264-CFTR (see below for a discussion of this vector) into the airways of four Rhesus monkeys at 0 and 30 days, followed by a single dose of green fluorescent protein (GFP) on day 60. Monkeys were sacrificed on day 90. No adverse events occurred, indicating that the AAV1 vector was safe. In this repeat-dosing study, our rAAV1-CFTR and rAAV1-GFP vectors were detectable in all lung sections and in the heart, liver, and spleen, suggesting that widespread gene transduction had occurred. This widespread transduction was expected because we had shown previously that AAV1 sprayed into the lung can be detected in the blood. 30
Interestingly, we noticed that AAV1 was able to transduce surface epithelial cells as well as basal cells, despite an increase in neutralizing antibody (Nab) and activated T cells. We concluded that AAV1 safely and effectively transduces monkey airway and basal cells, and that repeat dosing with AAV1-based vectors is achievable. However, we did notice that gene transfer was diminished by the third dose, potentially hampering the gene transduction achievable with repeat dosing.
The goal of this study was to provide a pathway for mitigating the ability of the immune system to reduce gene transfer and transduction with repeated dosing, and to determine the extent to which introduction of AAV1 into the airways can have the added benefit of transducing organs outside of the lung that are affected in CF, 31,32 particularly the liver and pancreas. For this study, we used Δ27-264 CFTR, which does not generate Cl− currents on its own, but binds to wt CFTR and enhances its trafficking and function (reviewed in Cebotaru and Guggino 33 ). Transcomplementation represents a novel combination of corrector and gene therapy.
METHODS
Viral particles
The AAV gene delivery vector AAV1-CBAA-Δ27-264 CFTR was the same as we used previously. 1 In brief, the vector contains a AAV serotype 1 capsid with inverted terminal repeats of AAV serotype 2. The expression cassette consisted of the CBAA transcription promoter, an intron, 21 the human form of the CFTR with the sequence encoding amino acids 27 to 264 deleted (Δ27-264 CFTR), 29 and a synthetic polyadenylation sequence. The GFP vector was rAAV-CBAA-GFP (UF11). 34 The vectors were manufactured at the Vector Core Laboratory, Powell Gene Therapy Center (PGTC), University of Florida, Gainesville. Titers are provided by the University of Florida using a dot-blot assay and secondarily checked using a secondary CFTR functional assay.
Study design
Juvenile Rhesus macaques (Supplementary Table S1) (five vector-treated, one control) were obtained from the Johns Hopkins University breeding colony, and housed according to Animal Care and Use Committee guidelines. All experimental protocols were reviewed and approved by the Animal Care and Use Committee, and complied with the standards associated with the ethical use of animals. The animals were preselected for a negative test for AAV1 Nabs. Two doses of 1013 vg (vector genomes) of AAV1-CBAA-Δ27-264 CFTR at 0 and 30 days, respectively, followed by a single dose of 0.8 × 1013 vg of AAV1-CBAA-GFP at day 60 were sprayed into the lungs through an endotracheal tube. 1 Monkeys were sacrificed on day 90 except for one monkey that was necropsied at 1 year. Control samples were obtained from an untreated animal.
Samples of blood and sera were taken before each vector administration. The animals were autopsied for detection of DNA and protein expression as well as pathologic analysis. Lungs were cut into regions as defined previously. 30 A portion of each lung region was flash frozen, and another was fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned and mounted on slides for analysis of vector transduction as discussed below.
Real-time DNA analysis
Quantitative polymerase chain reaction (PCR) with LightCycler (Applied Biosystems QuantStudio 6 flex) was used to test the various lung regions for CFTR- and GFP-specific DNA. Genomic DNA samples were prepared using a PureLink Genomic DNA Mini Kit (#K1820-02; Invitrogen). Vector genomes per microgram of lung DNA were measured using the qRT-PCR/Fast SYBR Green Master Mix (#A25742; Applied Biosystems) in genomic DNA obtained from various parts of the body (lung: trachea, bronchus, carina, cranial, middle, and caudal lobes; liver and pancreas).
Sample values were calculated using a standard curve of arbitrary fluorescence units plotted versus serial dilutions of plasmid DNA ranging from 200 to 0.0002 ng. The number of fluorescence units obtained from the untreated tissue samples was subtracted to obtain specific gene expression. The primer sequences used are as follows: CFTR-AAV-forward: GAC AGG GTG AAG CTC TTT CC and reverse: CTC CAT CAC TAG GGG TTC CT; GFP-AAV (UF11) forward: GTG CAG GAG AGA ACC ATC T and reverse: GCC ATT CTT TTA CTT GTC GGC.
Real-time mRNA analysis
CFTR mRNA expression was assessed as relative quantification method. The product was amplified from 300 ng cDNA. Quantitative PCR with LightCycler (Applied Biosystems QuantStudio 6 flex) was used to test the various regions for CFTR- and GFP-specific DNA. Total RNA was extracted from tissue using RNeasy Plus mini kit (Catalog No. 74134; Qiagen) following the manufacturer's instructions. Residual DNA contamination was avoided by performing the DNase digestion step during the RNA extraction. The RNA concentrations in the samples were measured using a NanoDrop 2000 Spectrophotometer.
The absorbance ratio 260 nm/280 nm was used to assess the purity of the samples, and ratios >1.9 were considered acceptable for quantitative reverse transcription PCR. AAV vector genomes were calculated using a standard curve of arbitrary fluorescence units plotted versus serial dilutions of plasmid DNA ranging from 0.002 to 200 ng. The primer sequences used are as follows: CFTR-forward: CTG GAG CAG GCA AGA CTT CA; CFTR and reverse: CTC CAT CAC TAG GGG TTC CT; GFP(UF11) forward: GTG CAG GAG AGA ACC ATC T and reverse: GCC ATT CTT TTA CTT GTC GGC.
Western blotting
Samples (20 mg) of flash-frozen lung tissue were cryopulverized with liquid nitrogen using a mortar and pestle, and then suspended in 1 mL of NP40 lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM ethylenediamine tetraacetic acid, 2 mM EGTA, 1.0% Nonidet P40) with complete protease inhibitor (Roche Diagnostics, Mannheim, Germany). Lysate samples were allowed to incubate on a rotator for 1 h at 4°C. Supernatants were collected by centrifugation at 15,000 g for 10 min. Protein concentrations were determined using a Bio-Rad protein assay dye reagent (#5000006), Bradford method-based colorimetric assay.
The supernatants (straight samples, 30–50 μg of proteins) were subjected to 4–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE, #4561084; Bio-Rad) and immunoblotting, followed by enhanced chemiluminescence (SuperSignal West Dura Extended Duration Substrate, Catalog No. 34075; Thermo Scientific). The chemiluminescent signal on the polyvinylidene difluoride membrane (Bio-Rad) was directly captured by an Amersham imager 600, GE system with a cooled CCD camera. CFTR protein was detected with monoclonal antihuman CFTR (217; 1:1,000) antibody provided by Dr. J. Riordan, Department of Biochemistry and Biophysics and Cystic Fibrosis Center of North Carolina. Vinculin, used as a loading control, was detected with antivinculin antibody.
Confocal microscopy
Paraformaldehyde-fixed and paraffin-embedded lung, liver, or pancreatic tissue sections were prepared on glass slides, deparaffinized with xylene, and rehydrated with a descending ethanol series into phosphate-buffered saline (PBS). Antigen retrieval was done by heat mediation in a citrate buffer (#S1699; Dako), and then sections were blocked with 4% serum for 1 h at 25°C. Sections were permeabilized with PBS containing 1% bovine serum albumin and 0.2% Triton X-100.
After blocking, sections were incubated with 5 μg/mL primary antibody in blocking buffer for 16 h at 4°C, and then processed for secondary antibody. After antibody staining and washes, slides were mounted on glass slides using Fluoro-gel (#17985-10; EMS) and coverslipped. Images were captured using a Zeiss LSM 880 confocal microscope with a 63 × oil-immersion objective (NIH Grant #S10OD016374). For quantification, intensity measurements were made using Zen 2012 software.
Reagents
Antibodies for Western blotting: monoclonal antibodies against CFTR (217) (from Dr. J. Riordan, Department of Biochemistry and Biophysics and Cystic Fibrosis Center of North Carolina), GFP (#11814460001; Roche), and vinculin (Sc#73614; Santa Cruz Biotechnology). For confocal studies: monoclonal antibodies against CFTR (596), GFP (#11814460001; Roche) and cytokeratin 5 (#ab52635; Abcam); antimouse-AlexaFluor 594 (#A11032; Life Technologies), antimouse AlexaFluor 488 (#A11001; Life Technologies), antirabbit AlexaFluor 488 (#A11008; Life Technologies), and antirabbit AlexaFluor 594 (#A11012; Life Technologies).
Nab experiments
Nabs were evaluated using AAV1-LacZ as reported previously. 1,35 The Nab titer was reported as the highest serum dilution that inhibited AAV1LacZ transduction by 50%, when compared with its own AAV1 vector-positive/serum-negative control.
ELISPOT assays
T cell responses to AAV1 were determined by evaluating the release of INF-γ after stimulation of T cells with transgene antigens from AAV using previously described protocols. 35
RESULTS
To begin to overcome the challenges associated with repeat dosing, we gave two male and three female monkeys each a total of 2 × 1013 particles of AAV1-CBA-Δ27-264 CFTR in two doses and 0.8 × 1013 particles in one final dose of AAV1-CBA-GFP, as we had done previously. 1 The difference here is that we treated the animals with methylprednisolone succinate 2 mg/kg intramuscular (IM) immediately before and ∼24 h after each of the three viral instillations.
After the final vector administration, the animals were followed for 4 weeks, and then four animals were autopsied; six lung regions were dissected from each monkey. One monkey was kept alive for evaluation of the duration of expression of the transgene as well as any possible long-term adverse events. The final monkey in this experimental group was necropsied after 12 months. One animal was not treated with any virus and served as a negative control.
It is well known that AAV vectors do not result in rapid transduction in nondividing cells, 36 requiring a period of time before maximum transduction is realized after vector instillation. For example, Chao et al. 37 injected an AAV1-factor IX vector into the muscle of mice, and found that the levels of factor IX in the blood reached their peak in ∼4 weeks. Although not directly comparable with lung delivery, we chose a 30-day interval between repeated instillations here, and in our previous study 1 to give the vector enough time to induce expression of CFTR or GFP.
The application of methylprednisolone in this study was inspired by a clinical study of AAV1-induced transduction of the alpha-sacroglycan gene, 38 where a 3-day course of intravenous (IV) methylprednisolone was administered IV. The question regarding whether the short-term immunotherapy increased gene transduction in that study was left open. The notion we are addressing here was that short-term immunotherapy administered IM would improve gene transduction.
Clinical parameters
All monkeys tolerated the vector instillation, methylprednisolone succinate, and anesthetics without adverse sequelae and remained healthy throughout the experimental protocol. The monkey (#22) kept for 1 year also remained without clinical sequelae throughout that time. The complete results for the clinical chemistry analyses are provided in Supplementary Tables 2–4. All the clinical values remained within the normal range. Thus, as we showed previously, 1 there were no clinical observations attributable to vector administration or methylprednisolone succinate treatment (Supplementary Tables S2–S4 and Supplementary Figs. S2 and S3).
Gene transfer
Samples were taken from six different lung regions: the trachea, carina, bronchus, cranial, middle, and caudal lobes, as done previously. 1 The trachea, carina, and bronchial tissues were selected to highlight the conducting airways, and the cranial, middle, and caudal lobes were selected to focus on alveolar regions. Figure 1 shows the vector genomes measured in each monkey and in each lung region at necropsy. Importantly, AAV1-CBA-Δ27-264-CFTR and -GFP vector genomes were detectable at ≥3 × 107 vg/μg genomic DNA in all lung tissues of all five vector-treated animals.

Distribution of AAV1-CBA-Δ27-264 CFTR and AAV1-CBA-GFP vector genomes in lung tissue. Vector-specific genomes per microgram of lung DNA were measured by real-time DNA PCR in the trachea, bronchus, carina, cranial, middle, and caudal regions of five Rhesus monkeys and one untreated control. The data from the untreated control were used as a tissue blank and subtracted from each of the experimental samples.
These data are remarkable because we detected a 10-fold higher vector genomes per microgram of AAV1-CBA-Δ27-264 CFTR and a 40-fold higher vector genomes per microgram of AAV1-CBA-GFP in the methylprednisolone succinate-treated animals than we previously found in the AAV1-infected monkeys in the absence of methylprednisolone succinate (Supplementary Fig. S1). 1 What is even more notable is that despite its being applied as a third dose, the vector genomes per microgram of genomic DNA were similar to those of AAV1-CBA-Δ27-264 CFTR. We had previously noted a 10-fold decrement in the vector genomes per microgram of genomic DNA when compared with that of AAV1-CBA-Δ27-264 CFTR. 1 Also notable is that vector genomes were still detectable in the monkey necropsied after 1 year.
Figure 1 also shows that AAV1-CBA-Δ27-264 CFTR and -GFP could be detected in the liver and pancreas after lung delivery. The detected levels in the liver were ∼1 log lower than those found in the lung and ∼2 logs lower in the pancreas than in the lung. These results are not surprising because we have shown previously 30 that after lung delivery, AAV1 vector genomes readily enter the blood, peaking at ∼3 h after treatment. By 48 h, the vector genomes in the blood were no longer detectable, suggesting (as we show here) that the vector genomes had entered the organs.
AAV vector transduction
To assess vector transduction, we measured vector-specific mRNA expression by using primers specific for AAV1-CBAΔ27-264 CFTR and -GFP (see Methods section). Note that in all lung tissues of all the Rhesus monkeys, mRNA expression generated by AAV1-CBA-Δ27-264 CFTR and rAAV1-CBA-GFP was detected (Fig. 2). The mRNA expression generated by CFTR containing AAV1 was ∼10 × higher than that observed for GFP.
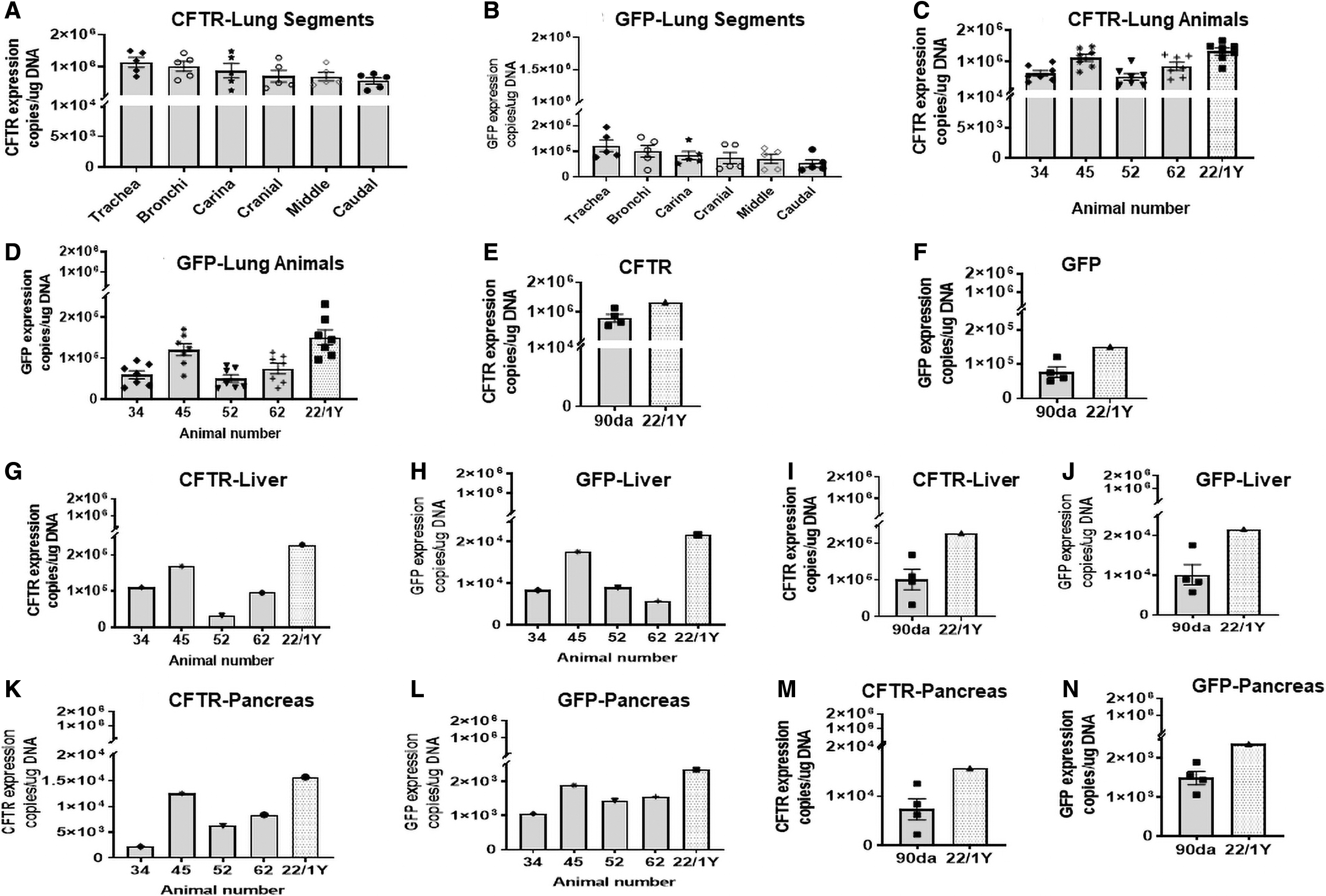
CFTR or GFP mRNA expression in lung tissue. Copies per microgram of lung DNA were measured by real-time DNA PCR in the trachea, bronchus, carina, cranial, middle, and caudal regions of five animals and one untreated control.
We also detected vector-specific mRNA expression in the liver and pancreas for CFTR and GFP. Levels in the pancreas were lower than those in the liver but still well above the values measured in the animal not treated with AAV1 vectors. Remarkably, in lung, liver, and pancreas the expression of both CFTR and GFP was sustained for 1 year at levels comparable with those observed in the animals sacrificed at 90 days.
Transgene expression
We measured the protein levels of both CFTR and GFP in lung sections. Figure 3A and B shows that CFTR is expressed in the normal lung that has not been treated with AAV1 vector-containing CFTR, and the levels are increased in the treated animals. Likewise, GFP protein expression (Fig. 3C, D) was also evident in the methylprednisolone succinate-treated CFTR-transduced lungs. These data are consistent with the mRNA data presented above, indicating that the AAV1 vectors express mRNA, which is indeed translated into protein.

CFTR expression in whole lung:
The vector genomes and mRNA and protein expression discussed in the previous sections were measured in tissue samples. However, since the lung, liver, and pancreas are complex organs with multiple cell types in which CFTR is expressed, 32,39 –41 we next performed immunofluorescent staining for CFTR and GFP to assess more precisely the localization of the protein expression from the transgenes.
Figure 3E and F shows the confocal images for CFTR. All conducting airways, including the trachea, carina, and bronchus, showed higher fluorescent staining for CFTR than did those from the control animal, indicating that CFTR had been expressed from the transgene. Monkeys contain wt CFTR in their airways; thus, staining for CFTR was detected in the untreated lungs at levels lower than those observed for the treated monkeys. We have shown previously that AAV1-CBA-Δ27-264 CFTR can increase the protein levels of wt CFTR through transcomplementation in both monkey lungs and cells in tissue culture. 19,29,42 Thus, the increased expression of CFTR noted in the treated animals is most likely the result of increased protein levels of wt monkey CFTR induced by transcomplementation through AAV1-CBA-Δ27-264 CFTR.
Likewise, GFP protein expression was also detected in the trachea, carina, and bronchi of all animals (Fig. 3G, H). As can be seen in the micrographs, GFP staining appears more uniform throughout the tissues compared with CFTR. Because CFTR is a membrane protein associated with the apical membrane in airway cells, 43 we would expect a localized staining compared with GFP.
In the conducting airways, basal cells function to rejuvenate the surface epithelium of the airway. 44 These cells characteristically express keratin 5/14. 45,46 To identify whether AAV1-induced transduction was present in the basal cells, we costained the tissue samples with an antibody detecting keratin 5. As we showed previously, 1 the basement membrane of the trachea, carina, and bronchus all contained K5-expressing cells, with positively stained basal cells showing a higher expression of CFTR in the treated monkeys than in the nontreated monkey (Fig. 3E, F). Likewise, we also observed positive expression of GFP within the K5-positive cells when compared with untreated cells (Fig. 3G, H).
The question then was whether there was still detectable expression of CFTR and GFP after a year. Figure 3 clearly shows that the expression of both CFTR and GFP in the 1 year animal in both the surface and basal cells persisted as long as 1 year.
To take this one step further, we asked the extent to which enhanced expression of CFTR and GFP is present in the liver and lungs of infected+ monkeys. For this purpose, we focused on detecting CFTR and GFP in the hepatic duct (Fig. 4) and pancreatic duct cells and pancreatic acinar cells (Fig. 5). We identified epithelial cells in the hepatic ducts by counterstaining with CK7 (Supplementary Fig. S4), a marker of epithelial cells, 47 and acinar cells using chymotrypsin (Supplementary Fig. 4).

CFTR or GFP expression in hepatic duct epithelial cells:

CFTR or GFP expression in pancreatic duct epithelial cells:
The figures demonstrate transduction of the surface epithelia of the hepatic and pancreatic ducts and pancreatic acinar cells after vector administration to the lung. Expression of both CFTR and GFP in the cells lining the ducts of both liver and pancreas and in the pancreatic acinar cells indicates that it may be possible after lung administration of AAV1 to rescue the pathology associated with CF in these organs as well as that in the lungs.
Neutralizing antibodies
The results of an evaluation of anti-AAV antibody are shown in Fig. 6. We selected animals for this study that had low-serum Nab titers (<5 reciprocal dilutions) before the experiment. Consistent with vector instillation, the serum levels increased dramatically in all the monkeys by 60 to 90 days post-treatment. Importantly, the Nab titer of the monkey treated long term, animal #22, declined nearly to basal levels during the year after vector instillation.

Neutralizing antibody: the neutralizing antibody titer increased significantly after the second vector instillation and continued to increase thereafter.
Immune responses to AAV capsid components
To quantitate the T cell responses to the AAV1 capsid proteins and CFTR, we performed interferon-γ ELISPOT assays on peripheral blood mononuclear cells using previously described protocols. 35 What is notable (Fig. 6B–E) is that the ELISPOT response was low in all animals tested, perhaps because of the methylprednisolone succinate treatment. However, we did detect a positive response to the capsid components, particularly at 90 days. Also notable was the decline in the interferon levels in monkey #22 at 365 days (Supplementary Table S5). On the contrary, we showed previously 1 that there was no response to CFTR, suggesting that our truncated form of CFTR is not recognized by the immune system.
DISCUSSION
CF gene therapy has been hampered by low expression of the transgene in appropriate locations, resulting from a series of barriers that prevent viral transduction. 48 –51 We show here, however, that it is possible to achieve transduction of the lung, liver, and pancreatic duct epithelial cells after repeated dosing of AAV1-based vectors, particularly when administered in conjunction with short-term methylprednisolone succinate treatment.
The historically low expression of the transgene has been attributed to several factors. For example, it is known that AAV2 does not efficiently transduce the apical surface airway cells, because of the lack of AAV2 surface receptors in airway cells. 52 Beyond the lack of receptors, particle entry of AAV2 through the apical route is impaired, and trafficking to the nucleus is not efficient but can be improved by using proteasome inhibitors. 53,54 On the contrary, Liu et al. have 52 shown that AAV1 is highly efficient in transducing ferret airway epithelial cells grown as polarized monolayers and shows less sensitivity to proteasome inhibitors than does AAV2, suggesting that both the presence of apical receptors and apical entry mechanisms are more efficient.
Interestingly, Yan et al. 55 have identified a secreted factor in the airway surface liquid of ferrets, which strongly inhibits rAAV1 and increases its sensitivity to proteasome inhibition by ≈1,600-fold. Along the same lines, Virella-Lowell et al. 56 have observed that bronchioalveolar lavage fluid (BAL) isolated from CF patients can significantly inhibit AAV transduction. They showed that the effect of BAL can be mitigated by adding alpha1-antitrypsin. Their data led them to conclude that factors associated with inflammation may be the culprits here.
Interestingly, when we applied AAV1-based vectors to both chimpanzees and Rhesus monkeys, we observed better transduction than with AAV2 or AAV5 without using proteasome inhibitors. 21,30 One possible explanation for the difference is that the ferret model may be more like the CF lung. 57
The immune response
One of the largest barriers to gene therapy for CF is the presence of anti-AAV Nabs. These Nabs are present in people previously exposed to AAV. 49 We have selected monkeys for these experiments who have low levels of endogenous anti-AAV1 antibodies. As expected, our experimental monkeys developed Nabs against the AAV1 capsid. AAV1 seroconversion occurs at ∼60 days, equivalent to 30 days after we applied the second dose of virus. Here in this study, the time to seroconversion and the magnitude of the Nab response were fairly consistent with our previous repeat-dosing study. 1 These data indicate that treating the animals with methylprednisolone succinate did not affect the development of Nabs. Importantly, the titer of the Nab diminished in the one animal examined after a year.
Because of the similarity of the AAV sequence among AAV serotypes, switching between them to avoid the Nab response is not a feasible long-term approach. On the contrary, short-term immunosuppression may be feasible, given that treatment of CF patients with AAV would be periodic. One approach that has been tried to overcome the effect of Nabs is B cell depletion with drugs already used in patients such as rituximab and sirolimus; B cell ablation with rituximab before AAV has been found to reduce the responsiveness to both capsid and transgene in a subject with Pompe's disease. 58
However, one of the most common manifestations of CF in patients is chronic lung infections. 59 Thus, aggressive immunosuppression to reduce the Nab response to AAV may not be clinically appropriate for CF patients. However, we have shown here that milder immunosuppression using methylprednisolone succinate might be a feasible approach for boosting vector transduction and increasing the longevity of the therapeutic effect, but human clinical trials will be needed to establish methylprednisolone succinate's feasibility conclusively.
The negative effects of T cells on the duration of gene transduction by AAV vectors were identified and have been studied extensively. 49 For example, Manno et al. 57 infused a recombinant AAV2 vector, including human Factor IX, into the liver of several human subjects and were able to achieve therapeutic levels of Factor IX without overt toxicity, but the levels were only sustained for 8 weeks and were then followed by a gradual decline. Concomitant with the decrease in Factor IX was a transient, self-resolving, increase in liver transaminase, suggestive of destruction of the transduced liver cells induced by CD8+ T cells. 60 Hui et al. 61 mapped the epitopes on AAV1 responsible for the T cell response and found them to be conserved among AAV serotypes.
In our previous study 1 in which three doses of AAV1 were administered in the absence of methylprednisolone succinate, we observed a positive ELISPOT response to AAV1 peptides, particularly after the third dose, which varied greatly among the animals studied. Although the numbers of animals are small, our ELISPOT assay here showed a lower response to AAV1 in the methylprednisolone succinate-treated animals than in our previous study.
This result highlights the potential role of CD8+ T cells in limiting the duration of transgene expression. Thus, it is likely that the higher levels of vector genomes, the lack of a dramatic decrease in GFP- versus CFTR-containing viral genomes after a third dose of AAV1, and the relatively persistent transduction with the CFTR vector in the monkey studied for 1 year are caused by a subdued T cell response in the methylprednisolone succinate-treated monkeys.
The need to ablate the antibody response is only one of the multiple challenges to the success of repeat dosing by AAV. As mentioned above, there is evidence that once AAV transduces a cell, CD8+ T cells can limit the duration of the transduction. On the contrary, CF patients with bronchiolitis, bronchial hyper-reactivity, aspergillosis, and mild-to-moderate obstructive pulmonary disease are already treated with prednisolone. 62,63 The recommendation followed in patients, as we have suggested, is to provide the lowest effective dose (estimated using Rhesus monkeys) and a short duration to minimize the risk of side effects. 63
Spread to other organs (liver and pancreatic basal cells)
When introduced into the airways, AAV1 can be detected in airway surface cells and basal cells by confocal microscopy, indicating that transduction has occurred in these locations. AAV1 penetrates the airway and can be readily detected in the blood for many hours after instillation. 30 Thus, it is not surprising to detect vector genomes in the liver and pancreas, as we show here. Consistent with the virus originating from airway delivery, the number of vector genomes detected in liver was ∼10-fold lower than that found in the lungs.
We saw a further 10-fold decrease within the pancreas. There can be several reasons for this observed difference between the liver and pancreas: one major reason may be vector accessibility, given the ample blood supply to the liver versus that to the pancreas. The other reason may be related to tropism. For example, it is well known that AAV8 and AAV9 transduce the liver quite efficiently, 64 when compared with AAV2 or AAV5. In human clinical trials in which liver transduction of Factor IX was the goal, AAV8 was used. 57
Others have shown that AAV1 and AAV5 will also transduce the liver and can be applied successfully with repeat dosing. 65 Quirin et al. have shown that both the serotype and the method of delivery are important for successful AAV-mediated transduction of the pancreas. 66 They found that AAV6, when delivered through retrograde pancreatic ductal delivery, is most efficient at transducing acinar, epithelial, and stromal cells when compared with systems delivery or other serotypes such as AAV8 and AAV9. Loiler et al. have demonstrated that AAV1 can indeed transduce the murine pancreas. 67
Although these aforementioned studies on liver and pancreatic transduction point to the need for head-to-head studies of the efficiency of various serotypes, one of the goals here was to determine whether a single serotype might be sufficient to transduce lung, liver, and pancreas with one instillation. To begin to address this question, we determined whether the vector genomes instilled through the lung could transduce liver and pancreas. We show here that there was indeed mRNA expression from the viral vectors for both CFTR and GFP. We also asked whether we could get expression of CFTR and GFP in cells that are normally affected by CF, namely the hepatic and pancreatic ducts. 31,68
Our results using confocal microscopy did demonstrate transgenic expression of both CFTR and GFP in both locations. The monkeys we used in this study were normal, so functional correction could not be validated. The challenge for the future will be to determine whether the expression levels we observed in the lungs, pancreas, and liver will provide therapeutic restoration of the functional defects associated with CF.
Long-term expression of AAV
Recombinant AAV is known to persist in cells as an episome, extending its persistence within cells. 69 This persistence was dramatically demonstrated by studies in which rAAV containing Factor IX was instilled into Rhesus macaques, 70 and expression from the transgene was detected in some monkeys 5 years post-instillation. Interestingly, Nabs did develop in the experimental animals in that study, but they were eradicated by immunosuppression induced with a combination of rituximab and cyclosporine. Thus, it is not unusual for us to detect vector genomes and transduction in one of our monkeys necropsied after a year.
Although we studied only one animal long term, it is possible that the short-term use of methylprednisolone succinate was one factor contributing to maintaining the levels of vector genomes and transgene expression at detectable levels after a year. More experiments are needed to verify our observations definitively. Airway cells are known for their long lifetimes and slow turnover, 71 –73 which in some cell types can take as long as a year. Also, the observation that AAV1 can transduce lung basal cells makes it even more likely that AAV gene therapy involving the lungs will persist over a long period.
In this study, we challenged the monkeys with repeated dosing with AAV1 at 30 and 60 days after the first dose and attempted to suppress the immune response with methylprednisolone succinate. Perhaps given the persistence of AAV1 transduction, this time period could be extended. Also, considering the slow increase in Nabs that we noted during this short-duration three-dose regimen, it may be feasible to achieve better transduction by using both methylprednisolone succinate and increasing the time between doses.
In conclusion, we show here that transduction occurs after repeated dosing with an AAV1-associated vector, and can be facilitated by using methylprednisolone succinate before and shortly after the instillation of the vector. We do appreciate that the studies reported on here from Rhesus macaques are from small numbers of animals, and thus should be taken as a road forward for future studies. However, our data suggest that with the proper intervals between doses to minimize overloading of the immune system with virus, coupled with some form of short-term treatment to reduce inflammation, may make repeat dosing with a therapeutic gene-based vector feasible for treating CF patients.
Footnotes
AUTHORs' CONTRIBUTIONS
M.K.Y., V.T., and C.V.C. contributed to laboratory investigation, methodology, and data analysis; W.B.G. contributed to data analysis, conceptual design, and article preparation; L.C. contributed to data analysis, overall administration, funding procurement conceptual design, and article preparation.
ACKNOWLEDGMENTS
We give special thanks to Dr. Robert Adams for providing the monkeys, anesthetizing them and drawing blood samples, and instilling the virus into the monkeys. We thank the Comparison Medicine Primate Facilities at JHU for the toxicology services provided for this study. We also acknowledge Dr. Jessica A. Chichester, PhD, Director of the Immunology Core, Gene Therapy Program, Perelman School of Medicine, University of Pennsylvania, for the neutralizing antibody and ELISPOT assays that were conducted by the Core. In addition, we acknowledge Mark Potter, PhD, of the Vector Core Laboratory, Powell Gene Therapy Center (PGTC), University of Florida, Gainesville, for manufacturing the virus and Deborah McClellan, PhD, for editorial assistance.
AUTHOR DISCLOSURE
L.C. has a license agreement with RA Capital, and W.B.G. is a consultant for Vertex and Sarepta Pharmaceuticals in areas that are unrelated to this study.
FUNDING INFORMATION
The work was funded by NIH grant R01 HL122267 to Liudmila Cebotaru and William B. Guggino.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.