
Abstract
Adeno-associated virus (AAV) vectors are proving to be clinically transformative tools in the treatment of monogenic genetic disease. Rapid ongoing development of this technology promises to not only increase the number of monogenic disorders amenable to this approach but also to bring diseases with complex multigenic and nongenetic etiologies within therapeutic reach. In this study, we explore the broader paradigm of converting the liver into a biofactory for systemic output of therapeutic molecules using AAV-mediated delivery of the endonuclease DNaseI as an exemplar. DNaseI can clear neutrophil extracellular traps (NETs), which are nuclear-protein structures possessing antimicrobial action, also involved in the pathophysiology of clinically troubling immune-mediated diseases. However, a translational challenge is short half-life of the enzyme in vivo (<5 h). This study demonstrates that AAV-mediated liver-targeted gene transfer stably induces serum DNaseI activity to >190-fold above physiological levels. In lupus-prone mice (NZBWF1), the activity was maintained for longer than 6 months, the latest time point tested, and resulted in a clear functional effect with reduced renal presence of neutrophils, NETs, IgG, and complement C3. However, treatment in this complex disease model did not extend lifespan, improve serological endpoints, or preserve renal function, indicating there are elements of pathophysiology not accessible to DNaseI in the NZBWF1 model. We conclude that a translational solution to the challenge of short half-life of DNaseI is AAV-mediated gene delivery and that this may be efficacious in treating disease where NETs are a dominant pathological mechanism.
INTRODUCTION
The clinical advent of gene transfer vectors derived from adeno-associated virus (AAV) is proving clinically transformative. 1 The technology has been applied successfully to produce long-term phenotypic correction in patients with monogenic disease-causing blindness, 2 –4 coagulation defects, 5 –7 and neuromuscular disorders. 8 In the case of gene therapy for severe spinal muscular atrophy, which is commonly fatal in infancy, a single infusion of vector can prevent disease progression and place infants on a normal developmental trajectory with potentially lifelong effect. 8 In addition, hemophilia patients treated with AAV vector continue to see benefits more than 10 years postvector treatment. 6,9
Due to limitations in AAV vector transduction efficiencies, early clinical trials sought to target cells located in a restricted space (eye) or where only a small proportion of the target organ (liver) required gene modification. However, the vector system continues to undergo significant improvements through the discovery or bioengineering of novel capsids with increasing efficiencies in gene delivery to target tissues. Using the human liver as an example, the 2–11% therapeutic increase in factor IX levels seen in an early hemophilia trial using vector packaged in AAV serotype 8 capsid 9 is likely to have transduced only a relatively small proportion of hepatocytes. 10
However, hepatotropic capsids have since been identified with >10-fold higher transduction efficiencies. 10 –13 Progression along this trajectory in concert with continued confirmation of vector safety brings into reach the prospect to treat genetic disease that requires higher frequencies of gene modification. In addition, it permits consideration of repurposing the liver to output transgene products that will correct not only simple monogenic diseases but also those with complex etiologies.
To this end, our laboratory has sought to harness the power of AAV-mediated gene delivery to convert the liver into a biofactory for systemic delivery of therapeutic proteins. In a previous proof-in-principle study, we have shown that this strategy can be used to increase plasma levels of the complement regulatory protein factor I to downregulate the proinflammatory activity of the alternative pathway (AP 14 ). Hyperactivity of the AP has been associated with autoimmune and/or degenerative disease caused by complement dysregulation. 15 In some instances, such as with age-related macular degeneration, disease can take many decades to manifest. A single intervention with AAV might provide a long-term solution to the challenge of permanently rebalancing complement activity. 16
There have been a growing number of reports supporting the concept that the endonuclease DNaseI might be used therapeutically to favorably alter the pathophysiology of multiple different conditions. 17 The enzyme is part of a family of endonucleases that catalyze the hydrolysis of phosphodiester bonds in double- and single-stranded DNA. 18 DNaseI has been clinically applied in cystic fibrosis patients to reduce airway viscosity caused by DNA released from dying cells. 19 In preclinical models, DNaseI has also been shown to clear neutrophil extracellular traps (NETs), which are DNA-protein structures released by neutrophils to combat infection. 20
Importantly, aberrant NET formation has been implicated in a number of pathophysiological contexts. DNaseI has been therapeutically applied in preclinical models to the prevention of thrombosis, 21 ischemia reperfusion injury, 22,23 atherosclerosis, 24 and cancer. 25 –28 It has also been hypothesized that DNaseI might clear nucleosomes believed to be the antigenic driver of antinuclear antibodies in systemic lupus erythematosus (SLE). 29 However, a considerable obstacle to the translation of DNaseI has been its short activity half-life in vivo (<5 h 30 ), caused, in part, by G-actin inhibition. 31
This study tested the potential of AAV vectors to systemically deliver recombinant DNaseI. We hypothesized that transduction of the liver would induce a high and persistent supply of enzyme through the circulatory system. We show that an AAV vector encoding DNaseI elevates serum enzyme activity to supraphysiological levels and that levels are maintained beyond the completion of the experiment at 6 months. Application of AAV-DNaseI in a lupus-prone mouse model (NZBWF1) decreases renal presence of neutrophils, NETs, complement, and IgG. However, this was insufficient in this complex disease model to preserve renal function or interrupt other serological endpoints. The implications of these findings are discussed.
MATERIALS AND METHODS
Mouse studies
All animal care and experimental procedures were evaluated and approved by the CMRI/CHW Animal Care and Ethics Committee. C57BL/6 mice were obtained from the Animal Resource Centre (Caningvale, Western Australia). NZBWF1 mice used in this study were bred in-house from breeding pairs obtained from The Jackson Laboratories (Bar Harbor, ME). Mice were housed in standard boxes and received normal food and water ad libitum for the duration of the experiments. Animals were injected through the intraperitoneal (i.p.) route at 8–12 weeks of age with 100 μL of the indicated vector after dilution in saline. NZBWF1 mice were sacrificed when moribund while C57BL/6 mice were sacrificed at the indicated time points through the approved method of cardiac puncture.
Blood was collected using either the tail vein nicking or cardiac puncture (terminal) method into uncoated collection tubes (Becton Dickinson). For cardiac puncture, mice were anaesthetized by isoflurane inhalation and cardiac puncture performed with a 25-gauge needle. Blood was left to clot for 30–40 min at room temperature, and then centrifuged at 3,000 g for 5 min. The serum was then taken and centrifuged again at 20,000 g for 5 min to eliminate any residual red blood cells. Serum was aliquoted and stored at −80°C.
Urine was collected for analysis of protein and creatinine levels. Mice were placed in metabolic cages lined with Whatman paper for 24 h. The filter papers with urine were then collected, dried overnight, and stored at 4°C. When required for analysis, 10 circular holes (∼3 mm in diameter) were punched into urine-stained areas on the filter paper and dissolved in 300 μL of water for ∼2 h at room temperature. The tubes were centrifuged for 1–2 min at 4,000 g and the supernatant collected for biochemical analyses. Creatinine and protein analysis were performed on urine and serum samples by The Department of Pathology, Westmead Children's Hospital.
Plasmid constructs and vector production
Vectors were produced using standard molecular biological techniques. The cDNA sequences encoding murine DNaseI or a hyperactive and salt-resistant variant of this enzyme were kindly provided by Robert Lazarus (Genentech). The natural leader sequence for DNaseI was replaced with the leader sequence from tissue plasminogen activator. Also, the variant DNaseI contained two amino acid changes (E35R; A136F, numbering as per wild-type DNaseI). Coding sequences were codon optimized for mammalian expression.
Sequences encoding either DNaseI or green fluorescent protein (GFP) were subcloned into an AAV construct under the transcriptional control of a liver-specific promoter 32 consisting of the mini human alpha-1-antitrypsin promoter (minhAAT), a single copy of the mini Apolipoprotein E enhancer (minApoE), and the simian virus 40 (SV40) intron.
The expression cassette also contains the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and a bovine polyadenylation signal. A stuffer sequence was included downstream of the polyadenylation sequence such that when the expression cassette was flanked by the AAV serotype 2 inverted terminal repeats (ITRs), the vector genome length would approximate wild-type virus (4.7 kb). All constructs were validated using Sanger sequencing. The AAV-GFP construct has been previously reported. 33
AAV vectors were packaged into AAV serotype 834 by triple transfection of HEK-293 cells. 35 HEK-293 cells were obtained from ATCC and cultured in Dulbecco's medium Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (complete DMEM) and passaged using trypsin.
Briefly, HEK-293 cells (American Type Culture Collection) were cultured in plastic culture dishes with complete medium (DMEM containing 10% FBS) and were passaged using trypsin. Cells transfected with packaging constructs were collected 48 h post-transfection and subjected to four freeze/thaw cycles in a dry ice/100% ethanol bath and centrifuged at 3,000 g for 15 min. Supernatant was treated with benzonase at 50 U/mL at 37°C for 30 min and vector was precipitated using ammonium sulfate, purified using CsCl purification, dialyzed against 5% glycerol in phosphate-buffered saline (PBS) (+/+), and stored at −80°C. The final viral titer was assigned by quantitative PCR (qPCR). 36
DNaseI activity assay
DNaseI activity was analyzed by radial enzyme diffusion (RED) assay as previously described. 30 Briefly, a DNA-agarose solution was prepared by adding 15 mL of RED buffer [0.05M Tris-HCl (pH 7.2), 0.05 M MgCl2, 0.05 M CaCl2, and 0.15 M NaCl] to 20 mL water and agarose was slowly dissolved into this solution with 2 μL of RedSafe nucleic acid staining solution (iNtRON biotechnology) to make a 1% (w/v) agarose gel. Once the solution cooled to ∼60°C, 5 mL of 2 mg/mL calf thymus DNA (D1501; Sigma-Aldrich, St. Louis, MO) was added and the solution poured into 15 cm round cell culture dish plates (Thermofisher). After the DNA-agarose solution was set, 1 mm diameter wells were cut into the gel using a glass pipette and suction apparatus.
Samples were diluted appropriately in RED buffer and 2 μL was added into each well. Double dilutions of DNaseI standards (Sigma) were also prepared in RED buffer from 1 μg/mL to 15 ng/mL and loaded at 2 μL per well. The gels were incubated overnight at 37°C, then overlayed with 0.05 M EDTA, and imaged under UV light on the Flour ChemTM 5500 (Alpha Innotch.). For analyses, the diameter of the halo around each well, representative of the DNA digested, was measured. A standard curve generated using the DNaseI standard was used to calculate DNaseI concentrations.
Quantitation of liver vector copy number and mRNA transcripts
Liver was snap frozen in liquid nitrogen and stored at −80°C for later assay. DNA was extracted using proteinase K digestion and phenol:chloroform extraction as previously described. 36 RNA was extracted using the Trizol® plus RNA purification kit (Ambion) following the manufacturer's instructions. DNA and RNA concentrations were determined by spectrophotometry using a Nanodrop 1000 (Thermo Fisher Scientific, Wilmington).
Total RNA was reversed transcribed into cDNA using the SuperScript® III Reverse Transcriptase kit (Invitrogen) according to the manufacturer's instructions. qPCRs were performed using a RotorGene 6000 (QIAGEN). Vector copy number was calculated from genomic DNA using primers specific to the WPRE region on the vector and was normalized to GAPDH levels quantified using SYBR Green (Takara). For WPRE mRNA transcript levels, cDNA samples using target-specific primers were normalized against β-actin levels by qPCR Logan. 37
Western blot
Western blot was performed on serum samples to detect murine DNaseI. After transfer, membranes were blocked with 5% milk/1 × PBST for 1 h and then incubated with polyclonal rabbit anti-mouse DNaseI (PA522017, dilution; Thermofisher) in 1% milk/1 × PBST overnight at 4°C, followed by washing and incubation with goat anti-rabbit secondary antibody (sc-2004, 1:5,000; Santa Cruz Biotechnology) in 5% Milk/1 × PBST for 1 h at 4°C. Signals were detected using SupersignalTM West femto chemiluminescent substrate (Thermofisher) and visualized on the Fujifilm LAS4000 (Berthold). Blots were stripped using a solution containing glycine (25 mM), SDS (1.5% SDS w/v) dissolved in distilled water, washed and probed using antibodies to vinculin (V9131, dilution; Sigma-Aldrich), and imaged.
Serum immune complex enzyme linked immunosorbent assay
Serum immune complexes were assayed as described previously. 30 Briefly, Maxisorb Nunc Plates (Thermofisher) were coated overnight with 2 μg/mL of bovine conglutinin in coating buffer [0.0125 M sodium borate, 0.0125 M boric acid, and 0.01 M CaCl2 (pH 8.2)] at 4°C. Plates were then washed 3 times with 200 μL of wash buffer [0.0125 M sodium borate, 0.0125 M boric acid, 0.01 M CaCl2, and 0.05% (v/v) Tween-20 (pH 8.2)] and blocked with 200 μL of 2% (w/v) bovine serum albumin (BSA) in coating buffer at room temperature for 2 h.
After washing, plates were incubated with serum diluted 1 in 130 in wash buffer for 1 h at room temperature. Plates were washed 4 times and incubated for 1 h at room temperature with goat anti-mouse IgG-AP (A3562; Sigma) diluted 1:1,000 in wash buffer. The plates were then washed four times and 100 μL of prepared Fast p-Nitrophenyl Phosphate Tablets (A3562; Sigma) was added to develop the assay, which was stopped by addition of 3 M NaOH, and the absorbance of each well was measured at a wavelength of 405 nm on the VersaMax microplate reader.
Anti-dsDNA enzyme linked immunosorbent assay
Anti-dsDNA antibodies were assayed as described previously. 30 Maxisorb Nunc Plates (Thermofisher) were coated overnight with 5 μg/mL of calf thymus DNA (Sigma) in coating buffer (1 mM EDTA, 1 × PBS) at 4°C. Plates were then washed 3 times with 200 μL of wash buffer [1 mM EDTA, 1 × PBS, and 0.05% (v/v) Tween-20] and blocked with 200 μL of 3% (w/v) BSA in wash buffer at room temperature for 2 h. After washing, plates were incubated with serum diluted 1 in 100 and serial dilutions of anti-dsDNA antibody (Abcam) from 0.2 ng/mL in wash buffer for 1 h at room temperature.
Plates were washed four times and incubated for 1 h at room temperature with goat anti-mouse IgG-AP (Sigma) diluted 1:1,000 in wash buffer. The plates were then washed four times and 100 μL of prepared Fast p-Nitrophenyl Phosphate Tablets (Sigma) was added to develop the assay, which was stopped by addition of 3 M NaOH, and the absorbance of each well was measured at a wavelength of 450 nm on the VersaMax microplate reader.
Neutrophil staining
Fixed mouse tissue in OCT embedding media was cut into 6 μm sections using the Leica CM1900 cryostat. Sections were allowed to adhere to the slides and then fixed in acetone for 10 min. Slides were air dried for 5 min, sealed with PAPEN (Sigma-Aldrich), and then blocked with 10% rat serum (Sigma) in 5%BSA/1 × PBS for 60 min. Blocking buffer was removed and the slides were incubated with Alexa Fluor® 594 anti-mouse Ly-6G Antibody (#127636; BioLegend) 1:200 diluted in 1% BSA/1 × PBS overnight at 4°C covered in foils in a humid chamber. Slides were washed two times for 5 min with PBS in the dark and to the third wash, 10 μL DAPI (Thermofisher) was added. Slides were washed, dried, and covered with two drops of Immunomount and a coverslip then imaged using Zeiss microscope under DAPI and Alexa Fluor 594. Images and quantification were performed using the ZEN software.
NETs immunofluorescence
NETs were detected as described previously. 38 Briefly, 2 μm sections of formalin-fixed, paraffin-embedded tissue were mounted on superfrost plus slide, dewaxed at 60c oven for 1 h, rehydrated, and placed into the antigen retrieval solution Tris-EDTA, pH 9, in a pressure cooker for 10 min at 95°C.
Sections were then allowed to cool, blocked for 30 min in 10% Donkey serum in 5% BSA/phosphate-buffered saline, and then probed with antibodies against myeloperoxidase (MPO) (R&D, AF3667 at 1:100), citrullinated histone H3 (H3Cit) (Abcam ab5103, 1:100), and protein arginine deiminase 4 (PAD4) (Abcam ab128086, 1:50 dilution) in 1% BSA/phosphate-buffered saline for 16 h at 4°C. On the next day, slides were washed and fluorescent detection was achieved by incubation with Donkey anti goat Alexa Fluor 594 at 1:200, Donkey anti rabbit Alexa Fluor 488 at 1:200, and Donkey anti-mouse Alexa Fluor 647 at 1:200 for 40 min at room temperature.
Slides were then washed and counterstained with Sudan black (Sigma-Aldrich) 0.1% in 70% ethanol for 30 min to quench tissue autofluorescence. Slides were washed with phosphate-buffered saline in the dark and to the third wash, 10 μL DAPI (Thermofisher) was added. Slides were washed, dried, and covered with two drops of prolong gold (Molecular Probes). Fluorescent images were acquired using a Zeiss microscope; 405, 488, 561, and 647nm lasers were used to specifically excite DAPI, Alexa 488, Alexa 594, and Alexa 647. Images were acquired using 40 × or 100 × objective. Images and quantification were performed using the ZEN software.
C3 and IgG staining
Fixed mouse kidney tissue in frozen OCT blocks was cut into 5 μM sections using the Leica CM1900 cryostat. Sections were re-hydrated with PBS for 10 min and then placed in a coplin jar with cold methanol (−20°C) on ice for 15 min to permeabilize the sections. Slides were washed PBS, then dried, and sealed with PAPEN (Sigma-Aldrich). Slides were blocked with 250 μL of 20% (v/v) goat serum (Sigma) per slide for 3 h.
Blocking buffer was removed and the slides were incubated with anti-mouse IgG (F0257; Sigma-Aldrich) or anti-mouse C3 (MP Biomedical 0855500) conjugated to FITC in blocking buffer at 4°C in a moist chamber for 1 h in the dark. Slides were washed 3 × 5 min with PBS and to the second wash, 10 μL DAPI (Thermofisher) was added. Slides were dried and covered with two drops of Immunomount and a coverslip then imaged using Zeiss microscope under DAPI and Texas Red fluorescence. Images and mean fluorescence intensities were taken using the ZEN software.
Fluorescence intensities for individual glomeruli (8–10 glomeruli per mouse, on average) were calculated using ImageJ software with mean background fluorescence subtracted.
H&E staining and grading
Formalin-fixed tissue was embedded in paraffin by Virginia James at Westmead Institute for Medical Research. Paraffin-embedded tissue was prepared as 5 μM sections using the Leica RM2235 microtome and sections were stained using standard staining protocols. Brightfield/light microscope imaging of sections was performed on an Axio Imager A1 using a Spot Insight Color camera for taking images using Spot Version 4.0 software. Kidney sections were assessed in a blinded manner and graded based on the Jablonski scale of injury. 39
Statistical analysis
Data were analyzed using GraphPad Prism Ver 8.4.0. Statistical analysis was performed using nonparametric two-tailed Mann–Whitney test to assess for significance. p-values <0.05 were considered significant.
RESULTS
AAV transgene delivery raises serum DNaseI levels
AAV vectors were constructed to encode murine DNaseI under the transcriptional control of a liver-specific promoter (Fig. 1A) and packaged into the AAV8 capsid for in vivo transduction of the murine liver. The vector was delivered by intraperitoneal (i.p.) injection into adult C57BL/6 mice (1 × 1011 vg/mouse). Six weeks postinjection, serum DNaseI activity levels were elevated 380-fold in AAV-DNaseI-treated mice when compared to physiological levels in AAV-GFP-treated mice (Fig. 1B). Analysis of the liver at the same time point demonstrated similar vector copy numbers for AAV-DNaseI and AAV-GFP-treated mice (Fig. 1C).
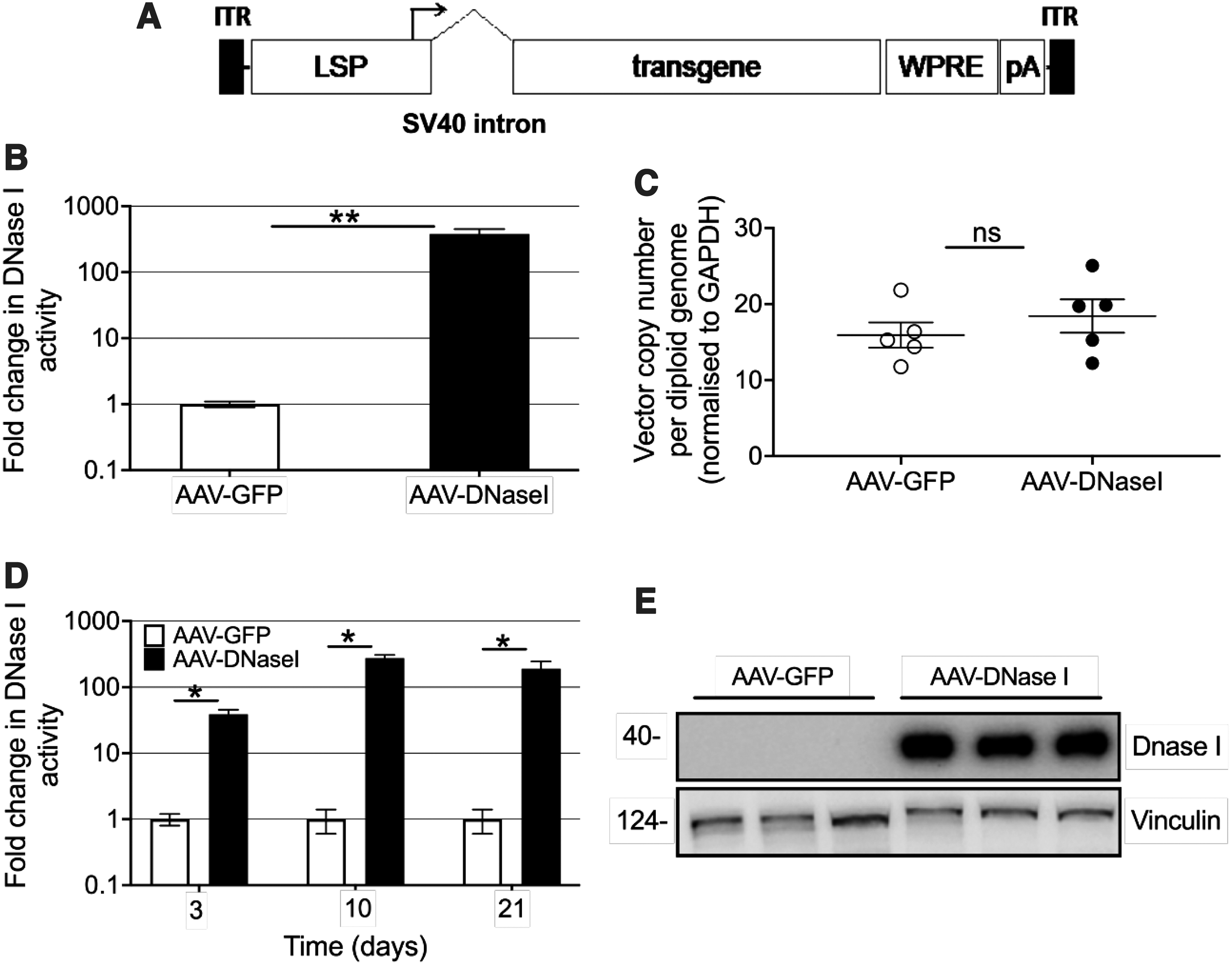
Liver-directed AAV gene delivery induces elevated serum activity of the DNaseI.
An in vivo time course was similarly conducted to understand the kinetics of vector transduction and transgene expression. Sera and livers were collected from mice at 3, 10, and 21 days postvector injection. On day 3, significantly higher levels of serum DNaseI activity were detected in AAV-DNaseI-injected mice when compared to AAV-GFP controls (40-fold higher, Fig. 1D).
By day 10, the activity was further increased 280-fold above the physiological levels observed in control mice and continued to remain high on day 21 (190-fold). Western blot analysis for DNaseI on sera from representative mice detected a ∼40 kDa signal in AAV-DNaseI-treated mice, but did not detect the low physiological levels present in control animals (Fig. 1E). Equivalent between groups throughout the time course were vector copy numbers and vector transcript levels (data not shown). There was also no difference between experimental groups with respect to histological analysis of liver (data not shown) or for serum AST/ALT levels (Supplementary Fig. S1) indicating there was no overt damage to hepatocytes, despite DNaseI overexpression.
Similar fold changes in DNaseI activity levels were observed 2 months after AAV-DNaseI vector delivery to the NZBWF1 mouse with a 200-fold increase in AAV-DNaseI-treated mice compared to AAV-GFP controls (Fig. 2A). Long-term monitoring showed the levels remained stable and high at 6 months postinjection (300-fold). Western blot analysis confirmed a 40 kDa signal in the AAV-DNaseI-treated mice, but did not detect the low physiological levels in control animals (Fig. 2B). Liver vector copy number at the end of life (∼10–12 months) was the same between groups, indicating long-term vector stability and transgene expression (Fig. 2C). Collectively, these data demonstrate that AAV-DNaseI can convert the liver into a biological factory for the long-term supraphysiological production of DNaseI.
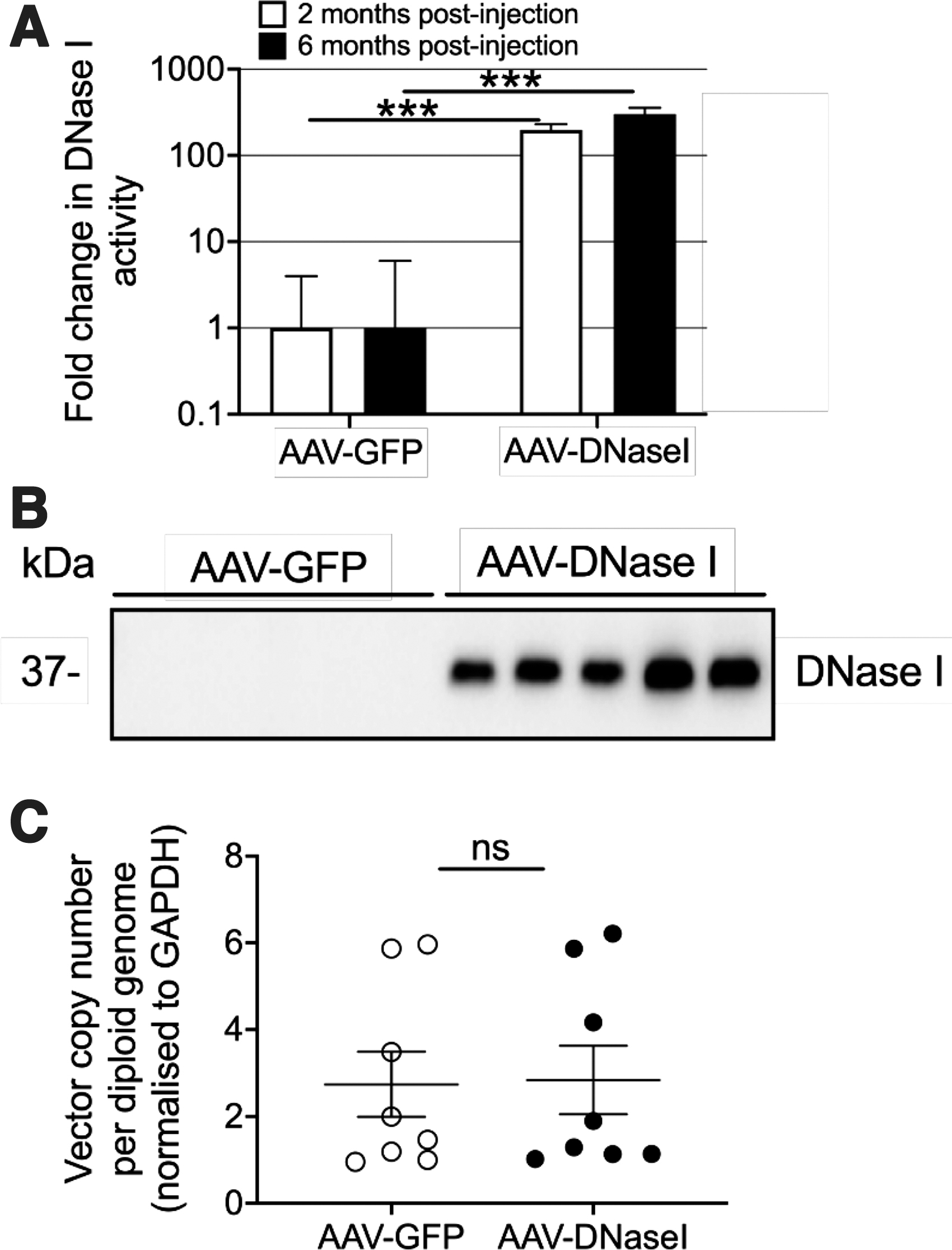
AAV-vector gene delivery induces stable long-term DNaseI transgene expression in lupus-prone NZBWF1 mice. Eight-week-old female NZBWF1 mice were injected with AAV vectors encoding either GFP or DNaseI (1 × 1011 vg per mouse, n = 8 per vector group). Serum was collected and analyzed for
Treatment of lupus-prone NZBWF1 mice using AAV-mediated DNaseI expression
DNaseI has been proposed to be a potential therapeutic for SLE 29 and a previous study has demonstrated that recombinant enzyme reduces disease endpoints in the NZBWF1 SLE mouse model. 30 The model bears a strong resemblance to the human condition with female mice spontaneously developing lupus-like disease evident by raised serum levels of antinuclear antibodies and immune complexes, as well as lupus nephritis (LN). 40
We tested whether the delivery of the AAV-DNaseI vector to female NZBWF1 mice (Fig. 2) ameliorated lupus endpoints in the model. AAV-DNaseI treatment did not affect animal survival or alter anti-dsDNA antibody levels or serum immune complexes (Fig. 3A–C). There was also no effect of the vector in improving renal function as measured by urinary protein levels or serum creatinine levels (Fig. 3D–E). Glomerular damage appeared equivalent in both groups as seen by histological examination of the kidneys (Fig. 3F and Supplementary Fig. S2).
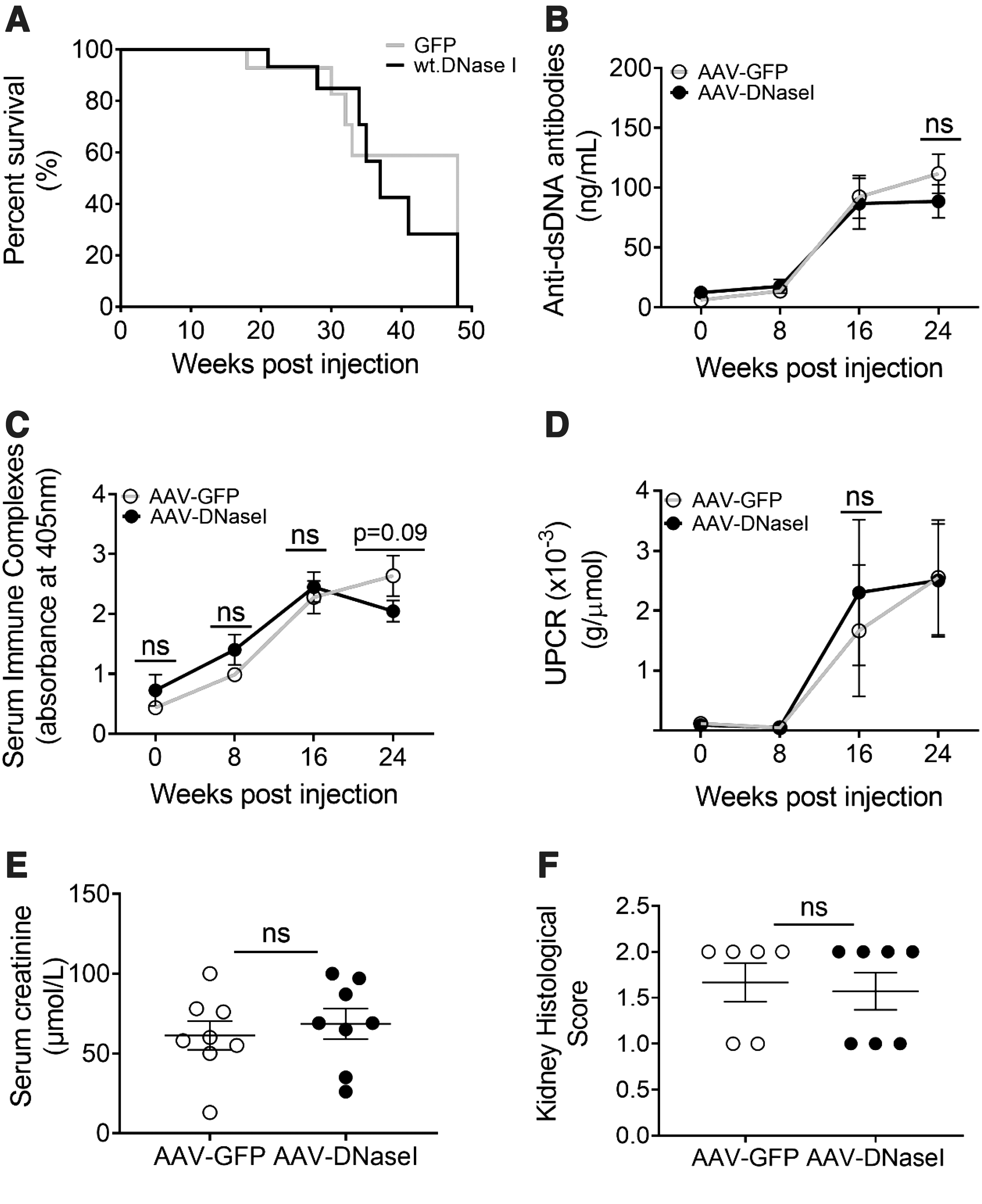
AAV-DNaseI does not alter disease progression in NZBWF1 mice. Mice depicted in Figure 2 were monitored for
We conducted further analysis to determine whether vector treatment may have altered renal presence of LN biomarkers in the model. DNaseI vector treatment was found to significantly lower the incidence of infiltrating neutrophils when compared to GFP-vector control-treated mice (Fig. 4A and Supplementary Fig. S3). Further analysis was therefore performed to determine whether there was also a reduction in the incidence of NETs. Preliminary studies with DAPI-stained kidney sections labeled for MPO, H3Cit, and PAD4, whose colocalization is indicative of NETS, showed that our protocol could discern between intact and NET-forming neutrophils (Supplementary Fig. S4). We also showed in untreated animals that NETs develop in the kidneys of female NZBWF1 mice by 7 months of age (Supplementary Fig. S5). Furthermore, analysis of the AAV vector treatment groups showed that AAV-DNaseI significantly reduced this pathological feature when compared to control-treated animals (Fig. 4B) with representative images shown in Fig. 4C.
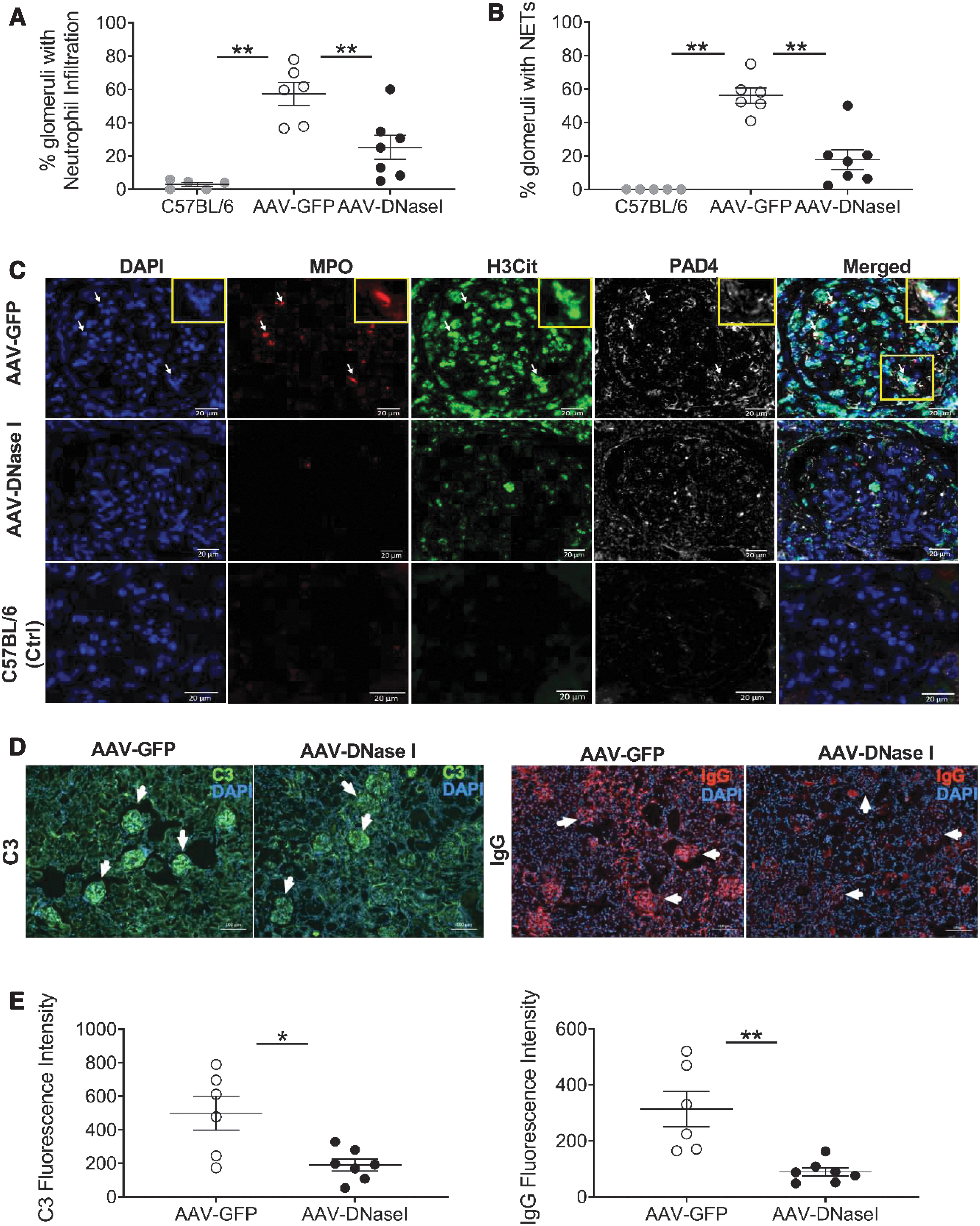
Long-term DNaseI expression in NZBWF1 mice reduces the renal presence of neutrophils, NETs, and C3/IgG.
Additional benefits of DNaseI treatment were seen after analyzing kidneys for other hallmarks of SLE pathology, namely IgG and C3 deposition. Immunolabeling revealed that the AAV-DNaseI treatment group exhibited significantly lower glomerular C3 levels when compared to AAV-GFP controls (Fig. 4D–E), and even further pronounced were the decreases in renal IgG deposition with AAV-DNaseI treatment (Fig. 4F, G). Collectively, these data demonstrate that long-term delivery of DNaseI by AAV-liver transduction in NZBWF1 mice reduces renal presence of neutrophils, NETs, C3, and IgG, but does not alter other endpoints of SLE.
DISCUSSION
This study demonstrates that liver-targeted AAV-mediated gene delivery induces sustained expression of DNaseI activity in the circulatory system at supraphysiological levels (>190-fold). Enzyme activity was rapidly induced within 3-days of gene delivery in the absence of detectable liver toxicity and was stably maintained for 6 months, the latest time point measured. These data therefore support AAV transgene delivery as a viable approach to overcome the issue of short half-life of the enzyme and provide a steady supply of DNaseI activity.
While clear evidence of DNAseI effects was observed in lupus-prone NZBWF1 mice, demonstrably stable elevation of DNaseI activity did not alter natural disease progression as determined by serological biomakers, renal function, or animal life span. It has been hypothesized that an antigenic driver of SLE might be prolonged survival of DNA nucleosomes present both in apoptotic cells or outside the cellular compartment and that this stimulus could be cleared by exogenous DNaseI. 29 Our data do not support this hypothesis.
However, SLE is a heterogenous and pathophysiologically complex disease influenced by multiple genetic and environmental factors, 41 so other factors are likely contributing to disease progression. Notwithstanding, the increased levels of DNaseI activity did reduce kidney presence of neutrophils, NETs, C3, and IgG, which are collectively a hallmark of LN. This is a significant finding in that it demonstrates that the DNaseI vector is biofunctional and might be more effective in preventing disease where NETs is a dominant pathological mechanism. Indeed, application of the vector in a model of Anti-Neutrophilic Cytoplasmic Autoantibody (ANCA) vasculitis has proven to be effective (O'Sullivan et al., article in preparation).
The reduction in renal presence of both NETs and complement/IgG deposition in the SLE model correlates with previous studies that have shown complement is required for NETs formation 42 and that NETs also induce complement activation, 26,43 presumably driving a feedback circuit that promotes inflammation. Importantly, NETs have been associated with SLE pathogenesis 44 –47 and in vitro studies using SLE patient sera have shown that DNaseI can degrade NETs 44,48 and prevent complement activation to short-circuit, resulting in amplification of inflammatory pathways. 48
Support for exogenous DNaseI as a therapy to counter deficits in DNaseI has come from studies showing DNaseI deficiency in humans and mice leads to vasculitis and LN-like symptoms. 49 –51 Also, elevated serum levels of G-actin, a DNaseI inhibitor, correlate with SLE biomarkers 31,52 and low serum levels of DNaseI are observed in SLE cohorts. 53 –56 Finally, progressive LN has also been associated with a decrease in renal production of DNaseI. 57,58 Despite this circumstantial evidence for exogenous DNaseI as a potential SLE therapy, our data add to a body of reports producing contradictory outcomes when recombinant DNaseI has been applied to mouse models of SLE. 30,59,60
One report found that daily DNaseI administration in NZBWF1 mice delays the rise in anti-DNA antibody titers and reduces kidney histopathology and proteinuria, as well as reduces renal presence of C3/IgG and extends animal life span. 30 Another almost identical study showed that the enzyme delayed the rise in anti-DNA antibody titers, but did not lead to clinical improvements. 59 In addition, the transgenic expression of a DNaseI variant in a lupus-prone mouse model protected from the development of anti-ssDNA and anti-histone antibodies, but did not afford renal protection. 60 As these earlier studies predate the discovery of NETs involvement in SLE, 44 –46,61 they did not investigate the effect of DNaseI on this pathological feature.
Our study indicates that AAV-DNaseI reduces NETs, but does not have utility to improve clinical endpoints of lupus. This is in keeping with observations in a derivative of the NZBW model that applied chemical inhibitors of NETS. 62 However, another recent study by Motwani et al. has shown that AAV-mediated gene delivery of DNaseI in a pristane-inducible lupus model decreased inflammation and antinuclear antibody levels and improved renal function, as indicated by decreased proteinuria. 63 The inconsistency in replicating DNaseI study outcomes across disease models is likely due to the complexity of pathophysiological mechanisms uniquely underlying each SLE model.
The study by Motwani et al. also differs from this report in its use of AAV capsid serotype (AAV9 vs. AAV8) and vector promoter (ubiquitous vs. liver specific), resulting in expression of transgene in the kidneys and, in all probability, other tissues, as well as the liver. A systematic appraisal of distinct vector configurations across different lupus models might identify further DNaseI sensitive models and determine the most effective tissue targets for vector transduction.
Testing the therapeutic potential of other members of the DNaseI family of enzymes is also worth exploration. 18 This includes DNase1L3, which differs from DNaseI at its c-terminus, rendering it more effective at degrading ecDNA in lupus-associated microparticles. 64,65 Deficits in DNase1L3 in humans have been associated with an aggressive form of SLE with associated pathology indicating that this enzyme has an important physiological role in preventing autoimmunity. 66,67 Exogenous DNase1L3 may therefore be therapeutically effective in the lupus setting either alone or in combination with DNaseI.
Our study also investigated the possible utility of AAV-mediated delivery of a hyperactive DNaseI variant (data not shown) that differs from the wild-type enzyme by only two amino acids (see Materials and Methods). Engineering and creation of hyperactive DNaseI have been undertaken to improve performance of the enzyme for therapeutic purposes. 60,68,69 In this study, it was anticipated that the hyperactivity of the variant along with its decreased inhibition by G-actin would increase vector potency. In contrast to the stable and prolonged transgene expression of wild-type DNaseI, expression of the variant proved hepatotoxic and led to vector loss within 3 weeks of transduction. This is consistent with observations of hyperactive DNaseI toxicity in the manufacturing industry where regulated expression systems have been required to produce stable transfectants. 70 A better understanding of enzyme biology is therefore required to capitalize on a DNaseI approach from a gene therapy perspective.
Should members of the DNaseI family of enzymes eventually prove therapeutic, the requirement for a short pulse of recombinant enzyme versus a steady supply delivered by AAV-mediated gene delivery will depend on the disease and associated risk-benefits. To this end, improvement in the utility of AAV vectors will come from the ongoing discovery of AAV capsids that more efficiently transduce human hepatocytes, 10 –13 as well as the development of drug-inducible genetic switches for regulated transgene expression. 71 Alternatively, transient enzyme expression could also be achieved using other emerging technologies, such as lipid nanoparticle delivery of mRNA. 72
In summary, our study demonstrates the feasibility of using AAV gene delivery to overcome the obstacle of short half-life of DNaseI to permit its long-term and persistent action. The supraphysiological levels of DNaseI activity induced in the circulatory system appear to be nontoxic and biofunctional in reducing NETS in the renal system of NZBWF1 mice. The observation that high levels of DNaseI activity do not reduce clinical endpoints of disease in NZBWF1 mice indicates there are other elements of pathophysiology that remain inaccessible to DNaseI. Further development of the AAV gene delivery vector or application in other DNaseI-sensitive disease models may ultimately lead to a novel and nontoxic therapeutic for a broad range of diseases.
Footnotes
AUTHORs' CONTRIBUTIONS
A.A., M.M., K.M.O., S.R.H, P.J.L., I.E.A., and G.J.L. designed the experiments. A.A., M.M., C.M.S., J.J.Y., H.D., and G.J.L. generated reagents, algorithms, and protocols and performed experiments. A.A., M.M., K.M.O., C.M.S., J.J.Y., M.C.P., P.J.L., I.E.A., and G.J.L analyzed and interpreted the data. A.A., M.M., I.E.A., and G.J.L. wrote the article and generated the figures. All authors reviewed, edited, and commented on the article.
ACKNOWLEDGMENTS
We are grateful to Virginia James (Westmead Institute for Medical Research) for embedding and staining tissue sections.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
G.J.L. received funding support from the Rebecca L Cooper Foundation (PG2019449).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.