
Abstract
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause cystic fibrosis (CF), a chronic disease that affects multiple organs, including the lung. We developed a CF ferret model of a scarless G551→D substitution in CFTR (CFTR G551D-KI), enabling approaches to correct this gating mutation in CF airways via gene editing. Homology-directed repair (HDR) was tested in Cas9-expressing CF airway basal cells (Cas9-GKI) from this model, as well as reporter basal cells (Y66S-Cas9-GKI) that express an integrated nonfluorescent Y66S-EGFP (enhanced green fluorescent protein) mutant gene to facilitate rapid assessment of HDR by the restoration of fluorescence. Recombinant adeno-associated virus (rAAV) vectors were used to deliver two DNA templates and sgRNAs for dual-gene editing at the EGFP and CFTR genes, followed by fluorescence-activated cell sorting of EGFP Y66S-corrected cells. When gene-edited airway basal cells were polarized at an air–liquid interface, unsorted and EGFP Y66S-corrected sorted populations gave rise to 26.0% and 70.4% CFTR-mediated Cl− transport of that observed in non-CF cultures, respectively. The consequences of gene editing at the CFTR G551D locus by HDR and nonhomologous end joining (NHEJ) were assessed by targeted gene next-generation sequencing (NGS) against a specific amplicon. NGS revealed HDR corrections of 3.1% of G551 sequences in the unsorted population of rAAV-infected cells, and 18.4% in the EGFP Y66S-corrected cells. However, the largest proportion of sequences had indels surrounding the CRISPR (clustered regularly interspaced short palindromic repeats) cut site, demonstrating that NHEJ was the dominant repair pathway. This approach to simultaneously coedit at two genomic loci using rAAV may have utility as a model system for optimizing gene-editing efficiencies in proliferating airway basal cells through the modulation of DNA repair pathways in favor of HDR.
INTRODUCTION
Cystic fibrosis (CF) is a lethal autosomal-recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
1
CFTR plays an important role in conducting chloride and bicarbonate ions across epithelia to regulate secretory volume, pH, and mucus viscosity of liquid secretions.
2,3
Nearly 2,000 distinct CFTR mutations have been identified, of which over 400 are causative for CF (
Although no prior gene therapy clinical trial for CF lung disease has achieved desired outcomes, more recent advances in vector development targeting the airways of large animal models that recapitulate the human CF lung phenotype have inspired a resurgence in gene therapy efforts for CF. 7 –13 While CFTR gene-replacement strategies still remain the primary approach for current and pending CF clinical trials, rapidly emerging gene-editing technologies present new opportunities to advance CF gene therapy from simple gene addition to precise gene correction. CFTR expression is highly regulated by individual cell types in the lung, each having its own potentially unique composition of channels and functions that coordinate clearance and innate immunity. 14 –17 Gene editing provides a therapeutic solution to restore CFTR function without altering its endogenous patterns of expression in the gene-corrected cells.
Clustered regularly interspaced short palindromic repeats (CRISPR) work with a CRISPR-associated protein, such as Cas9 to generate a double stranded DNA break at a desired site in the genome. 18,19 Acting as a specific programmable nuclease, the CRSIPR/Cas9 approach enables genetic modification of a gene of interest via homologous recombination (HR), exon skipping, gene disruption, or targeted insertion. 20 CRISPR-based gene therapy has shown promise against sickle cell disease by correcting the mutation in hematopoietic stem cells ex vivo, 21 and transthyretin amyloidosis by disrupting the disease-causing allele in vivo. 22 Gene editing theoretically solves the complexities of CFTR regulation at the cellular level in the lung.
While successful correction of CFTR mutations in postmitotic airway epithelial cells likely leads to durable treatment in CF patients because of the relatively slow turnover rate of these cell types, 23 permanent correction is only achievable by targeting self-renewing stem/progenitor cells in the airways. Basal cells are the major multipotent somatic stem cell population in the tracheobronchial airways and responsible for maintenance and repair of the airway epithelium. 24 –26 In this study, we tested CRISPR/Cas9-mediated HR using recombinant adeno-associated virus (rAAV) to correct the G551D gating mutation in the CFTR gene of proliferating airway basal cell cultures derived from CFTR G551D-KI ferrets (GKI ferrets).
MATERIALS AND METHODS
Generation of CFTR G551D-KI ferret model using CRISPR/Cas9-mediated HR
Animal experimentation was performed according to protocols approved by the Institutional Animal Care and Use Committees of the University of Iowa. The mutagenesis template for HR was a 192-mer single-stranded DNA oligonucleotides (ssOND). The sgRNA, gRNA-G551D was designed using online tool CRISPOR (
Sequence of mutagenesis template, sgRNA targeted site and polymerase chain reaction primers
The oligo mutagenesis template: Upper case indicates the nucleotides different from the original wild-type allele, bold fonts are the D551 codon and underlined nucleotides indicate the BclI site created in the HR template for easy genotypic assay of the G551D mutation.
Cas9/gRNA complex targeted DNA sequences with the PAM in ferret CFTR locus or integrated EGFP Y66S in ferret genome: upper cases are the PAM, // mark the cleavage site.
CFTR, cystic fibrosis transmembrane conductance regulator; EGFP, enhanced green fluorescent protein; HR, homologous recombination; PAM, protospacer adjacent motif; ssOND, single stranded DNA oligonucleotides.
Viral vectors and primary cell cultures
Lentiviral vectors
LentCas9-Blast (Addgene plasmid #52962) carries an EF1α core promoter driven SpCas9-p2A-BSD expression cassette. 28 SpCas9-p2A-BSD is a self-cleaving fusion protein of a SpCas9 (with nuclear localization sequence) and BSD (product of the blasticidin S resistance gene). Lenti-Y66SeGFP-Puro was produced from a derivative of pLenti-CMVGFP-Puro (658-5) (Addgene plasmid #17448), 29 in which the EGFP cDNA was replaced by the mutant EGFP Y66S, through a single-nucleotide substitution at the Y66 codon. 30 VSV-G pseudotyped lentiviral vectors were generated from transfection in HEK293T cells and the functional titers were quantified using TaqMan PCR quantification of viral genome integration following infection of HEK293T, as previously described. 31 Titer were expressed as transduction units (TU)/mL.
Recombinant adeno-associated virus
AAV2/6.tempG551Y66-gRNA(2) was produced as previously described by pseudopackaging an rAAV2 genome into AAV6 capsid. 32 Titer was determined by TaqMan PCR as DNase I-resistant particles (DRP) per microliter. The rAAV genome carries two sets of homology templates and sgRNAs, enabling a dual-correction of the mutations of G551D in CFTR and Y66S in EGFP by homology-directed repair (HDR). The expression of sgRNAs was driven by the human U6 snRNA promoter. gRNA-G551D was the one used in the generation of the CFTR G551D-KI ferret model. gRNA-S66 (sequence is presented in Table 1) specifically recognized the sequence of the EGFP Y66S with the protospacer adjacent motif (PAM) placed within the S66 codon, which prevented cleavage at the wild-type (WT) EGFP sequence (Y66).
The HR template for G551D correction was an 811 bp sequence containing CFTR exon-12 with the non-CF G551 codon at the center. Silent mutations within the HR donor fragment substituted three nucleotides near and within the PAM sequence targeted by the gRNA-G551D; this created a new restriction enzyme site (BspEI), which was used to confirm HDR and prevented the CRISPR-mediated cleavage of the HR template and repaired sequence. The HDR template for Y66S correction was a shortened EGFP (enhanced green fluorescent protein) sequence harboring a 90 bp truncation at 3′ end (EGFP-630), as previously described. 33 This sequence does not produce a fluorescent protein product. 33
Proliferating ferret airway basal cells
As previously described, 34 primary tracheal cells were dissociated and collected from the trachea of homozygote GKI ferret (GK/GK143, male, 84 days old). Airway basal cells from GKI ferret were expanded in PneumaCult™-Ex Plus medium (StemCell Technologies, Vancouver, Canada) for six continuous passages before being cryopreserved as the P6 frozen stocks for future studies.
Polarized ferret airway epithelial cultures
1.5 × 105 basal cells were seeded onto 6.5-mm, polyester Transwell® inserts (Corning, Corning) that were precoated with collagen IV (Sigma, St. Louis). Seeding occurred in PneumaCult-Ex Plus medium. At 24 h postseeding, medium was replaced with PneumaCult ALI medium (StemCell Technologies) in both apical and basal chambers. Cultures were then air-lifted the following day to facilitate differentiation of the polarized ferret airway epithelium (FAE) at an air–liquid interface (ALI) for at least 3 weeks. 35 Matured FAE-ALI cultures (transepithelial electric resistance >1,000 Ω/cm2) were used for Ussing chamber assays. 36 The basal chamber medium was replaced twice a week during the culturing period.
Short-circuit current measurements in Ussing chambers
The ALI culture inserts were placed under VCC MC8 voltage/current clamps in self-contained P2300 Ussing chambers (Physiologic Instruments, San Diego, CA) for short-circuit current (Isc) measurement using an asymmetric chloride buffer system, as previously described. 37 One hundred micromolars amiloride, 100 μM 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid, 100 μM 3-isobutyl-1-methylxanthine (IBMX)/10 μM forskolin (Forsk), and 50 μM GlyH-101 were sequentially added to the apical chamber, and the changes in current were recorded during the experiment.
Establishment of the Cas9-expressing airway basal cell pools
Cas9-GKI cell pool
The P6 GKI airway basal cells were infected with Lenti-Cas9-Blast at the multiplicity of infection (MOI) of 1.5 TU/cell. Selection for blasticidin S-resistant cells was started 2 days after lentiviral infection in PneumaCult-Ex Plus medium supplemented with 10 μg/mL blasticidin S. All the cells of mock-infected control died within 2 days after exposure to blasticidin S, allowing for rapid selection of a blasticidin S-resistant polyclonal pools of Cas9-expressing cells within a week.
Y66S-Cas9-GKI cell pool
Cas9-GKI basal cells (5 days after blasticidin S selection) were infected with Lenti-Y66SeGFP-Puro at an MOI of 1.5 TU/cell to enable the expression of Y66S-EGFP. Selection with puromycin at 1 μg/mL was started 2 days after infection. The double-resistant cell pool, Y66S-Cas9-GKI, was obtained after exposure to blasticidin S and puromycin for 1 week.
Gene editing and assessments
Of the Cas9-GKI, 2 × 105 cells or Y66S-Cas9-GKI basal cell pool were seeded onto a well of a six-well plate. The next day, cells were infected with AAV2/6.tempG551Y66-gRNA(2) at an MOI of 5 × 104 DRP per cell. The rAAV infected Cas9-GKI cells were then lysed at 3 days postinfection. Genomic DNA was extracted for PCR to assess gene editing at the CFTR G551D locus. The rAAV-infected Y66S-Cas9-GKI cells were subjected to limited expansion before the assessment of dual-gene editing of EGFP Y66S and CFTR G551D loci. At 3 days postinfection, cells were transferred to two 100 mm dishes for expansion. The culture attained confluency after 1 week, from which ∼3 × 107 cells were obtained.
Ten days postinfection, 9/10th of the total cell population (2.7 × 107 cells) was used for fluorescence-activated cell sorting (FACS) to collect the EGFP Y66S-corrected cells (green) and to determine the gene correction efficiency. The viable green cells obtained from FACS and the remaining portion (1/10th) of unsorted cells were put back into culture. Two days later (12 days post-rAAV infection), cells from both sorted and unsorted populations were used for genomic DNA extraction and polarization of FAE-ALI cultures.
The PCR primer set HR-Fw/Rev, located outside the CFTR homology sequence carried in the rAAV-targeted vector, was used to amplify an 887 bp product from the genomic DNA obtained from rAAV-transduced cells. The PCR products were cloned into pCR4blunt-Topo (Invitrogen by Thermo Fisher Scientific, Waltham, MA) and 48 plasmid clones were randomly picked for Sanger sequencing. A 243 bp amplicon that covered exon-12 was obtained by PCR using the 887 bp product as template and an internal primer set G551DtgFw/G551DtgRev. This 243 bp amplicon was evaluated by deep sequencing to quantify variants using an Illumina platform; sequencing was performed by BGI Americas Corporation (Cambridge, MA).
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 8. Values and error bars show means ± standard error of the mean, statistical significance was determined using a Student's t-test with p < 0.05 being significant.
RESULTS
Generation of the CFTR G551D ferret model via CRISPR/Cas9-mediated gene editing in ferret zygotes
CFTR G551D is the third most common CF-associated mutation found in 2–5% of patients with CF. The G551D codon resides in the ATP-binding pocket within the nucleotide-binding domains-1 of CFTR protein. The substitution of aspartate (D) for glycine (G) abolishes ATP-dependent gating of the channel. The CFTR potentiator VX-770 (ivacaftor; KALYDECO®) restores channel gating of the mutant protein. We have previously described the generation of a G551D ferret model (CFTR G551D-GM) by somatic cell nuclear transfer (SCNT). CFTR G551D-GM Ferret recapitulates the human CF disease phenotype and is also responsive to the CFTR potentiator VX-770. 38 While this model is advantageous for testing CFTR gene replacement therapies, its use in testing gene editing is limited due to an ∼50% reduction in expression of G551D-CFTR mRNA from the targeted locus.
This reduction was caused by the insertion of the neo (neomycin resistance) expression cassette located in intron-13, which was needed to select HR-targeted mutant fibroblasts for SCNT following rAAV transduction. Thus, we developed the second generation ferret model (CFTR G551D-KI) containing a scarless G551D knock-in without an integrated antibiotic resistance gene using CRISPR-mediated mutagenesis in ferret zygotes. Of note, to maintain our capacity to use the same gRNA-G551D for the later gene editing studies and reduce the chances of altering mRNA secondary structure that might otherwise impact CFTR expression, we decided not to disrupt the gRNA-G551-targeted sequence in the mutagenesis oligo template.
In the mutagenesis template, there were three of the nucleotide differences from the WT CFTR sequence. Two of the base changes were used to covert the G551 codon to D551, a third base for silent mutation was placed in the G550 codon. These substitutions generated a new BclI restriction endonuclease site not present in the original G551 allele and facilitated genotype of G551D mutation by PCR and BclI digestion (Fig. 1A). The CRISPR approach demonstrated much higher efficiency to knock in the mutation than the previous method using rAAV-mediated HR. 38 A total of 81 kits were born in 10 litters, of which 21 kits (25.9%) were positive for the G551D mutation. As expected, six kits that bore mutations in both CFTR alleles (G551D/G551D or G551D/indel) suffered from meconium ileus (complication of CF) and were euthanized.

Generation of scarless G551D knock-in ferret model.
PCR products of the remaining 15 surviving kits were cloned into a plasmid pCR4blunt-Topo for Sanger sequencing. All of the 15 kits sequenced had at least one G551D mutations, and 10 kits (66.7%) also had undesired indels generated by nonhomologous end joining (NHEJ). For example, the kit I8 was a compounded heterozygote containing G551D and knockout genotypes, which evidently was a chimera and did not suffer from meconium ileus (Fig. 1B). Kit K7 was determined to have a heterozygote G551D mutation (CFTR G551D-KI/WT) with roughly equal reads of G551D-KI and WT sequences obtained and no undesired off-target mutations in either allele (Fig. 1B).
Of the total 21 kits born, there was a high frequency (76.2%) of simultaneous modification of both CFTR alleles (16 of 21 kits). Kit K7 animal was male and thus chosen as the founder for breeding expansion with WT ferrets (CFTR WT/WT). Germline transmission of the G551D mutation was confirmed in the F1 offspring. Further crossbreeding of F1 CFTR G551D-KI/WT male and female animals successfully generated F2 homozygote CFTR G551D-KI/G551D-KI ferrets (GKI ferrets).
To assess the function of G551D-CFTR in FAE, we seeded primary tracheal GKI basal cells onto Transwell inserts and polarized differentiated these cultures at an ALI. Following differentiation, short circuit currents (Isc) were measured in Ussing chambers following the sequential addition of non-CFTR ion channel antagonists, cAMP agonists (to activate CFTR), and CFTR inhibitor GlyH-101. Similar to CFTR G551D-GM cultures, 38 GKI FAE-ALI cultures demonstrated a low basal level of Cl− current upon Forsk and IBMX stimulation, which was inhibited after the addition of GlyH-101. Treatment of GKI FAE-ALI cultures with VX-770, cAMP-induced Cl− current was enhanced 5.9-fold and achieved 66% that observed in WT FAE-ALI cultures (Fig. 1C, D). Thus, VX-770 potentiates gating of ferret G551D-CFTR in a similar manner to the human mutant.
HDR corrects the G551D mutation in CF ferret airway basal cells
Our main objective was to develop a model system for optimizing editing the CFTR G551D mutation in primary ferret airway basal cells for applications in autologous cell therapy. rAAV vectors are widely used as a gene transfer agent for human gene therapy. Although rAAV cannot accommodate all the components required for CRISPR-mediated HDR, due to its limited package capacity, it may be practical to use a dual-AAV delivery system 39,40 (e.g., one AAV vector encoding Cas9 and second encoding the sgRNA expression cassette with donor HR template). To simplify variables that impact cotransduction and maximize the efficacy, we incorporated an spCas9 expression cassette into primary GKI basal cells using a lentivirus (LentCas9-Blast). This enabled studies to use a single rAAV vector [AAV2/6.tempG551Y66-gRNA(2)] for the gene-editing experiments (Fig. 2A).

Correction of the G551D CFTR mutation in ferret airway basal cells.
rAAV6 was the most efficient at transducing primary ferret airway basal cells in comparison to other AAV serotypes (rAAV1, rAAV2, rAAV5, rAAV8, and rAAV9, data not shown), with ∼90% transduction efficiency at an MOI of 5 × 104 DRP per cell. We infected Cas9-GKI cells with AAV2/6.tempG551Y66-gRNA(2) and evaluated gene editing by PCR of genomic DNA compared to noninfected control cells. We used two primer sets that amplified CFTR exon-12 using flanking intronic sequences not contained in the rAAV HR template (Fig. 2A).
The two primer sets HR-Fw/Rev and Fw1/Rev produced 887 and 714 bp products, respectively, with gain of a BspEI site diagnostic for HDR events and loss of a BclI site diagnostic for HDR or indels produced by NHEJ (Fig. 2B). A small proportion of HDR was observed as evident by BclI cutting, while the fraction of PCR product that lost BclI cutting appeared more abundant was likely caused by indels at the target site (Fig. 2B).
To confirm correction of the G551D codon, we cloned the 887 bp PCR product and randomly picked 48 clones for Sanger sequencing. Of the total 43 readable sequences, 62.79% were unmodified, 23.26% contained indels surrounding the CRISPR cut site, and 13.95% had undergone HDR (Fig. 3A). Within the 13.95% of clones that converted the D551 codon to G551, there were no undesired mutations in exon-12 or the flanking intronic sequence and all retained the six nucleotide changes associated with the new BspE1 site. Of the 10 NHEJ/indel sequences, 4 were identical with one extra “T” insertion right at the CRISPR cut site, 2 sequences contained a 15 bp deletion, and other 4 harbored deletions of 1, 2, 4, and 11 bp.

Next-generation sequence analysis G551D locus in gene editing Cas9-GKI airway basal cells.
To further analyze sequence details of gene editing, a 243 bp amplicon containing CFTR exon-12 from the combined products of three biologic replicates was used for targeted gene next-generation sequencing (NGS) on an Illumina platform. NGS generated 27,755,676 reads for alignment with the reference sequences using CRISPResso2 (
Because of small sample size, Sanger sequencing was not able to reveal the imperfect G551 correction that accounted for 0.07% of the total aligned reads. This analysis also detailed the types of mutations that occurred with imperfect HDR (Fig. 3C) and NHEJ (Fig. 3D). Both were composed of various deletions, insertions, and substitutions, as well as the combinations of insertions and substitutions or deletions and substitutions. However, no combination of insertions/deletions or deletions/insertions/substitutions were found by NSG.
Of the total 6,098,062 sequences that incurred NHEJ, the majority were insertional mutations, which was 3.5-fold higher than those with deletions (76.26% vs. 21.71%). Very few reads (1.03%) contained substitutions. A recurrent +1 bp insertion at the CRISPR/Cas9 cut site was the most frequent genotype (75.31%) in all NHEJ mutants sequenced and accounted for nearly all (98.42%) of the insertional only mutants. The second most frequent NHEJ genotype (7.62%) was a −15 bp deletion. These +1 and −15 genotypes were also the majority of NHEJ mutants seen from Sanger sequencing.
By contrast, the mutations, including substitution (34.90%), deletion (32.48%), and their combination were more frequently seen in the imperfect HDR reads, while insertion only mutations accounted for only 18.54%. As there was no major recurrent genotype found for the imperfect HDR reads, we reasoned the cause to be random errors from DNA synthesis during HDR or artifacts from the PCR amplification, although the error rate using high fidelity Taq polymerase was expected to be minimal.
Dual-gene editing in CF ferret airway basal cells
The high rate of NHEJ mutations suggested the need to improve CRISPR-mediated gene correction of airway basal cells. As CFTR expression is silent in basal cells, an easily assessable reporter would be extremely useful for approach optimization. We hypothesized that coediting at a second reporter locus would be enriched in basal cells correctly edited at the CFTR locus. Previously, we used the EGFP
Y66S as reporter to indicate the gene correction using RNA-mediated and rAAV-mediated HR in transformed cells and primary cells of transgenic mice.
30,33
This reporter harbors a single base substitution at the Y66 codon [T
In this study, Cas9-GKI basal cells were transduced with Lenti-Y66SeGFP-Puro lentivirus (Fig. 4A) and double-resistant polyclonal pools of cells (Y66S-Cas9-GKI) were obtained following a week selection with blasticidin S and puromycin. To validate the gene-editing approach, Y66S-Cas9-GKI basal cells were then transduced with the rAAV vector harboring both CFTR-G551 and EGFP-Y66 homology templates and two gRNA expression cassettes targeting both mutations [AAV2/6.tempG551Y66-gRNA(2)]. Restoration of EGFP fluorescence was visible 1 day following infection and ∼10% cells recovered fluorescence by 3 days postinfection (Fig. 4B). At 10 days postinfection, FACS analysis demonstrated that 8.02% of Y66S-Cas9-GKI basal cells had recovered EGFP fluorescence (Fig. 4C). The viable green cells (EGFP Y66-corrected) were collected by FACS were put back in culture.
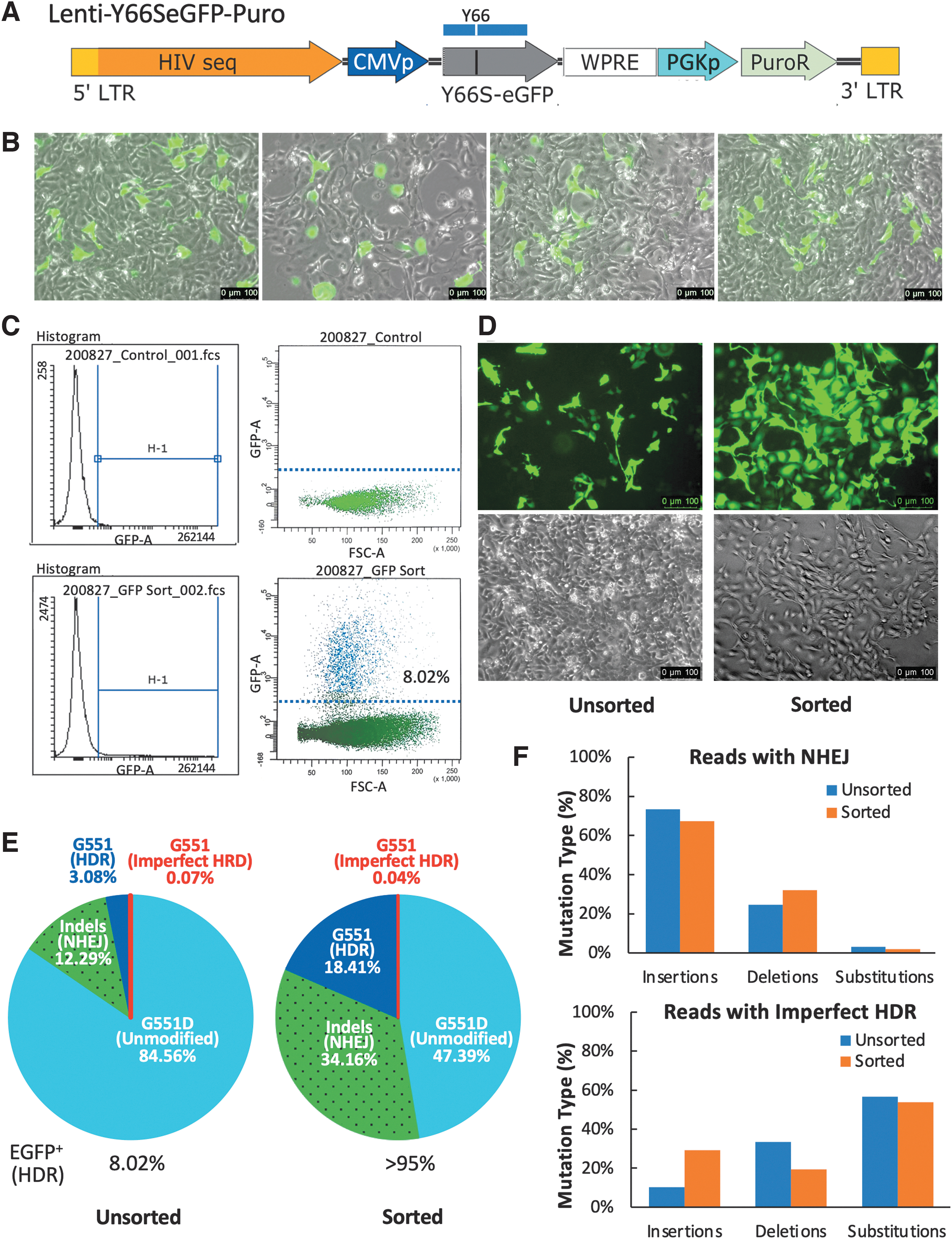
Dual-correction of the CFTR
G551D and EGFP
Y66S in ferret airway basal cells.
We also continued to culture a small fraction (1/10th) of unsorted cells. The purity and expansion of sorted fraction were checked visually 2 days after sorting. As expected, >95% cells were strongly fluorescent (Fig. 4D). There was no notable difference of EGFP-positive cells at 3 and 12 days postinfection for unsorted cells (Fig. 4B vs. D). Both sorted and unsorted populations were then seeded onto separate Transwell inserts to generate polarized FAE-ALI cultures and noninfected Y66S-Cas9-GKI FAE-ALI cultures were also generated as controls. Genomic DNA was extracted from the remaining portion of unsorted and sorted AAV2/6.tempG551Y66-gRNA(2)-infected basal cells for PCR to obtain the 243 bp amplicon for NGS as described above for rAAV-infected Cas9-GKI cells.
Analyses of the deep sequencing data revealed that the G551 (HDR-corrected, including imperfect HDR) sequences accounted for 3.15% of the total reads from the unsorted cell population and 18.45% of the total aligned reads from FACS-sorted EGFP fluorescent populations (Fig. 4E). Similar to the analyses of the Cas9-GKI cells, insertional mutations at the G551D locus were most common results of NHEJ and substitution mutations were the most common result of imperfect HDR reads, in both the sorted and unsorted cell populations (Fig. 4F). The ∼6-fold enrichment of the G551 sequences in the EGFP fluorescent basal cells suggests a high frequency of simultaneous HDR at the two genetic loci. However, in both the unsorted (U) and sorted (S) populations, HDR correction of EGFP Y66S (U 8.02% vs. S > 95%) was more efficient than that at the CFTR locus (U 3.15% vs. S 18.45%, including imperfect HDR sequences) (Fig. 4E).
The lower correction rate of CFTR G551D in unsorted Y66S-Cas9-GKI basal cells (3.15%), compared to unsorted Cas9-GKI basal cells (13.21%), suggests the possibility of HDR substrate competition, since one rAAV genome was used for HDR at both sites. This hypothesis is supported by a larger difference in the abundance of NHEJ-related versus HDR-related sequences in the unsorted Y66S-Cas9-GKI basal cells (3.9-fold; 12.29% vs. 3.15%, respectively), compared to gene editing of just CFTR in Cas9-GKI basal cell (1.7-fold; 22.57% vs. 13.21%, respectively). With a 11.8-fold enrichment in EGFP fluorescent HDR-corrected basal cells by FACS and a 3.15% frequency of CFTR HDR events in unsorted basal cells, if all the CFTR HDR events were within EGFP+ sorted basal cells, the frequency of HDR at the CFTR locus would have been 37% (11.8-fold enrichment × 3.15% CFTR HDR).
Given that the observed frequency of CFTR HDR in EGFP+ sorted basal cells was 18.45%, we estimate that ∼50% of the CFTR HDR events occurred in nonfluorescent basal cells, and the HDR efficiency at the CFTR gene was ∼50% less than that at the eGFP gene.
Correction of CFTR function in CRISPR-edited differentiated ferret airway basal cells
To assess functional correction of CFTR in gene-edited basal cells, rAAV-infected Y66S-Cas9-GKI basal cells (FACS-sorted and unsorted) were used to generate differentiated FAE-ALI cultures. FAE-ALI cultures generated from noninfected Y66S-Cas9-GKI basal cells were used as control. CFTR-mediated short circuit current (Isc) was then assessed on differentiated cultures in Ussing chambers. Typical traces of the Isc from three different types of FAE-ALI cultures are presented in Fig. 5A. These traces demonstrated expected responses to non-CFTR ion channel antagonists, cAMP agonists, and CFTR inhibitor, indicating that the manipulations of lentiviral and rAAV vectors infections in ferret airway basal cells did not alter their ability to differentiate into a functional pseudostratified epithelium.

Partial functional correction of CFTR-mediated Cl− transport in polarized epithelial cultures derived from gene-edited Y66S-Cas9-GKI basal cells.
CFTR-mediated Cl− conductance was quantitated as the change in Isc (ΔIsc) following the addition of cAMP agonists and the CFTR inhibitor GlyH101 (Fig. 5B). ΔIsc-GlyH101 responses from noninfected Y66S-Cas9-GKI cultures were 11.3% that of non-CF FAE-ALI cultures. This low level of Cl− channel activity was similar to that observation in FAE-ALI cultures derived from the original GKI basal cell stock (Fig. 1C). We observed increased CFTR-mediated Isc in FAE-ALI cultures from both two populations of the rAAV-infected Y66S-Cas9-GKI basal cells. FAE-ALI cultures derived from unsorted Y66S-Cas9-GKI edited basal cells demonstrated 2.3-fold higher ΔIsc-GlyH101 responses than control unedited cultures. Although the rate of G551D correction in this cell population was only 3.08%, this gave rise to 26.0% of the ΔIsc-GlyH101 observed in non-CF cultures.
FAE-ALI cultures derived from sorted EGFP Y66-corrected basal cells with 18.4% G551 correction demonstrated 70.4% functional rescue in CFTR activity (ΔIsc-GlyH101) compared to non-CF cultures. The 6.2-fold increase ΔIsc-GlyH101 responses was comparable to the improvement achieved by treatment with the CFTR potentiator VX770 (5.9-fold, Fig. 1C).
DISCUSSION
Previously, we used the CRISPR/Cas9 approach to manipulate the ferret genome in zygotes, through which we successfully knocked in a CRE-responsive reporter expression cassette into a predetermined site at the ROSA locus via homology-independent targeted integration (HITI), which utilizes a NHEJ repair mechanism. 27 In this study, the CRISPR/Cas9 approach was utilized to facilitate the mutagenesis by HDR to generate a scarless CFTR G551D-KI knock-in ferret. Of note, we observed a high rate of simultaneous modification in both two CFTR alleles, with NHEJ-mediated mutations in two-thirds of the G551D-KI kits. These undesired mutations were found in both the non-HDR allele and the allele bearing the G551D mutation, indicating that NHEJ-mediated mutagenesis was highly efficient even when the HR template was provided.
Ongoing studies are evaluating the phenotype of homozygous CFTR G551D-KI ferrets. Here, we sought to generate vector tools and model systems to evaluate the efficiency of gene editing ferret CFTR G551D with the eventual goal of applying this technology in vivo in the ferret model.
While HDR was effective in proliferating airway epithelial cells, the observed 13.21% correction rate of the G551D mutation by HDR (including imperfect HDR events) was lower than the undesired mutagenesis rate (22.57%) caused by NHEJ in the Cas9-GKI basal cells. Thus, basal cell NHEJ mechanism appears to dominate over HR when rAAV is harboring the HR template. This was similar to that observed in RNP/ssODN-injected ferret zygotes, where HDR corrected the G551D mutation at one allele, but mutated the second CFTR allele by NHEJ. Modulation of cellular DNA repair pathways to favor HDR over NHEJ could improve gene editing efficiencies. Several approaches using chemical and genetic modulation have proposed for enhancing HDR pathways, 41 such as inhibiting the key enzymes and proteins involved in NHEJ, 42 –44 directly modulating the HDR pathway, 45,46 and overexpressing RAD52 and dn53BP1, 47 or RAD18 (a ubiquitin ligase involved in HDR and post-DNA replication repair). 48
Semi high-throughput combinatorial screening using chemical and/or genetic agents that modulate these NHEJ and HDR pathways would benefit from the mutant EGFP reporter system we reported here. Thus, we created a system capable of rapidly evaluating HDR efficiencies at two loci in primary basal cells, while reducing the variables associated with codelivery of a Cas9-expressing rAAV vector. The rAAV vector and Y66S-Cas9-GKI reporter basal cells developed here constitute such a model system for the optimization HDR approaches.
Our study was conducted in polyclonal pools of Cas9-expressing GKI basal cells, a requirement for the maintenance of CFTR function following differentiation. The difference in HDR correction rates for G551D (3.15%) and Y66S (8.02%) in the total population of unsorted rAAV-infected Y66S-Cas9 GKI basal cells could be due to many factors, but not the level of Cas9 expression or rAAV infection, given that a Cas9 expression cassette was integrated into the genome and a single rAAV vector harbored both HR templates. The high frequency of simultaneous gene editing at two loci (52.61% of CFTR mutagenesis rate in the EGFP Y66S-corrected cells, of which 18.45% was from HDR) suggests that inefficient cleavage by Cas9/gRNA-G551D was unlikely responsible for the lower HDR rate.
Factors that may have impacted lower HDR rates of CFTR may include target gene location in the genome, its epigenetic landscape, and/or nuclear three-dimensional topology, which could differentially impact access of the HR template to the EGFP and CFTR loci. Of note, the lentiviral integration of the Y66S-EGFP cassette was random and studies have shown that lentiviruses preferentially integrate into regions of transcriptionally active chromatin. 49 Thus, FACS isolation of EGFP fluorescent basal cells could have enriched for HDR events if they occurred more efficiently at transcriptionally active loci. However, since the CFTR gene is silent in basal cells and HDR events at the CFTR locus were enriched sixfold in EGFP fluorescent basal cells, this cannot completely explain the discrepancy in editing at the two loci.
Previous studies have attempted to site-specifically integrate a CFTR minigene via HDR in airway epithelial cells using helper-dependent adenoviral vectors (HD-Ad) to deliver both the donor template and programmable nuclease (CRISPR/Cas9 or TALENs) within the same vector. 50,51 These studies have shown that the vector genome is unstable following nuclease excision of the donor template, thus limiting availability for effective HDR. This is also likely true for the application of rAAV vectors. Thus, a rAAV genome harboring two HR templates may not be capable of participating in two HDR events. Indeed, when AAV2/6.tempG551Y66-gRNA(2) was used to correct only the G551D mutation in Cas9-GKI cells, a 13.21% (including imperfect HDR events) correction rate was achieved—a rate much higher than the 3.15% correction of CFTR G551D observed in Y66S-Cas9-GKI cells where dual correction was conducted using the same construct.
However, in Y66S-Cas9-GKI cells, the combined HDR rate at both CFTR G551D and EGFP Y66S loci (11.17%) was more similar to the single CFTR targeting approach in Cas9-GKI cells (13.21%).
Another hypothesis to explain differences in editing efficiency of EGFP and CFTR could be substrate competition, which may have led to more favorable homology recombination at the EGFP Y66S loci in the same infected cell because the GC content of the HR template for EGFP correction is much higher than that for CFTR (74% vs. 38%). In support of this hypothesis, higher GC content has been reported to elevate mutation and recombination rates in the yeast and the efficiency of gene editing positively correlates with GC content at the target sites. 52,53
A third possible explanation of the higher correction rate of EGFP Y66S is the potential for multiple lentiviral vector integration events. We used an MOI of 1.5 TU/cell for lentiviral infection and would thus expect EGFP Y66S alleles to be less than the two CFTR copies per cell. However, it is possible that puromycin selection enriched cells with multiple copies of the EGFP Y66S expression cassette.
A previous study in primary human airway basal cells derived from ΔF508/ΔF508 CF airways has reported that Zinc-finger nucleases-mediated HDR using transfected ssOND or rAAV6-delivered as the HR templates can achieve ∼10.6% and ∼31.0% gene editing efficiency at the ΔF508-CFTR locus, respectively, for edited cells. 54 When differentiated into ALI cultures, these cells produced ∼15% (ssOND) and ∼40% (rAAV) cAMP-inducible CFTR Cl− currents observed in non-CF ALI cultures, suggesting a direct correlation between gene-editing frequency and restoration of CFTR current. 54 By contract, our study comparing unsorted and sorted (EGFP-targeted enriched) populations of CFTR gene-edited ferret basal cells demonstrated greater CFTR functional correction than gene editing frequency. For example, ALI cultures derived from unsorted basal cells with a 3.1% gene-editing frequency produced 15.6% and 16.7% of the WT ΔIsc Cl− responses to cAMP and GlyH101 treatment, respectively, when baseline currents in unedited parental CF cultures were subtracted from values.
This represents a 5.1- to 5.4-fold greater functional correction than gene-editing frequency. This was also true for sorted ALI cultures, for which functional restoration of CFTR-mediated Cl− transport was 3.0- and 3.6-fold greater than gene-editing frequency for cAMP and GlyH101 responses, respectively. The reason for this difference in the correlation of gene editing frequency versus CFTR functional correction in our study versus those by Suzuki et al. 54 remains unclear, but could involve the persistence of certain CFTR-expressing lineages in human versus ferret primary basal cell cultures and their mechanisms of involvement in CFTR-mediated anion conductance. 11
Consistent with our findings, others have shown that ALI cultures derived from mixtures of non-CF and ΔF508/ΔF508 primary human airway epithelial cells at a 2:8 ratio (WT:CF) restore ∼70% CFTR-mediated Cl− transport of those 100% WT cultures. 55
The ability to correct mutations at an endogenous locus is leading to a new era of personalized medicine. Although gene-editing therapy for CF lung disease is still at a very early stage, many promising approaches are being pursued. Such approaches include HDR, 54,56 HITI, 54 base editors, 57 prime editing, 58 and programmable nuclease-mediated integration of a CFTR minigene expression cassette into a genome safe harbor or the CFTR locus. 50,51 These approaches are being applied in vitro in primary airway basal cells, iPSCs, and organoid cultures derived from CF patients and demonstrated proof-of-concept for gene therapy. While the CF animal models are ready to facilitate this research toward in vivo gene editing of the lung, the availability of effective airway vectors for delivery of the CRISPR components remains a challenge.
Additional challenges include the fact that HDR is highly cell cycle dependent. Most vector-accessible epithelial cells in the airways are mitotically quiescent and do not support efficient HDR. HITI, base editors, and prime editing are possible solutions to correct mutations in nondividing cells, including quiescent differentiated airway epithelial cells. Although the turnover rates of the human airway epithelial cells remain unclear, terminally differentiated ciliated cells of the mouse airways have a half-life of ∼17 months. 23 Since ciliated cells do not express CFTR, 16 they are not a viable target for CFTR gene editing. However, if vectors were available to efficiently target secretory cells or pulmonary ionocytes, CFTR gene editing could produce durable functional complementation if these cell types have similar lifespans as ciliated cells.
Nevertheless, permanent correction will only be achieved if stem cell compartments are efficiently targeted (i.e., basal cells in the proximal airways and club cells in the bronchioles).
59
Efficient in vivo rAAV transduction of basal cells has yet to be achieved, due to a lack of an exposed apical membrane. However, method of transiently disrupting the columnar cell tight junctional barrier with
Efficient HDR-based ex vivo CFTR gene editing in airway basal cells is the first step in applying autologous cell-based therapy to the CF airway. Additional requirements for efficient cell-based therapy include maintenance of multipotency during ex vivo expansion, tractable methods of engraftment, and long-term persistence in the recipient airways. Toward this goal, we present the creation of a new CFTR G551D ferret model for which differentiated airway cultures are responsive to the CFTR potentiator VX-770. Our study demonstrates that CFTR gene-editing correction in a small fraction of airway basal cells can restore ∼3- to 6-fold greater functional correction of CFTR than the gene-editing frequency.
Given that NHEJ dominated HR in the presence of a donor rAAV template and CRISPR/Cas9 components, improvements in the current gene-editing approach of proliferating airway basal cells are required before cell therapy applications in the G551D ferret model. The developed primary cell system with an HDR reporter may be useful in optimizing and studying the cell-intrinsic properties responsible for efficient gene editing in primary basal cells.
Footnotes
AUTHORs' CONTRIBUTIONS
Z.Y. and J.F.E. conceived and designed the experiments. K.V., Z.F., S.Y.P., S.C., Y.Z., M.W., and X.S. performed the experiments. X.S. contributed to zygote microinjection and NSG data analysis. Z.Y. and J.F.E. wrote the article. All authors contributed to data analysis, edited the article, and approved submission.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by grants from the National Institutes of Health P30 DK054759, P01 HL152960, R01 HL165404, and Federal Contract #75N92019R0014 to John F. Engelhardt, a grant from the Cystic Fibrosis Foundation to John F. Engelhardt, and a research grant YAN19XX0 from the Cystic Fibrosis Foundation to Ziying Yan.