
Abstract
Chimeric antigen receptor (CAR) T cell therapy has transformed the treatment of hematological malignancies but has yet to achieve similar success in solid tumors due to a lack of persistence and function in the tumor microenvironment. We previously reported the augmentation of CAR T cell therapy in an engineered solid tumor model through the secretion of anti-PD-1 single-chain fragment variable region (scFv), as shown by enhanced CAR T cell antitumor efficacy, expansion, and vitality. We have since improved the platform to create a superior cellular product—CAR T cells secreting single-chain trimeric 4-1BB ligand fused to anti-PD-1 scFv (αPD1-41BBL). 4-1BB signaling promotes cytotoxic T lymphocyte proliferation and survival but targeting 4-1BB with agonist antibodies in the clinic has been hindered by low antitumor activity and high toxicity. CAR T cells using 4-1BB endodomain for costimulatory signals have demonstrated milder antitumor response and longer persistence compared to CAR T cells costimulated by CD28 endodomain. We have, for the first time, engineered CD28-costimulated CAR T cells to secrete a fusion protein containing the soluble trimeric 4-1BB ligand. In vitro and in vivo, CAR19.αPD1-41BBL T cells exhibited reduced inhibitory receptor upregulation, enhanced persistence and proliferation, and a less differentiated memory status compared to CAR T cells without additional 4-1BB:4-1BBL costimulation. Accordingly, CAR19.αPD1-41BBL T cell-treated mice displayed significantly improved tumor growth control and overall survival. Spurred on by our preclinical success targeting CD19 as a model antigen, we produced mesothelin-targeting CAR T cells and confirmed the enhanced solid tumor efficacy of αPD1-41BBL-secreting CAR T cells.
INTRODUCTION
Cancer immunotherapy has recently caused a paradigm shift in the treatment of cancer. 1 In contrast to the three traditional pillars of cancer therapy, namely surgery, radiation, and chemotherapy, which directly target or remove cancer cells, immunotherapy harnesses the power of the immune system to eradicate cancer. To date, the centerpiece for successful cancer immunotherapies is engaging the T lymphocyte of the immune system and improving its antitumor efficacy, especially through the use of immune checkpoint inhibitors or by engineering T cells to express chimeric antigen receptors (CARs). 2,3
CARs are composed of an antigen-recognizing extracellular moiety, a hinge, a transmembrane domain, and intracellular T cell activating and costimulatory domains. 4 CAR T cells recognize tumor associated antigen on tumor cells through the extracellular domain, which triggers intracellular domains to activate the T cells to attack tumor cells. CAR T cell therapy has achieved remarkable success in hematological malignancies, but its clinical efficacy in solid tumors requires further improvement. 5 –7 A critical challenge encountered by CAR T cells in both hematological malignancies and solid tumors is the lack of function and proliferation upon prolonged antigen exposure. 8 The immunosuppressive solid tumor microenvironment (TME) only worsens the problem with a multitude of inhibitory molecules and receptors present in the TME suppressing effector cell functions. 9
Substantial efforts have been aimed at improving CAR designs to generate CAR T cells with better resistance to exhaustion and hypofunctionality, focusing on (1) providing additional signals to promote T cell activation or costimulation, 10,11 (2) engineering CAR T cells to transgenically express cytokines or constitutively active cytokine receptors that support proliferation, 12,13 (3) silencing or knocking-out molecules that restrict T cell activation, 14 and (4) modulating transcription factors in CAR T cells. 15,16 Among them, 4-1BB signaling components have been prominent candidates given the costimulatory role of 4-1BB:4-1BBL in supporting the clonal expansion, survival, cytokine release, and effector functions of immune cells. 17 Besides, the use of 4-1BB endodomain for costimulation in CARs, which favors CAR T cell persistence, 18 transgenic expression of 4-1BB ligand on CD28 costimulated CAR T cells provided significantly better therapeutic efficacy compared to both 4-1BB-costimulated and CD28-costimulated CAR T cells. 19
Another method of targeting 4-1BB involves the use of monoclonal antibody agonists. Although strong antitumor efficacy has been observed in preclinical models, their clinical development was restricted because of either low efficacy or severe liver toxicity. 20 Compared to 4-1BB agonist antibodies, 4-1BB ligand has demonstrated better immunomodulatory activity and safety profile, 21 but the soluble ligand on its own is inactive and requires crosslinking for T cell costimulatory activity. 22 Wyzgol et al. confirmed that the activity soluble trimeric 41BBL can be strongly increased by oligomerization or cell surface immobilization, and targeted immobilization of the tumor necrosis factor ligand could be achieved by genetic fusion to a single-chain fragment variable region (scFv) Ab fragment. 23
We previously engineered CAR T cells to secrete anti-PD-1 checkpoint inhibitor. 24 The secreted anti-PD-1 scFv protected CAR T cells from inhibition by PD-1:PD-L1 interaction and significantly enhanced the antitumor efficacy of the CAR19.αPD1 T cells compared to parental CAR T cells, especially in aH292 tumor model where PD-L1 is upregulated. Despite the clinical success of PD-1/PD-L1 blockade across multiplemalignancies, PD-1 blockade only benefits a minorityof patients (20–30%) and several mechanisms lead to both primary and acquired resistance to PD-1 blockade. 25,26 One prominent mechanism is the loss of intratumoral T cell function characterized by a lack of memory T cells and re-exhaustion of effectors cells invigorated by PD-1 blockade.
Since 4-1BB costimulation favors T cell expansion and persistence and the development of T cell memory, and 4-1BB ligand requires crosslinking for costimulatory activity, we hypothesized that fusion of the current anti-PD-1 scFv with 4-1BB ligand would provide additional benefits to CAR T cells and is potentially of translational value in the management of tumors resistant to PD-1 blockade due to insufficient T cell function. Therefore, we engineered CAR T cells to secrete a novel immunomodulatory fusion protein consisting of anti-PD-1 scFv linked to a single-chain format of trimeric 4-1BB ligand. The single-chain format is based on a previous design that connects three extracellular domain units with polypeptide linkers. 27 In the current study, we uncovered potent αPD1-41BBL-mediated effects on protecting human CAR T cells from the upregulation of inhibitory receptors and that αPD1-41BBL secretion significantly enhanced CAR T cell preclinical antitumor activity.
MATERIALS AND METHODS
Protocols used for retroviral vector production, T cell transduction and expansion, surface immunostaining analysis, and intracellular cytokine staining analysis are based on protocols in a previous publication. 28
Antibodies
Primary antibodies used in this study include biotinylated goat anti-mouse Fab antibody (Invitrogen, Carlsbad, CA, USA); PE-anti-hCD45, PerCP-anti-hCD45, PE-Cy5.5-anti-hCD3, FITC-anti-hCD4, PerCp-anti-hCD4, Pacific BlueTM-anti-hCD8, FITC-anti-hCD8, PE-anti-hCD8, PE-anti-IFN-γ, Brilliant Violet 421™-anti-hPD-1, PE-anti-hPD-L1, PerCP/Cy5.5-anti-hLAG-3, PE-anti-hTIM-3, BV-anti-hCD25, APC-anti-IL-2, PerCP-anti-TNFα, PE-anti-FOXP3, APC-anti-CD62L, PE-anti-CD45RA (BioLegend, San Diego, CA, USA), recombinant Anti-HA tag antibody (Abcam, Cambridge, United Kingdom), and beta-actin mouse monoclonal antibody (LI-COR, Lincoln, NE, USA). The secondary antibodies used were APC-conjugated streptavidin (BioLegend), IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody (LI-COR), and Goat anti-Rabbit IgG antibody with HRP conjugate (Sigma-Aldrich, St. Louis, MO, USA).
Cell lines and cell culture
SKOV3 and 293Tcells (ATCC) were maintained in Dulbecco's Modified Eagle Medium with 10% fetal bovine serum (FBS) (Gibco), 2 mM
Mice
Female 6–8 weeks old
Plasmid design
The retroviral vector encoding anti-CD19 CAR (CAR) was constructed based on the MP71 retroviral vector kindly provided by Prof. Wolfgang Uckert. The vector encoding anti-CD19 CAR with anti-PD-1 scFv secretion and αPD1-4-1BBL fusion protein secretion were then generated from the anti-CD19 CAR. The CD19 CAR with anti-PD-1 scFv secretion consisted of the following components in frame 5′ to 3′ end: anti-CD19 CAR, P2A linker, human IL-2 leader sequence, and aPD1 scFv with HA tag. The CD19 CAR with fusion protein secretion consisted of the following components in frame 5′ to 3′ end: anti-CD19 CAR, P2A linker, human IL-2 leader sequence, aPD1 scFv, GS linker, 4-1BBL extracellular domain 1, 2nd GS linker, 4-1BBL extracellular domain 2, 3rd GS linker, and 4-1BBL extracellular domain 3 with HA tag. For producing mesothelin-targeting CAR T cells, the scFv SS1 was cloned in place of the anti-CD19 scFv (FMC63).
Protein isolation and characterization
HEK-293T cells were transduced with CAR19.αPD1 or CAR19.αPD1-41BBL retroviral vectors for the stable expression of HA-tagged anti-PD-1 scFv or fusion protein aPD1-41BBL. Following successful transduction and expansion, the engineered cells were seeded in 10 mL plates in D10. Sixteen hours later, the cells were rinsed twice with phosphate buffered saline (PBS) and then cultured for 48 h in 10 mL serum free media. Supernatants were subsequently collected, clarified, and then centrifuged in 10 kDa isolation columns (Sigma) for 1 h at 5,000 g 4°C. The remaining supernatant was purified for HA-tagged protein using Dynabeads (Thermo) according to the manufacturer's instructions, and standard BCA assay (Sigma) was used for quantification of the purified proteins.
Binding assays were conducted by testing the affinity of purified proteins to recombinant human PD-1 or recombinant human 4-1BB. Ninety-six-well plates (Maxisorp) were coated with 10 ng/mL human PD-1 Fc chimera or human 4-1BB Fc chimera (Genscript, Piscataway, NJ, USA) in PBS for 4 h at 4°C, after which the coated wells were blocked with 1% bovine serum albumin in PBS with .05% Tween 20 (PBS-T) for 1 h at room temperature (RT). Then, wells were washed with PBS-T and incubated with increasing concentrations (0, 0.098, 0.39, 1.56, 6.25, 25, and 100 nM in triplicates) of aPD1 scFv or αPD1-41BBL fusion protein for 1.5 h at RT. After three washes with PBS-T, wells were incubated for 1 h at RT with rabbit anti-HA (Abcam) antibody, followed by washes and 1-h RT incubation with secondary HRP-conjugated anti-rabbit antibody.
After washes, enzyme concentrations were detected with tetramethylbenzidine substrate by measurement of the absorbance at 450 nm with subtraction of the absorbance at 570 nm. The apparent Kd values were obtained by nonlinear regression analysis in Prism software (version 7; GraphPad) assuming one-site–specific binding.
For western blotting, 1ug of each purified protein was used per group for SDS-PAGE using 7.5% Mini-PROTEAN precast gel (Bio-Rad, Hercules, CA, USA) together with Chameleon® 700 Prestained Protein Ladder (LI-COR) and transferred to a nitrocellulose membrane (Thermo Scientific, Waltham, MA, USA) for western blot analysis. The membrane was stained with Rabbit anti-HA antibody (Abcam) at 1:1,000 in 5% milk in TBS-T and IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody (LI-COR) at 1:10,000, and its chemiluminescence detected with Odyssey® Fc imaging unit (LI-COR).
Preclinical tumor models
Six to eight weeks old NSG mice were injected subcutaneously with 2.5 × 106 SKOV3.CD19 cancer cells in 50 μL. Tumor volume was determined by caliper measurement (L × W 2 /2). Physical examination of the implanted tumors revealed a distinct “fixed” phenotype of the tumors in response to lateral displacement of the overlying skin, indicating successful subcutaneous tumor formation. 29 Once tumors reached an average size of 50–80 mm3, CAR T cells were injected intravenously at the dose of 1.5 × 106 CAR+ cells per group. Tumor sizes were measured three times a week and mice were euthanized when they displayed obvious weight loss, ulceration of tumors, or tumor size larger than 1,000 mm3.
For investigation with mesothelin-targeting CAR T cells, mice were inoculated with 2.5 × 106 SKOV3.Meso cancer cells and treated with 3.5 × 106 CAR+ T cells intravenously. Tumor growth was monitored with calipers and tail bleeds were performed on days 5, 10, 15, 20, and 25. Whole blood specimen was placed into a heparin tube and kept on ice. Red blood cells were lysed with chilled ammonium chloride buffer, after which cells were prepared for flow cytometry analysis.
Organ harvest for T cell analysis
Tumor, spleen, bone marrow, and blood were harvested from mice. The resected tumors were weighted, and the tumor, spleen, and bone marrow were minced and filtered through 70 μm nylon strainers (BD Falcon, Franklin Lakes, NJ, USA) for single-cell suspensions. Blood from cardiac puncture was immediately transferred to ammonium chloride buffer for red blood cell lysis. The filtered cells and blood samples were washed and incubated with ammonium chloride buffer if necessary, and then blocked with 1% BSA in PBS. Cells were then prepared for fluorescent-activated cell sorting.
Statistical analysis
Statistical analysis was performed in GraphPad Prism version 5.01. Tumor growth curves were analyzed using two-way analysis of variance (ANOVA) with Tukey's posttest for multiple comparisons and mice survival curves were evaluated by log-rank Mantel–Cox test analysis. Other statistical differences were determined with two-tailed unpaired t-tests, using the Holm-Sidak method for multiple comparisons.A p-value <0.05 was considered statistically significant. Significance of findings were defined as: ns = not significant, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001. In vitro assays are performed in triplicate and representative of at least two independent experiments. For in vivo studies each figure specifies the number of mice studied (n = 3–6 mice/group).
RESULTS
Design of the CAR and protein characterization
We previously engineered anti-PD-1 scFv secreting CAR T cells (CAR19.αPD1) from a traditional CD19 targeting, CD28 costimulated second-generation CAR construct. For the new CAR19.αPD1-41BBL construct, we linked anti-PD-1 scFv with 4-1BBL trimer of a single-chain format to create the fusion protein αPD1-41BBL (Fig. 1A). Three units of ectodomain of the 4-1BB ligand are linked by flexible GS linkers and aschematic of the fusion protein is shown in Fig. 1B, illustrating the 1:1 ratio of αPD1 to 41BB-trimerized ligand.

CAR and fusion protein design and characterization.
Following the successful cloning of the CAR constructs, retroviral vectors containing the CARs were generated and used to transduce activated human peripheral blood mononuclear cells (PBMCs). All three CAR constructs were expressed in T cells (Fig. 1C) and CAR T cells were expanded for 12 days followed by cryopreservation. CAR T cells were thawed, and CAR expressions were normalized with nontransduced (NT) T cells for all the in vitro and in vivo studies. 41BB is upregulated on activated T cells, 30 which we confirmed by coculturing CAR T cells with antigen presenting cells (Supplementary Fig. S1).
The two modified CAR T cells secreted their respective immunomodulatory proteins. Anti-hemagglutinin (HA)-stained immunoblot identified anti-PD-1 scFv at a molecular weight of ∼25 kDa, and αPD1-41BBL at ∼90 kDa (Fig. 1D). We assessed the binding kinetics of secreted anti-PD-1 scFv and fusion protein to recombinant human PD-1 (Fig. 1E) and recombinant human 4-1BB (Fig. 1F) using ELISA. Similar to previously reported, 27 fusion of antibody scFv to the single chain format of 4-1BBL significantly reduced scFv binding affinity, as shown by the increase in EC50 from ∼2.5 to ∼100 nM for recombinant human PD-1. The fusion protein had nanomolar affinity for recombinant human 4-1BB with EC50 ∼ 6.25 nM.
CAR T cells secreting αPD1-41BBL maintain cytotoxicity but have reduced cytokine production
We conducted in vitro cytotoxicity and cytokine production assays to evaluate CAR T cell effector functions. For specific cell lysis analysis, CAR T cells were cocultured with SKOV3.CD19 cells at effector to tumor cell (E:T) ratios of 0.1:1, 0.5:1, 1:1, 3:1, and 5:1 for 16 h, followed by flow cytometry analysis. The CAR T cells were highly efficient killers of SKOV3.CD19 cells and there was no significant difference in the tumor killing ability between CAR groups at the tested E:T to ratios (Fig. 2A).
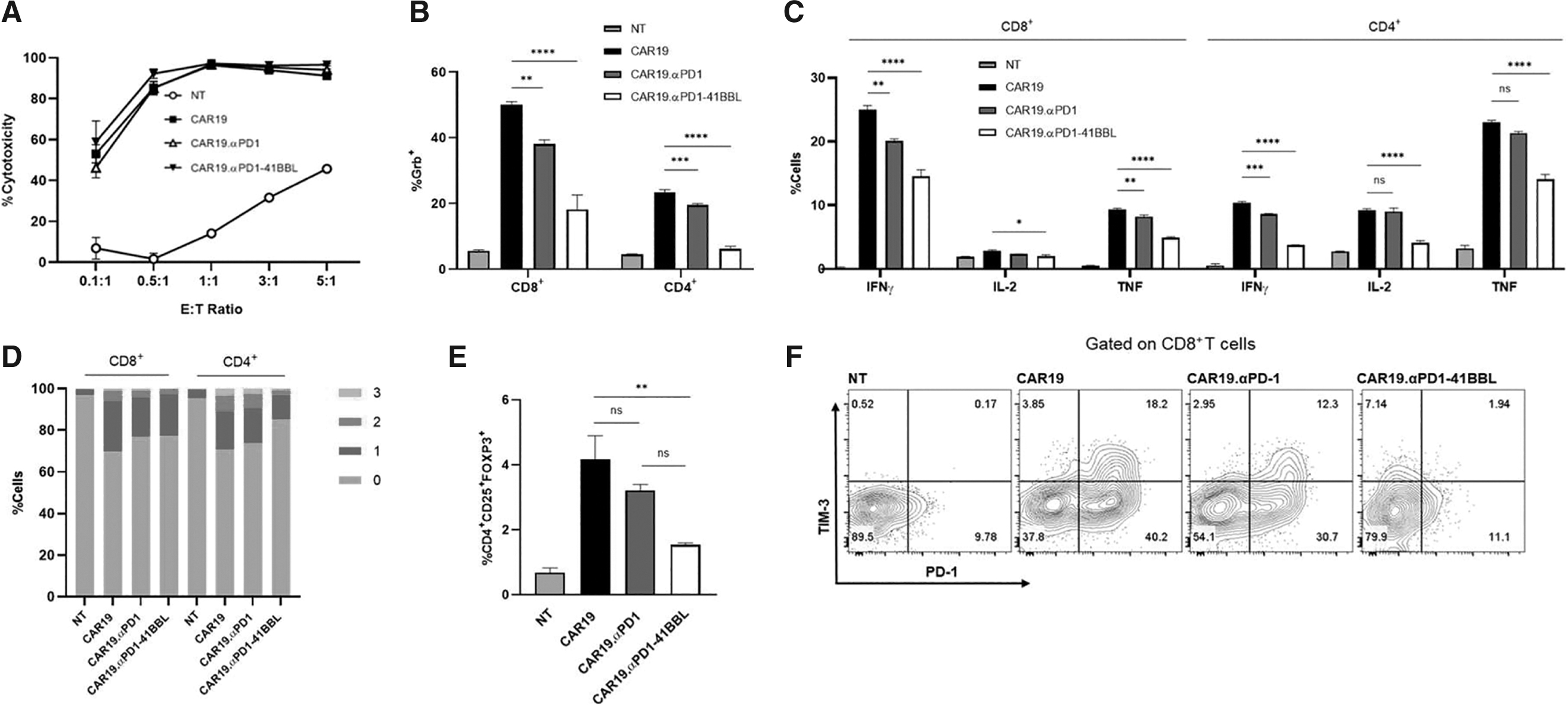
In vitro analysis of CD19-targeting CAR T cells.
Following 24 h coculture with antigen presenting tumor cells, in which Brefeldin A was added at hour 18 to block protein transport, CAR T cells were stained for intracellular cytokine expression. Interestingly, CAR19.αPD1-41BBL T cells had the lowest percentage of T cells positive for effector cytokines. Figure 2B and C shows the expression levels for granzyme B, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-2 for NT and CAR T groups. CAR T cells with protein secretions expressed less granzyme B, IFN-γ, and TNF-α compared to parental CAR19 T cells for both CD8+ and CD4+T cells. CAR19.αPD1 expressed commensurate IL-2 level compared to parental CAR19 T cells, and CAR19.αPD1-41BBL T cells showed lower IL-2 expression level.
Similarly, cytokine coexpression analysis for IFN-γ, TNF-α, and IL-2 revealed less polyfunctional CAR T cells in the αPD1-41BBL group, which contained a smaller percentage of CAR T cells expressing two or three cytokines simultaneously (Fig. 2D). The reduction in effector cytokine production was not due to T regulatory
Anti-PD1-41BBL protects CAR T cells from inhibitory receptor upregulation
NT and CAR T cells were assessed for inhibitory receptor expression following 24 h coculture with antigen presenting tumor cells (Supplementary Fig. S2A). In accordance with our previous report, 24 CAR19.αPD1 T cells showed less PD-1, LAG-3, and TIM-3 expression levels compared to CAR19 T cells. CAR19.αPD1-41BBL T cells showed even less PD-1, LAG-3, and TIM-3 expression levels than CAR19.αPD1 T cells, with a 70% reduction in PD-1 expression on CD8+ T cells, as well as 15% and 55% reductions in LAG-3 and TIM-3 expression, respectively. We further assessed T cell inhibitory marker upregulation by measuring the coexpression of PD-1, LAG-3, and TIM-3 after 48 h coculture (Fig. 2F, G). In both the CD8+ and CD4+ compartments, the fusion protein-secreting CAR T cells had significantly lower percentages of cells expressing multiple coinhibitory receptors compared to parental CAR19 T cells and CAR19.αPD1 T cells.
To validate that the secreted proteins are the source of CAR T cell protection from inhibitory receptor upregulation, we collected supernatants from CAR19, CAR19.αPD1, and CAR19.αPD1-41BBL cell cultures and put parental CAR19 T cells in the supernatants with or without antigen-presenting tumor cells. Twenty-four hours later, T cells were again assessed for the expression of coinhibitory receptors. When cultured with CAR19.αPD1-41BBL supernatant, CAR19T cells showed significantly lower expression of PD-1, TIM-3, and LAG-3 after antigen stimulation (Supplementary Fig. S2B). CAR19.αPD1-41BBL supernatant also reduced the percentage of T cells expressing multiple inhibitory receptors in cultures with (Fig. 2H left) or without target cells (Fig. 2H right), with a much more distinct reduction observed in the presence of target cells (Fig. 2H left).
Culturing parental CAR19 T cells in CAR19.αPD1 T cell supernatant and CAR19.αPD1-41BBL supernatant did not affect cytotoxic functions of parental CAR19 T cells (Fig. 2I), indicating that the reduced exhaustion marker upregulation does not come at the cost of cytolytic potency.
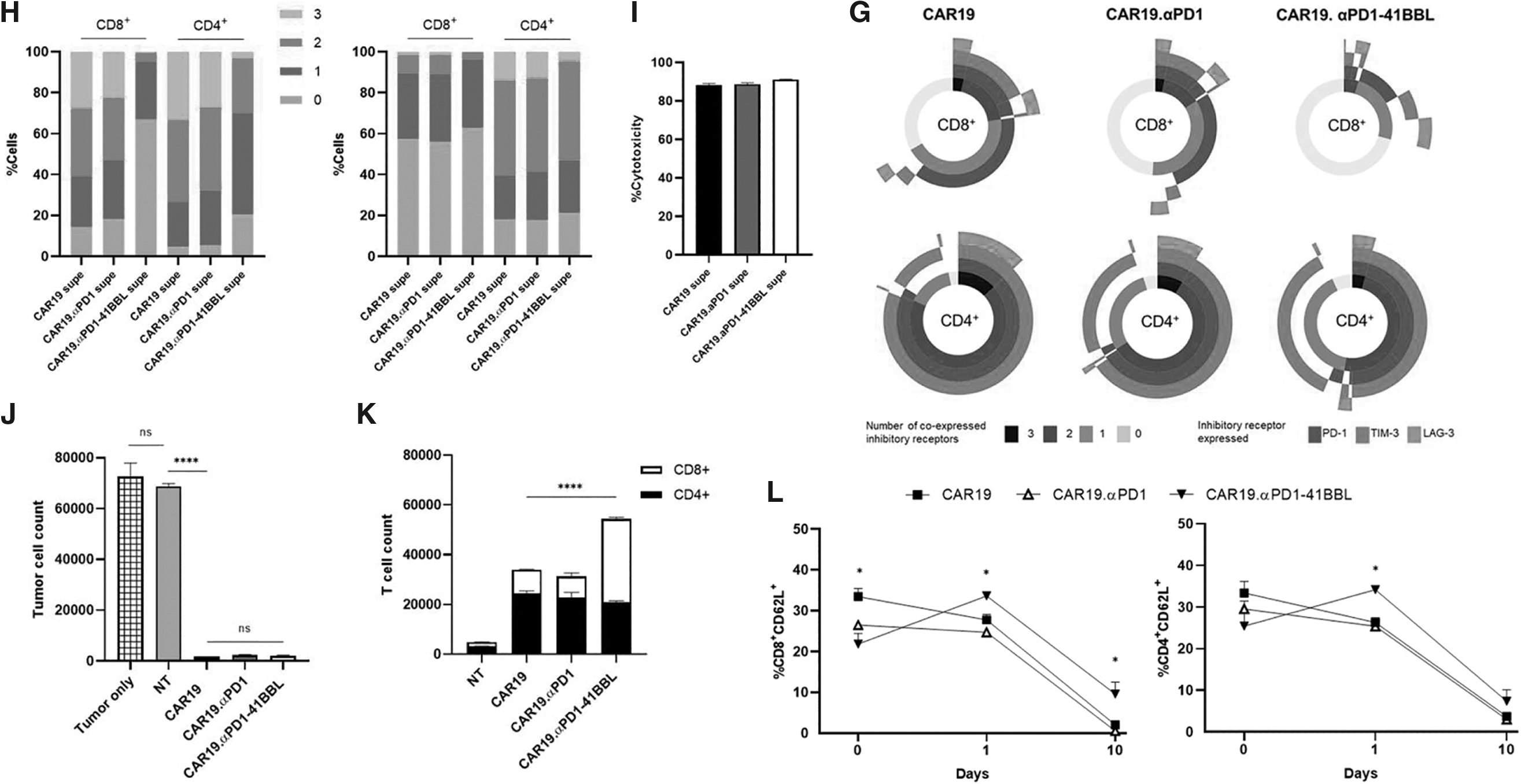
41BBL fusion enhances T cell persistence and preserves T cell memory status
We performed repeated tumor challenge assays to investigate the long-term functionality and persistence of the CAR T cells. On days 0, 2, 5, and 8, tumor cells were added to T cell cultures. On day 10, cytotoxicity of target cells and T cell persistence were quantified. The CAR T cell groups were highly effective target cell killers over the 10-day period (Fig. 2J), lysing almost all the tumor cells introduced and highlighting the robustness of the parental CAR19 T cells. Only CAR19.αPD1-41BBL T cells experienced significantly enhanced T cell proliferation (Fig. 2K). The larger T cell number was due to enhanced expansion of CD8+ cells, supporting 41BB:41BBL axis promotion of CD8+ T cell persistence.
The reduced exhaustion marker upregulation and cytokine production, and enhanced persistence of our αPD1-41BBL-producing T cells led us to hypothesize that the fusion protein reduced CAR T cell differentiation. 31 CD62L is a marker of T cell differentiation and has been used to determine CAR T cell memory status. 32 Using the same repeated tumor challenge, CAR T cells were stained for percentages of CD62L+ cells before the addition of tumor cells (Day 0) and after coculture with SKOV3.CD19 cells for 24 h and 10 days (Fig. 2L). CAR19.αPD1-41BBL T cells did not express higher levels of CD62L compared to CAR19 T cells and CAR19.αPD1 T cells before antigen exposure, as shown by the baseline staining (day 0).
With antigen exposure, CAR19.αPD1-41BBL T cells showed a less differentiated state compared to both CAR19 and CAR19.αPD1 T cells since CAR19.αPD1-41BBL T cells had a significantly higher percentage of CD62L+ cells. Specifically, the CD8+ T cells in the fusion protein group displayed a higher percentage of CD62L+ cells on day 1 and 10 than other CAR T cell groups, and the CD4+ T cells displayed higher CD62L expression on day 1.
Anti-PD1-41BBL secretion enhances CAR T cell antitumor efficacy
The in vivo antitumor activity of CAR19.αPD1-41BBL T cells was evaluated in a xenograft tumor model. Once SKOV3.CD19 tumors in NSG mice reached a volume of 50–80 m3, we adoptively transferred 1.5 × 106 CAR T cells into mice by intravenous injection (Supplementary Fig. S3A). Tumor growth and body weight (Supplementary Fig. S3B) in all mice were monitored two-three times per week. The CAR19.αPD1-41BBL group started showing significantly better tumor growth control at day 9 compared to other groups (Fig. 3B). As shown by the individual tumor growth curves in Fig. 3A, CAR19.αPD1-41BBL T cells reduced tumor burden for all six mice in the group by day 12, while other CAR T cell groups only showed control of tumor growth rather than tumor burden reduction after treatment.
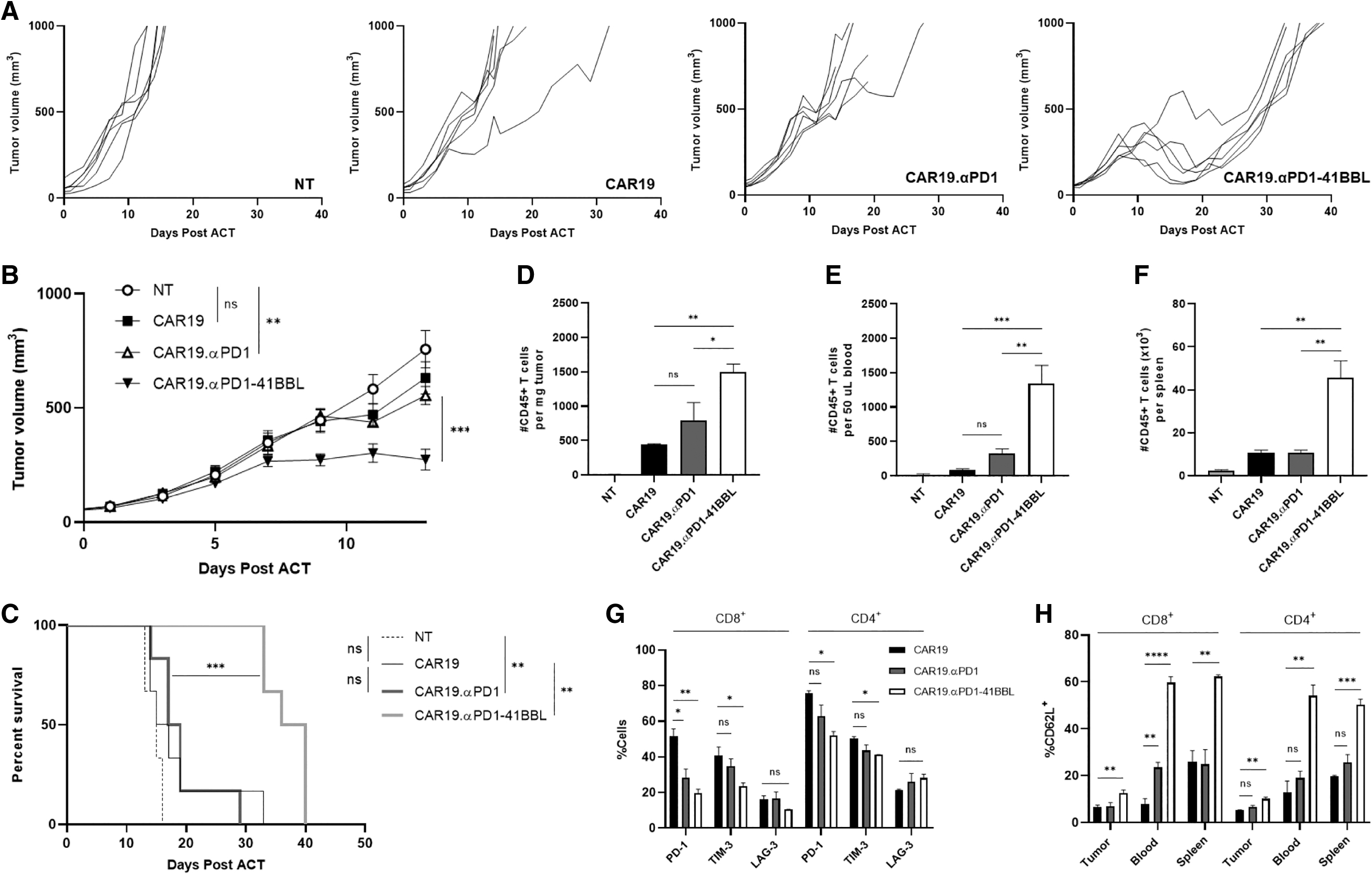
Anti-PD-1-41BBL secretion enhances CAR19 T cell in tumor control and expansion in a xenograft mouse model. Mice bearing SKOV3.CD19 tumor were treated with T cell variants and monitored for
All NT-treated mice were euthanized by day 17, and 2 days later, five of six mice in the CAR19 and CAR19.αPD1 groups had reached experimental endpoint, whereas 100% of the CAR19.αPD1-41BBL group were alive (Fig. 3C), with an average tumor size of 180 mm3. Despite the distinct tumor regression witnessed in all mice in the CAR19.αPD1-41BBL group, the mice eventually succumbed to cancer progression.
Anti-PD1-41BBL secretion augments CAR T cell in vivo expansion and exhaustion resistance
As in the tumor growth study, mice were inoculated subcutaneously with 2.5 × 106 SKOV3.CD19 tumor cells (Supplementary Fig. S3B). When the tumor sizes reached 50–80 mm3, mice were treated with 1.5 × 106 CAR T cells. Ten days after treatment, the mice were sacrificed, and their organs homogenized and analyzed for human T cell proliferation, phenotype, and exhaustion markers. CAR19.αPD1-41BBL T cells expanded significantly more than CAR19.αPD1 T cells and CAR19 T cells, as manifested from the number of CD45+ cells per mg of tumor, per 50 μL of blood, and per spleen (Fig. 3D–F) and shown by the percentage of CD45+ cells in the harvested tissues (Supplementary Fig. S3C, D).
There was no significant difference in CD8+:CD4+ T cell ratios between the CAR groups in the tissue harvested (Supplementary Fig. S3C). In the CAR19.αPD1-41BBL T cell treatment group, the tumor infiltrating lymphocytes expressed less PD-1 and TIM-3 compared to the tumor infiltrated lymphocytes in the parental CAR19 T cell (Fig. 3G). There were also significantly higher percentages of CD62L+ T cells in tumor, blood, and spleen (Fig. 3H) of the CAR19.αPD1-41BBL T cell treatment group, illustrating the earlier memory state of human T cells circulating in these mice.
Mesothelin-targeting CAR T cells enhanced by αPD1-41BBL secretion
Spurred on by the noteworthy preclinical success of αPD1-41BBL expressing CAR T cells targeting CD19 as a model antigen, we switched the scFv in the CAR T cell groups to target mesothelin (meso) (Supplementary Fig. S4). Mesothelin is a cell-surface glycoprotein with limited expression on normal tissue such as the pleura, peritoneum, and pericardium, but is overexpressed in many solid cancers, including ovarian, lung, and pancreatic carcinomas, making mesothelin a promising target for cancer therapies, including CAR T cells. 33 Meso-targeting CAR T cells, although proved safe in clinic, have resulted in minimal objective responses, and multiple ongoing trials are testing meso-targeting CAR T cells designed for improved antitumor activity. 34 –37
Using the SS1 anti-meso scFv, we produced CARMeso, CARMeso.αPD1, and CARMeso.αPD1-41BBL T cells (Fig. 4A) and confirmed the binding of secreted proteins to activated CARMeso T cells (Fig. 4B). The CAR T cells specifically lysed meso-expressing cancer cells (Fig. 4C, D) and displayed similar in vitro trends to CD19-targeting CAR T cells. Anti-PD1-41BBL secretion reduced cytokine production, although to a lesser extent given solely granzyme B production was significantly lessened (Fig. 4E). Similarly, anti-PD1-41BBL secretion protected CAR T cells from inhibitory receptor upregulation (Fig. 4F, G), prevented T regulatory cell differentiation (Fig. 4H), preserved earlier memory status of both CD8+ and CD4+ populations (Fig. 4I), and enhanced CAR T cell expansion and persistence in long-term in vitro repeated challenge studies (Fig. 4J).
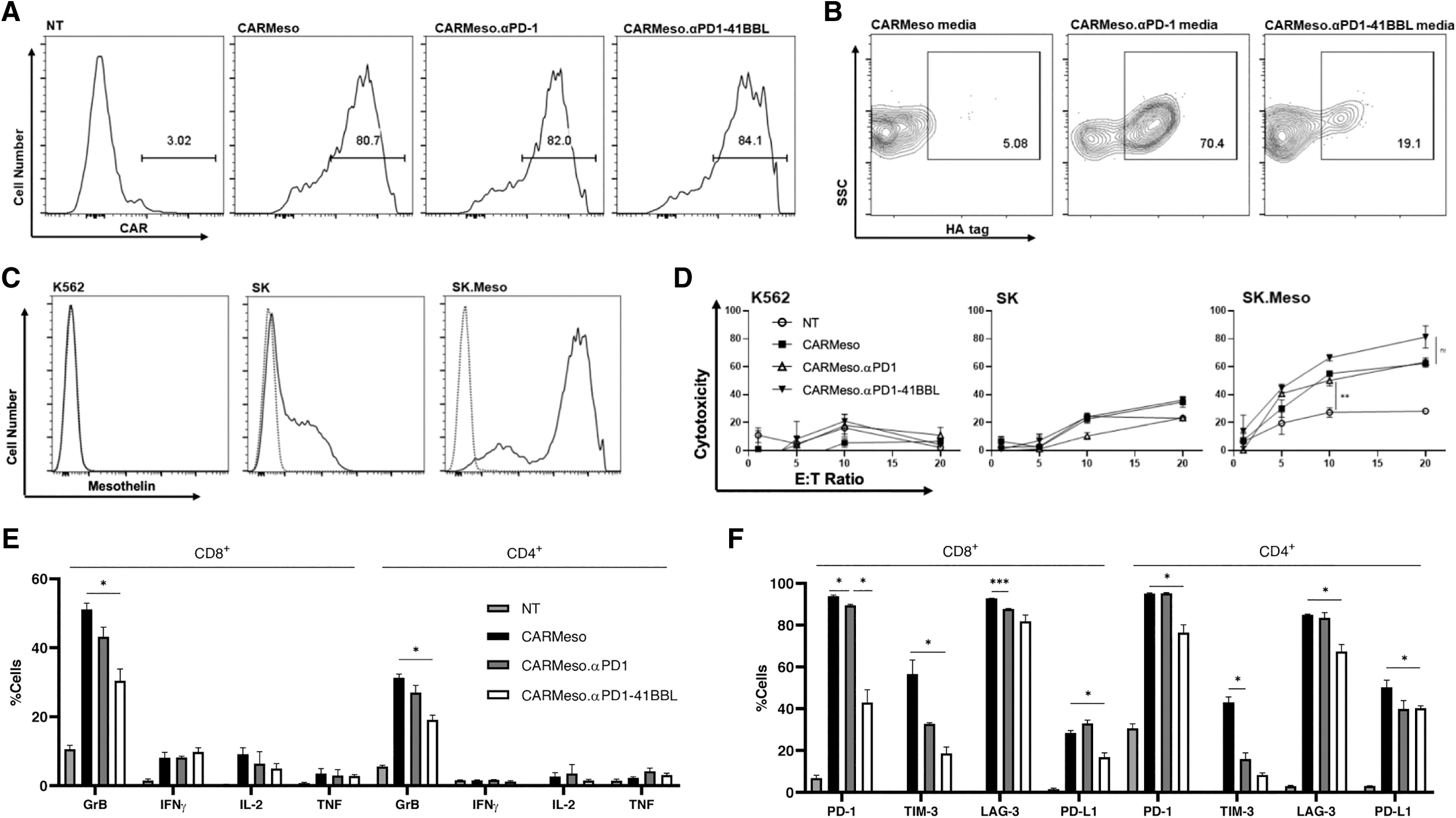
In vitro characterization of mesothelin-targeting CAR T cells.

PD-L1 upregulation was also assessed given the recently elucidated role of T cell PD-L1 in the promotion of self-tolerance and suppression of neighboring effector T cells in cancer, 38 and the fusion protein reduced PD-L1 expression (Fig. 4F). CD62L and granzyme B costaining analysis demonstrated that CD62L− cells have higher expression of granzyme B for both CD8+ and CD4+ cells (Fig. 4K). Since CARMeso.αPD1-41BBL T cells had a larger proportion of CD62L+ T cells, and for all the CAR groups, the CD62L+ cells produced less granzyme B, the costaining results confirm the correlation between higher CD62L+ population and lower granzyme B expression upon antigen exposure.
Given the requirement of high mesothelin expression and high effector:target ratio for significant CAR-mediated 16 h cytotoxicity, we elected to use SKOV3.Meso tumor cells (SKOV3 cells stably transduced to express mesothelin) and a CAR T cell dose of 3.5 × 106 cells for our in vivo study (Fig. 5A). At this dose, all the CAR groups significantly controlled tumor growth when compared to control (Supplementary Fig. S5A) without significant body weight loss in any treatment group (Supplementary Fig. S5B), although it was the αPD1-41BBL-secreting CAR T cells that had superior long-term tumor control compared to all other groups (Fig. 5B–D). Tail bleeds of the mice on days 5, 10, 15, 20, and 25 were performed to study T cell in vivo expansion and persistence (Fig. 5E–G).

Anti-PD-1-41BBL secretion augments mesothelin-targeting CAR T cell in vivo persistence and antitumor efficacy.

CARMeso T cells achieved the highest maximum expansion in peripheral blood (day 10), whereas CARMeso.αPD1-41BBL T cells had enhanced persistence as shown by increased CD8+ T cell percentages in peripheral blood on days 15, 20, and 25. Phenotypic analysis on days 10 and 15 revealed that CARMeso.αPD1-41BBL T cell-treated mice had higher proportions of central memory T cells compared to parental CARMeso T cells. The results indicate that the enhanced persistence and preserved memory status caused by aPD1-41BBL secretion enabled CAR T cells to have better long-term tumor control.
DISCUSSION
The secretion of our novel αPD1-41BBL protein—single-chain trimerized 41BBL fused to anti-PD-1 scFv—potentiatedCAR T cell preclinical solid tumor efficacy. 4-1BB is an established axis for T cell stimulation. Utilizing the natural ligand, which requires crosslinking for activity, can potentially improve 4-1BB modulation compared to anti-41BB antibody treatment and avoid the toxicity associated with the current 4-1BB antibody clinical candidates. 21 –23,39,40 In vitro assays revealed that CAR19.αPD1-41BBL T cells exhibit comparable cytotoxicity, reduced T cell exhaustion marker upregulation, and a less differentiated phenotype compared to parental CAR19 T cells and CAR19.αPD1 T cells. In an engineered solid tumor model using SKOV3 cells transduced to express CD19 (SKOV3.CD19), CAR19.αPD1-41BBL T cell-treated mice experienced significantly enhanced tumor control and overall survival and demonstrated much better expansion of CAR T cells in vivo compared to other groups.
Interestingly, while αPD1 secretion alone enhanced the eradication of modified H292 (
The TME is notorious for its abundance of inhibitory signaling pathways that bind receptors such as PD-1, TIM-3, LAG-3, and CTLA-4 on T cells, which impair T cell functions and result in insufficient immune response. 41 Analysis of clinical samples has identified a population of less differentiated CD8+ CART cells without PD-1 expression that plays an important role in mediating tumor control. 42 It was also observed that lower expressions of PD-L1, PD-1, LAG-3, and TIM-3 in lymphoma patients correlate with better responses to anti-CD19 CAR T cell treatment. 43 In particular, higher expressions of fatigue-related inhibitory receptors PD-1 and TIM-3 on CD8+ T cells is associated with defects in T cell proliferation, degranulation, and cytokine production. 44 Protecting infused CAR T cells from inhibitory receptors signaling is thus crucial to unleash the potentials of CAR-mediated killing effects.
The previous anti-PD-1 scFv secreting CAR T cells protected cells from PD-1/PD-L1 interactions and other inhibitory receptor signaling, and the αPD1-4-1BBL fusion protein mediated even stronger protection than anti-PD-1 scFv alone. Such protection from inhibitory receptors signaling prevents impairment in T cell function and contributes to the robust antitumor efficacy of αPD1-41BBL CAR T cells.
The lower cytokine expression levels of CAR T cells secreting αPD1-41BBL after short antigen exposure (∼24 h) is contradictory to literature findings that 4-1BB signaling markedly enhanced IFN-γ production from CD8+ T cells. 17 Since effector function is increased upon CD8+ T cell differentiation, 31 it is likely that the αPD1-41BBL groups exhibited less cytokine expression due to the less differentiated state compared to parental CAR T groups upon the one-day antigen exposure, and our CARMeso studies confirmed that CD62L+ cells had lower granzyme B production. A growing body of work indicates that the memory state of CAR T cells influences their clinical efficacy and less differentiated products bring about better clinical results. 45 –47
Central memory T cells or stem cell-like memory T cells for adoptive cell transfer are known to have sustained in vivo responses because they simultaneously display persistent effector functions as well as maintain expansion potential. 48 There are several methods for making this possible, such as selecting for certain T cell populations before CAR T cell production, producing CAR T cells in the presence of different cytokines or inhibitors, or altering CAR signaling domains. 49 Anti-PD1-41BBL-secreting T cells maintained a less differentiated state upon coculture with target cells and in animal models, which correlated with greater in vivo persistence and antitumor efficacy compared to control CAR T groups. Furthermore, several recent studies characterizing T cell exhaustion using single-cell RNA sequencing, mass cytometry, and ATAC-seq found that less differentiated, less exhausted CD8+ cells have reduced expression of effector cytokines. 50 –52
Our study demonstrates that downregulation of exhaustion markers and maintenance of memory status, despite reduced immediate cytokine production, results in enhanced CAR therapeutic efficacy.
Anti-PD1-41BBL secreting CAR T cells exhibit significantly smaller regulatory T cell population upon antigen exposure. Previous studies have illustrated how the balance between effector T cells and regulatory T cells can influence the success of immunotherapy, 53 and the infiltration of CD4+ regulatory T cells into solid tumor decreases the antitumor activity of second-generation CAR T cells costimulated by CD28 endodomain. 54 Therefore, the fusion protein's downregulation of T regulatory differentiation may also have contributed to the better performance of αPD1-41BBL secreting CAR T cells in treating solid tumors.
Despite the enhanced antitumor efficacy of αPD1-41BBL secreting CAR T cells against solid tumors, several aspects could be addressed with future efforts to optimize the design of the secreted fusion protein. For example, in our current design, the crosslinking of the single-chain trimerized 4-1BB ligand with anti-PD1 scFv compromised the binding affinity of anti-PD1 scFv to PD1, which may impair the blocking effect of the αPD1 side of αPD1-41BBL. Alternative designs of the flexible linker between the trimerized 4-1BB ligand and the anti-PD1 scFv that better preserve the binding affinity of anti-PD1 scFv to PD1 could potentially further improve the therapeutic efficacy of the secreted fusion protein.
The strategy of combining CAR T cell therapy with immune checkpoint inhibitors has received great attention in the hope that such combination would overcome some limitations of CAR T cells in treating hematological malignancies and solid tumors. 55 Besides the direct combination of PD-1/PD-L1 monoclonal antibodies and CAR T cells, there are also a number of candidates in clinical trials evaluating the antitumor efficacy of PD-1 knockout or PD-1 negative receptor CAR T cells. 56 Given the significantly better therapeutic efficacy of αPD1-41BBL expressing T cells over αPD1 expressing T cells, we believe that it is of high translational value to adopt secretion of αPD1-41BBL fusion protein to improve CAR T cell solid tumor efficacy, especially given the large number of patients who are PD1/PD-L1 therapy resistant.
Footnotes
ACKNOWLEDGMENTS
We thank the University of Southern California animal facility for providing animal support.
AUTHORs' CONTRIBUTIONS
Z.S.D. and Y.Q.: study design, performing experiments and data analysis, and drafting of the articles. M.M.: involved in data analysis; X.C.: study design, involved in data analysis; G.C.: conducting of study-related assays. P.W.: study design, study oversight, data analysis, and drafting of the article.
AUTHOR DISCLOSURE
P.W. is a scientific advisor to Grit Biotechnology, a stockholder of Simcere Pharmaceutical Group, and a cofounder and stockholder, and advisory board member of Appia Bio, TCRCure Biopharma, and Simnova Bio. None of the decared companies contributed to or directed any of the research reported in this article.
FUNDING INFORMATION
Z.S.D. is a USC Rose Hills Foundation Fellowship recipient.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.