
Abstract
The mucopolysaccharidoses (MPS) are a group of recessively inherited conditions caused by deficiency of lysosomal enzymes essential to the catabolism of glycosaminoglycans (GAG). MPS I is caused by deficiency of the lysosomal enzyme alpha-L-iduronidase (IDUA), while MPS II is caused by a lack of iduronate-2-sulfatase (IDS). Lack of these enzymes leads to early mortality and morbidity, often including neurological deficits. Enzyme replacement therapy has markedly improved the quality of life for MPS I and MPS II affected individuals but is not effective in addressing neurologic manifestations. For MPS I, hematopoietic stem cell transplant has shown effectiveness in mitigating the progression of neurologic disease when carried out in early in life, but neurologic function is not restored in patients transplanted later in life. For both MPS I and II, gene therapy has been shown to prevent neurologic deficits in affected mice when administered early, but the effectiveness of treatment after the onset of neurologic disease manifestations has not been characterized. To test if neurocognitive function can be recovered in older animals, human IDUA or IDS-encoding AAV9 vector was administered by intracerebroventricular injection into MPS I and MPS II mice, respectively, after the development of neurologic deficit. Vector sequences were distributed throughout the brains of treated animals, associated with high levels of enzyme activity and normalized GAG storage. Two months after vector infusion, treated mice exhibited spatial navigation and learning skills that were normalized, that is, indistinguishable from those of normal unaffected mice, and significantly improved compared to untreated, affected animals. We conclude that cognitive function was restored by AAV9-mediated, central nervous system (CNS)-directed gene transfer in the murine models of MPS I and MPS II, suggesting that gene transfer may result in neurodevelopment improvements in severe MPS I and MPS II when carried out after the onset of cognitive decline.
INTRODUCTION
Mucopolysaccharidosis Types I (MPS I) and II (MPS II) are recessive lysosomal diseases caused by deficiency of alpha-L-iduronidase (IDUA) or iduronate-2-sulfatase (IDS), respectively, resulting in systemic accumulation of the glycosaminoglycans heparan sulfate and dermatan sulfate. 1,2 Severe deficiency is manifested early in life with organomegaly, skeletal abnormalities, neurologic deterioration, and death by age 10. Milder forms of disease are associated with partial retention of enzyme activity and a later time course of onset. Enzyme replacement therapy provides systemic relief from accumulated glycosaminoglycan storage but has limited effect on neurologic disease since neither of these enzymes cross the blood–brain barrier. 3,4
Hematopoietic stem cell transplantation (HSCT) is frequently used to treat MPS I, and although it carries the risk of significant mortality and morbidity, when carried out early in life it reduces the rate of cognitive decline. 5 –7 HSCT is also used experimentally for the treatment of MPS II, and although a clear record has yet to be established, there are several studies ongoing to assess neurologic benefit. 8,9
A common theme in the application of HSCT for MPS disease, particularly MPS I, is that patients are most benefitted when transplanted before the onset of neurologic decline. 1,10 Several studies have reported lack of neurologic effectiveness when patients are transplanted later in life, and that after neurologic function has been lost, it is not recovered post-transplant. 11 –13 For this reason, transplant is recommended as early in life as possible, a circumstance facilitated by the advent of newborn screening. 14
Since normal cells and tissues express a relatively low level of IDUA activity, the limited effectiveness of allogeneic HSCT in recovery of neurologic function could be due to the low level of enzyme delivered to the brain and other relevant tissues post-transplant. Studies in animal models of MPS I 15 –27 and MPS II 28 –32 have shown that remarkably high levels of IDUA and IDS enzymes are achieved in host tissues by gene transfer in vivo or by transplantation of hematopoietic stem cells that have been engineered for enzyme overexpression. 33 –38 We previously reported supraphysiological levels of systemic enzyme, including central nervous system (CNS) tissues, in mouse models of MPS I and MPS II after intracerebroventricular (ICV) delivery of AAV9 vectors transducing the human IDUA and IDS genes, respectively. 16,31 Glycosaminoglycans (GAG) storage of treated animals was normalized in all tissues, including the brain, and AAV vector treatment prevented emergence of neurocognitive deficit exhibited in untreated affected control animals at 5–6 months of age. Recognizing the overwhelming level of systemic enzyme achieved after AAV vector administration, we wondered whether this would be sufficient to rescue animals from neurocognitive deficit by administration of vector at an age that we had previously demonstrated the animals to have already developed neurologic impairment.
In this study, we demonstrate that MPS I and MPS II mice administered 0.5 to 1.1 × 1011 vector copies (2.5–5.5 × 1012 vector copies/kg) of AAV vector transducing IDS or IDUA expression sequences, respectively, at 5–6 months of age exhibit high levels of enzyme in the plasma and tissues of the treatment groups, normalization of GAG storage materials, and neurobehavior indistinguishable from normal unaffected animals 2 months after AAV vector infusion. These results suggest that achieving such high levels of enzyme in the CNS may provide sufficient correction of metabolic disease to support recovery after the onset of neurologic decline in human MPS I and MPS II.
MATERIALS AND METHODS
AAV vector assembly and packaging
The miniCAGS-regulated human IDUA and CB7-regulated human IDS expression constructs have been previously described. 15,31 These plasmids were packaged into AAV9 virions at the University of Pennsylvania Vector Core, generating recombinant (r)AAV9-IDUA and AAV9-IDS with a titer of 1.1 × 1013 genome copies per milliliter.
Animal care
All animals used in this study (MPS I, MPS II, normal, and heterozygote controls) were bred on the C57BL/6 strain. Animals were bred and maintained under specific pathogen-free conditions and provided food and water ad libitum. Mouse care and experimental procedures were conducted under University of Minnesota Institutional Animal Care and Use Committee (IACUC) approval. The C57BL/6 IDUA deficient mouse strain (MGI: 2651471) was kindly provided by Dr. Elizabeth Neufeld. Experimental, and control animals were produced by breeding IDUA−/− males with IDUA−/− or IDUA+/− females. Dr. Joseph Muenzer kindly provided the C57BL/6 IDS deficient mouse strain. Male IDS+/0 and IDS−/0 mice were generated for this study by breeding IDS+/− females to wild-type males. Animals were genotyped by PCR as described. 39
To avoid anti-IDUA immune response, cyclophosphamide (CP, Sigma-Aldrich Co., St Louis MO) was administered weekly to experimental MPS I mice at a dose of 120 mg per kg intraperitoneally starting on day 3 after vector infusion. 40 CP immune suppression of MPS II mice was not required. 31
ICV vector infusion
AAV vector was administered to 6 or 5 month of age IDUA-/− or IDS−/0 animals, respectively, via ICV infusion as previously described. 16 In brief, animals were anesthetized with isoflurane and placed in a stereotactic frame. The right lateral ventricle was located (AP, +0.4 mm anterior to bregma; ML, +0.8 mm right from midline; depth, 2.4 mm deeper from dura) and 10 μL of AAV9-IDUA or AAV9-IDS vector was infused using a Hamilton syringe over a period of 10 min. 41 Preoperative care consisted of 5 mg/kg of Enrofloxacin intraperitoneal injection, and postoperative care was 5 mg/kg of Ketoprofen subcutaneous injection daily for 3 days.
Sample collection and preparation
Blood was collected monthly by submandibular puncture into heparinized tubes, processed for plasma, and stored at −20°C until assayed. Animals were euthanized at 5 months (MPS I) or 7 months (MPS II) post-AAV injection. Animals were transcardially perfused with 0.9% sodium chloride. Brains were dissected into right and left hemispheres and then microdissected into olfactory bulb, cortex, hippocampus, striatum, cerebellum, and thalamus with brainstem, snap frozen, and stored at −80°C until processed. Spinal cords and livers were also harvested. Organs were homogenized using a Bullet blender STORM bead homogenizer (Next Advance) and clarified by centrifugation. Tissue lysates were stored at −80°C until assayed. 19,31
Enzyme assays
The IDUA assay was carried out as previously described. 42 In brief, activity was determined fluorometrically using 4-methylumbellferyl α-L-iduronide as substrate (4MU-iduronide; Glycosynth) in 0.4 M sodium-formate buffer, pH 3.5 at 37°C for 1 h. 0.2 M glycine carbonate buffer, pH 10 was added to stop the reaction. Fluorescence was measured at 365 nm excitation and 460 nm emission using a Bio-Tek plate reader.
The IDS assay was carried out as previously described in a two-step reaction, using 4-methylumbelliferyl-α-L-iduronide-2-sulphate disodium as substrate (4-MU-αIdoA-2S; Toronto Research Chemical Incorp.) 29,31 In brief, plasma or tissue lysates were incubated with 1.25 mM 4-MU-αIdoA-2S at 37°C for 90 min, then the reaction was stopped using PiCi buffer (0.2 M Na2HPO4+0.1 M citric acid, 0.02% Na-azide, pH 4.5). The reaction was supplemented with 1 μg/mL iduronidase (#4119-GH; Bio-Techne), incubated overnight at 37°C, and then stopped (using 0.5 M Na2CO3, 0.5 M NaHCO3, 0.025% Triton X-100, pH 10.7). Reaction tubes were centrifuged, supernatants were transferred to black 96-well plates, and fluorescence was measured at 365 nm excitation and 450 nm emission. IDUA and IDS enzyme activities are expressed as nmol/h/mL for plasma and nmol/h/mg protein for tissue extracts. Protein in tissue samples was determined using the Pierce assay.
Glycosaminoglycan assay
Urine was collected monthly and frozen at −20°C until assayed for GAG content. Urine and tissue GAG assays were carried out as previously described. 15,31 Tissue lysates from liver, spinal cord, and microdissected brain were incubated with proteinase K overnight at 55°C followed by overnight digestion with DNaseI and RNase. GAG contents were measured using the Blyscan Sulfated Glycosaminoglycan Assay kit as per the manufacturer's instructions (Biocolor Life Science Assays; Accurate Chemical, NY). Creatinine was assayed according to manufacturer's instructions (Sigma-Aldrich no. MAK080). Tissue GAG levels were normalized to protein and are expressed as μg GAG/mg protein, and urine GAG levels are expressed as μg GAG/mg creatinine.
Quantitative polymerase chain reaction
DNA was extracted from tissue homogenates using the 5 PRIME Archive Pure DNA Purification kit according to manufacturer's instructions. For IDUA QPCR, reactions contained 60 ng of DNA template, 2 × IQ SYBR Green Supermix (Bio-Rad), 5 pm/μL of forward primer (5′-GGAGCAGGAGAAGGTCGTC-3′), and 5 pm/μL of reverse primer (5′-GTCGTTGTAAATGGGGGTGT-3′). PCR conditions were: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min in a Bio-Rad C1000 Touch Thermo Cycler. The standard curve consisted of serial dilutions of plasmid pTR-MCI linearized by digestion with restriction enzyme SnaBI or XbaI. Linearized DNA was purified by gel extraction using the Monarch DNA gel extraction Kit (NEB). IDS QPCR conditions were the same as those used for IDUA QPCR and contained 5 pm/μL of forward primer (5′-TCCCTTACCTCGACCCTTTT-3′), 5 pm/μL of reverse primer (5′-CACAAGGTCCATGGATTGC-3′). Serial dilution of plasmid pENN.AAV.CB7.hIDS was used to generate a standard curve and results are expressed as vector copies per genome equivalent.
Barnes maze
At 11 months of age (MPS I) and 9 months of age (MPS II), treated animals and controls were analyzed for memory and spatial navigation using the Barnes maze as described previously. 19,31 In brief, animals were released in the center of a 40-hole circular maze and were given 3 min to explore and find the single hole open to the escape box under the maze. If the mouse did not find the escape box, it was guided there and left in the escape box for 30 s before being returned to its home cage. The time taken to enter the escape box was recorded as latency to escape. Animals were given six trials a day for 4 days (MPS I) or four trials a day for 5 days (MPS II). Latency to escape in seconds was recorded and analyzed.
Fear conditioning
AAV9-IDS-treated mice were also tested by fear conditioning, a neurocognitive test. This associative learning and memory test measures a fear response (i.e., time spent freezing) to a conditioned stimulus (cue) that predicts an unconditioned stimulus (mild foot shock) presented during training trials. Data collection and analysis are semiautomated via a video-monitoring fear-conditioning apparatus (Med Associates, Inc.). On the conditioning day (training day; day 1), the test chamber was sprayed with a solution of Simple Green as an olfactory cue, and mice were exposed to a series (five pairings; 60-s intertrial interval) of cue (80-dB white noise tone and light) presentations (15 s in duration) coterminating with a mild foot shock (0.7 mA, 1 s in duration).
Twenty-four hours later, contextual fear conditioning was carried out in a test chamber sprayed with Simple Green. Mice were placed in the chamber for 3 min with no sound or light cues, and no foot shock. Freezing response was assessed for 3 min. After 1–2 h of the contextual test, cued fear testing was conducted in a test chamber with altered contextual elements (white floor and walls with vanilla odor) and consisted of a 3 min baseline (nonspecific freezing behavior) and a 3 min light and sound cue exposure (cued fear) period. Percent freezing time was assessed during all sessions.
Statistical analysis
GraphPad Prism (GraphPad software) was used for all statistical analyses. For plasma activities and Barnes maze, data were compared to normal (IDS) or heterozygote (IDUA) levels and untreated mice, respectively, using two-way ANOVA, followed by Dunnett's multiple comparisons test. Tissue IDUA and IDS activities and GAG levels were compared to heterozygote levels using the Kruskal Wallis test. Significance cutoff of <0.05 was used.
RESULTS
We have previously reported prevention of MPS I disease when MPS I mice are treated at 2–3 months of age or younger by administration of AAV vector transducing human IDUA. 15,16,18,19 We have similarly described prevention of MPS II manifestations in a mouse model of Hunter syndrome when treated at 2 months of age with AAV transducing human IDS. 31 Our goal here was to determine whether AAV9-IDUA/IDS restores neurocognition in MPSI and MPS II mice when administered after the onset of neurological dysfunction. IDUA deficient mice were treated at 6 months of age by ICV administration of AAV9-IDUA vector. MPS II mice were treated at 5 months of age by ICV infusion of AAV9-IDS. We have previously demonstrated that MPS I and MPS II mice exhibit neurocognitive deficiency at 6 and 5 months of age, respectively. 17,31
In-life studies of metabolic correction
Several studies have shown that after infusion of AAV vector into the CSF, there is substantial release, resulting in systemic distribution and circulation of secreted gene product. 28,43 Blood was therefore collected from animals to test for IDUA or IDS levels in plasma. IDUA activity in the plasma of AAV9-IDUA treated MPS I mice ranged from 1,714 to 7,965 nmol/h/mL, 1,000 times the level of normal controls, starting at 4 weeks post-treatment and lasting until the end of the experiment (Fig. 1A). IDS activity in AAV9-IDS-treated MPS II mice was roughly 450 times higher than the wild-type level, ranging from 12,700 to 39,200 nmol/h/mL (Fig. 1B).

Plasma enzyme activities in MPS I and MPS II mice administered AAV vector intracerebroventricularly.
To monitor in-life correction of metabolic storage disease, urine was collected monthly and assayed for excretion of GAG. Untreated MPS I mice exhibited a several-fold increase in GAG excretion compared to normal IDUA heterozygotes. In contrast, the high levels of IDUA activity observed in AAV9-IDUA treated mice were associated with normalization of GAG excretion. AAV9-IDUA-treated animals exhibited significantly lower levels of GAG excretion by 1 month post-treatment and remained at or near normal GAG levels for the duration of the experiment (Fig. 2A). Similarly, for AAV9-IDS-treated MPS II animals, urine GAG levels decreased following vector administration and were normalized by 1 month post-treatment. Normalized levels of GAG excretion were then sustained throughout the duration of the experiment (Fig. 2B).
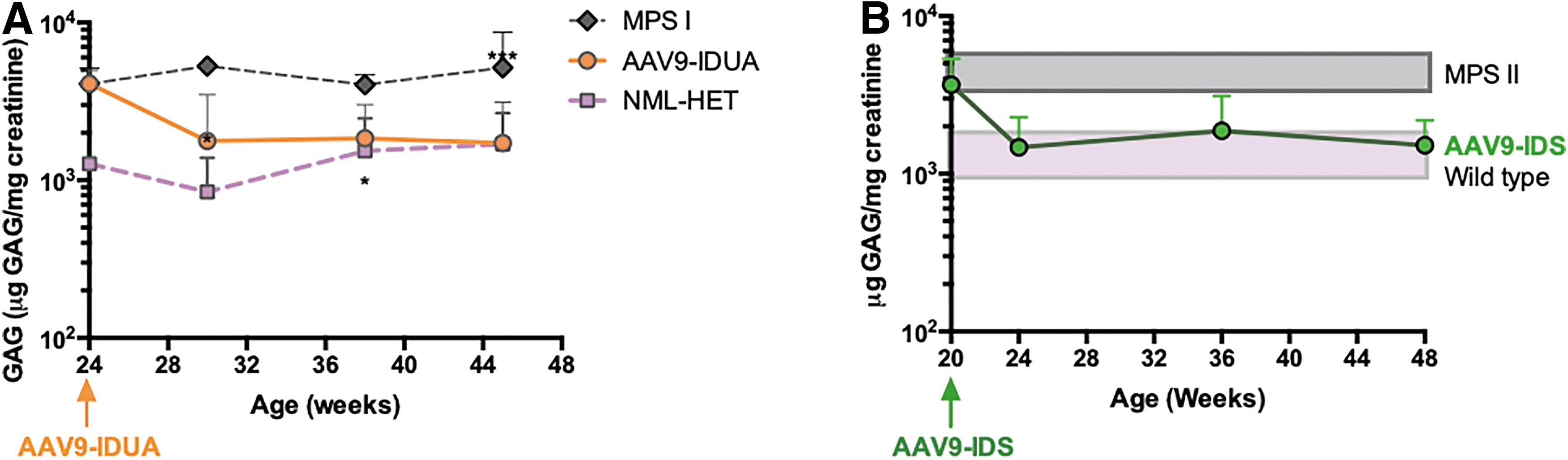
Urinary GAG excretion in MPS I and MPS II mice after AAV9 vector administration. Mean GAG content+S.D. is shown for each time point.
High levels of enzyme in tissues
MPS I and MPS II animals were euthanized at 11 and 9 months of age, respectively. Brains were harvested and microdissected, and along with spinal cord and liver were extracted and assayed for enzyme activity, GAG storage, and vector copy number. IDUA enzyme levels in spinal cord of treated MPS I animals were normalized, and IDUA enzyme activity in all sections of the brain and liver ranged from 100 to 1,000 times higher than heterozygous control samples. Levels of enzyme activity were similar in both hemispheres of the brain, with slightly higher levels in the right hemisphere. The highest levels of IDUA activity were observed in the hippocampus (average of L and R Hippocampus 4,820 ± 3,110) and olfactory bulb (average 1,550 ± 842), with lowest levels in the striatum (330 ± 653) (Fig. 3A).
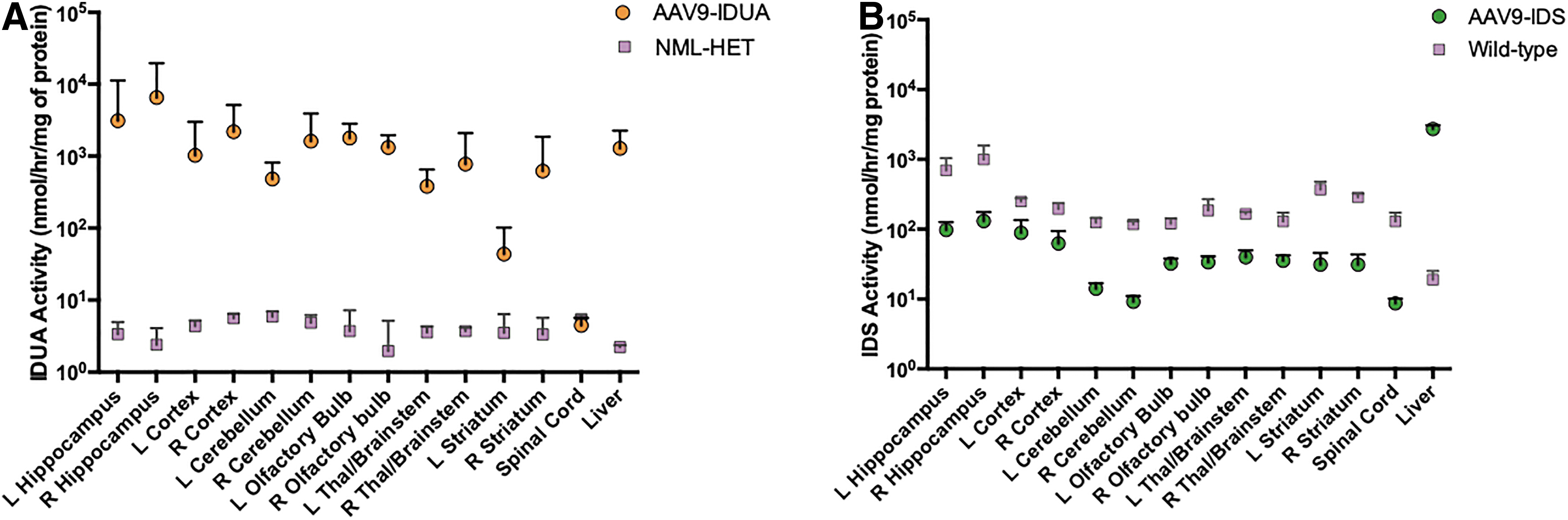
Enzyme activities in the brain and liver 4–5 months after ICV administration of AAV9 vector in MPS I and MPS II mice.
In AAV-IDS-treated animals, levels of IDS in the liver were 100-fold higher than normal controls. Consistent with our previously reported results, 31 we saw a high level of endogenous IDS expression in all parts of the brain and spinal cord (129 ± 74) in normal C57BL/6 mice, while enzyme activities in the brain and spinal cord (9 ± 4) of AAV9-IDS-treated mice ranged from approximately 50% to 60% to 7% that of normal controls, respectively (Fig. 3B).
Normalization of tissue GAGs
Harvested tissues were assayed for GAG accumulation. The high levels of IDUA found in all tissues of MPS I mice treated with AAV-IDUA corresponded with GAG levels that were either reduced (left olfactory bulb, left striatum, liver) or normalized (all other areas of the brain and spinal cord) (Fig. 4A). IDS enzyme never reached wt levels in the brains of MPS II animals treated with AAV-IDS (Fig. 3B), but GAG levels were nonetheless consistently reduced to wt levels (∼10 μg/mg protein) (Fig. 4B). These results demonstrate the effectiveness of AAV-administered ICV at 5–6 months of age in correction of metabolic storage disease both in the CNS and in the periphery (i.e., the liver).

Tissue glycosaminoglycan storage in brain and liver 4–5 months after ICV administration of AAV9 vector in MPS I and MPS II mice.
Vector biodistribution
Tissue homogenates from treated MPS I and MPS II animals were extracted for DNA and assayed for the presence of hIDUA or hIDS vector sequences, respectively, by QPCR. In AAV-hIDUA-treated MPS I animals, vector copy number ranged from 0.01 to 1 copies/cell in brain and spinal cord, except for one animal which had a higher level of distribution in all tissues, ranging from 10 to 500 copies. Copy numbers were similar in both hemispheres. Liver samples from treated MPS I animals had an average of 50 copies/genome equivalent (except for one animal which was below the level of detection) (Fig. 5A). AAV9-hIDS-treated MPS II animals showed a greater variation in copy number throughout the brain, ranging from 0.01 to 300.

Vector biodistribution by quantitative PCR. DNA was extracted from the indicated tissues and assayed for the presence of
Similar to MPS I-treated animals, liver samples from AAV9-hIDS-treated MPS II animals had an average of 50 copies per genome equivalent (Fig. 5B). Considering these biodistribution studies plus the high level of enzyme observed, the results show a substantial release of AAV vector from the CSF into the peripheral circulation with subsequent high-level transduction and enzyme expression in the liver for both MPS I and MPS II mice.
Restoration of neurocognitive function
Treated MPS I and MPS II animals were evaluated for neurocognitive function at 11 and 9 months of age, respectively, using the Barnes maze, a test of spatial navigation and memory. For MPS I mice, animals were evaluated in four trials a day for 4 days with a maximum trial time of 180 s. Heterozygote controls and AAV9-hIDUA-treated MPS I animals showed improvement in spatial navigation over time, locating the escape hole in 50 s by day 4, while untreated MPS I mice showed a significant deficit achieving an escape time of only about 140 s on day 4 (Fig. 6A).
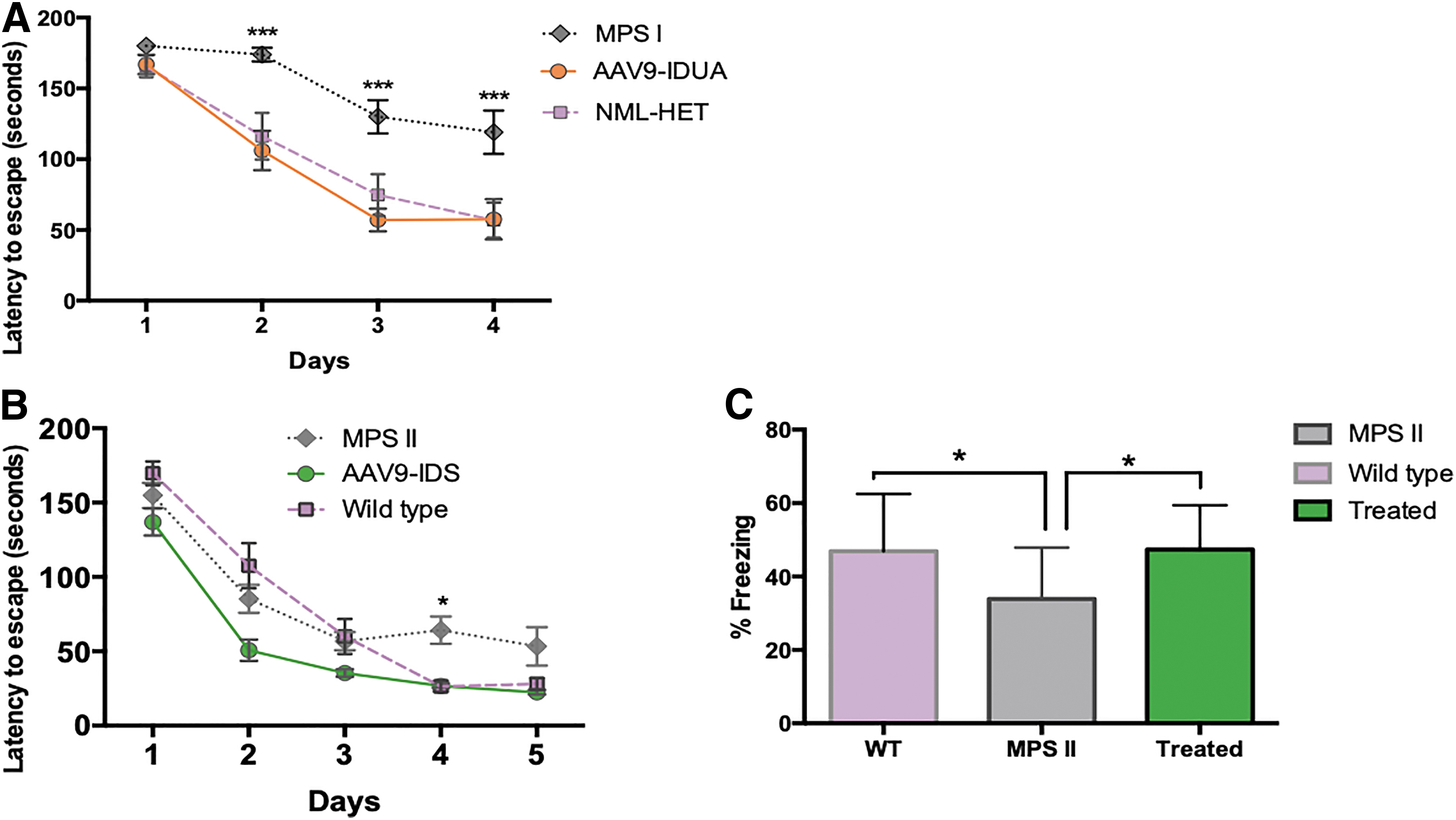
Neurocognitive evaluation of AAV9 vector-treated MPS I and MPS II mice. Animals were evaluated 4–5 months after ICV injection of AAV9 vector at 5–6 months of age (a time previously characterized to be after the onset of neurocognitive dysfunction).
MPS II animals were evaluated in four trials a day for 5 days. By day 4, both AAV9-hIDS-treated MPS II mice and heterozygote controls were able to locate the escape hole in about 30 s, while untreated MPS II mice demonstrated a significant delay on day 4 at about 50 s (Fig. 6B). Results from the Barnes maze thus show that AAV9 vector restored cognitive function in both MPS I and MPS II mice even when administered ICV after the onset of neurologic disease.
AAV9-hIDS-treated MPS II mice were also evaluated for neurocognitive deficit by fear conditioning. This associative learning task involves measuring a fear response (time spent freezing) to a stimulus (foot shock) presented during training trials. Training was carried out in a test chamber sprayed with an olfactory cue, and mice were exposed to a series of five light-sound cues, each of which were followed immediately with a mild foot shock 1 s in duration.
On the next day, animals were placed in the same cage with altered contextual elements (floor, wall, olfactory cue), and were allowed to explore the cage for 3 min (nonfreezing baseline), followed by an exposure to the light-sound cue, but without the foot shock. Freezing response was assessed videographically during both the baseline and the cue sessions (Fig. 6C). Untreated MPS II animals showed significantly reduced freezing responses, compared to AAV9-IDS-treated MPS II mice, which demonstrated freezing responses similar to normal controls. These results further demonstrate restoration of cognitive function in MPS II mice when administered AAV9-hIDS vector after the emergence of neurologic manifestations.
DISCUSSION
MPS I and MPS II in the severe forms are progressive neurologic diseases with cognitive decline and death in the first few years of life. 1 HSCT has thus far been the only recourse to impede the loss of neurologic function for MPS I, 6,10 with studies on MPS II in progress. 8,9 HSCT is thought to restore enzyme in the CNS by engraftment of donor-derived cells of the monocytic lineage (microglia) from which enzyme diffuses and cross-corrects host cells. 7
Experience in HSCT for MPS I has shown the importance of transplanting early in life, at least in the first year and even in the first few months, the treatment to minimize neurologic decline. 11 –13 HSCT carried out later in life, in the second or third years, has not been associated with effective neurologic outcome, and it is generally observed that while transplant can abate neurologic decline, any neurologic function that has been lost is not restored by HSCT.
A key limitation of HSCT is the low level of enzyme expressed from donor-derived cells. 33,44 Preclinical studies of MPS I have shown that much higher levels of enzyme can be achieved by either in vivo gene transfer 15 –26 or by ex vivo transduction of hematopoietic stem cells. 33 –35 This raises the question: how does the high level of enzyme expressed after in vivo or ex vivo gene transfer affect the timing of neurologic outcomes? We and others have conducted MPS I and MPS II gene transfer studies in a manner to prevent the emergence of neurologic disease by administration of test agent at birth or shortly after weaning.
In the present study, we evaluated the effectiveness of AAV vector administration on restoration of neurocognitive function when delivered after development of neurologic deficit in MPS I and MPS II mice. As previously demonstrated in younger animals, we observed supraphysiological levels of IDUA and IDS enzyme activity in the plasma and the brain after ICV delivery of vector, and normalization of urine and tissue GAGs. Remarkably, postsymptomatic administration of vector also restored neurocognitive function as assessed by the Barnes maze in MPS I mice, and by the Barnes maze and fear conditioning in MPS II mice. These results raise the provocative question as to whether recovery of neurologic function might similarly be restored in humans with MPS I or MPS II by administration of vector after the onset of neurologic decline.
Previous studies in which vector was delivered either neonatally or to young MPS I/MPS II mice (2 months of age or younger) showed therapeutic effectiveness that prevented the onset of neurologic dysfunction. 15,28,31,43 In contrast, Pasqualim et al. showed that urine and organ GAGs were normalized following late administration (6 months) of enzyme replacement therapy, compared to untreated MPS I animals. 45 They also observed improvements or normalization in some but not all cardiac parameters and varied effects in neurocognitive behavior in the open field test. Early studies in the MPS VII mouse model showed histologic and cognitive restoration after intrastriatal infusion of feline leukemia virus 46 or AAV4 vectors, 47 or ICV infusion of AAV5 vector 48 transducing ß-glucuronidase.
In a study carried out to assess treatment efficacy in MPS III, Fu et al. treated MPS III mice with AAV9 vector and found that late administration at 6 and 9 months improved disease outcomes such as longevity and GAG storage pathology in the CNS. 49 Here, we extend these findings to MPS I and MPS II by demonstrating that a single ICV administration of AAV9-vector after the onset of neurologic dysfunction restored normal levels of GAGs and normalized neurobehavioral outcomes in MPS I and MPS II mice.
ICV infusion introduced AAV9 vector directly into the cerebrospinal fluid of the right lateral ventricle. Supraphysiological levels of IDUA were detected in the plasma and all areas of brain, spinal cord and liver, resulting in metabolic correction of accumulated GAGs. IDS levels were also detected in the plasma, and all areas of brain, spinal cord, and liver. However, IDS enzyme activities were below wild type in the brain and spinal cord, while they were at supraphysiological levels in the plasma and liver. The reason for the stark difference between IDUA and IDS activities in the brain after AAV9 vector treatment is unknown. 31 GAG storage was nonetheless reduced in all tissues. Such normalization of GAG storage and neurocognition in mice expressing below wild-type IDUA or IDS levels has been previously reported. 15,28,31
Overall, the global distribution of vector detected in brain tissue from both AAV9-treated MPS I and MPS II mice at 5–6 months of age was similar to that previously observed after ICV infusion in younger mice. The left and right hemispheres showed a similar vector distribution after infusion into the right ventricle 15,31 which was reflected in the studies described here. Previous studies have shown that a substantial amount of vector is released into the periphery after ICV injection into MPS I and MPS II mice, and that livers of these animals exhibit the highest quantity of vector. 31,50 In this study, the livers of MPS I and MPS II mice treated postsymptomatically had an average copy number similar to those observed after ICV injection into young MPS mice, 31 further indicating that the age of the animal did not noticeably influence AAV9 biodistribution after ICV injection.
Preclinical studies have shown that syngeneic transplant using IDS+/IDUA+ donor marrow did not prevent the emergence of neurocognitive deficit in MPS I 33 or MPS II 51 mice, suggesting that CNS-directed AAV9 may be a superior approach compared to hematopoietic stem cell transplant in addressing neurologic manifestations of disease. The supraphysiologic levels of enzyme we observed in AAV9-administered MPS I and MPS II mice in this study and in previous studies 16,31 are also observed in animals that have been transplanted with lentiviral transduced hematopoietic stem and progenitor cells 33,51 and a treatment currently in clinical trials for MPS I. 35
However, there is significant risk associated with the preconditioning regimen required for engraftment of lentiviral-transduced HSPC, 52 and while the frequency of insertional oncogenesis has been reduced in comparison with gamma-retroviral vectors, 53 any randomly integrating element has the potential to interrupt normal growth regulation in a dominant manner. 54 By comparison, the primary risk of AAV administration is immune response against the gene product or against the vector capsid, 55 which can be mitigated by controlling vector dose and patient monitoring to see if there is a need for immunomodulation. 56
Both MPS I and II are single gene recessive disorders for which clinical studies testing the safety and effectiveness of in vivo 57 –60 and ex vivo 35 gene transfer in humans are advancing. Both of these lysosomal enzymes are secreted into the extracellular space and then taken up by mannose 6-phosphate receptors, 61,62 resulting in cross-correction of the enzyme deficit. Our results from postsymptomatically treated MPS II animals demonstrate that a fraction of normal IDS activity in the brain (8–10%) is sufficient to bring about a reduction in GAGs and also restore normal neurocognitive function.
While these results in mouse models of disease may not replicate what will be seen in humans, they nonetheless provide evidence that neurological damage in MPS patients might be reversible. As currently there are limited treatment options for MPS patients with severe cognitive impairment, the results reported in this study suggest the prospect of restoring neurologic function after the progression of disease in patients with late diagnosis or persistent residual disease post-HSCT.
Footnotes
ACKNOWLEDGMENTS
The authors thank Dr. Joseph Muenzer for providing the IDS KO strain and Dr. Elizabeth Neufeld for providing the IDUA KO strain. Behavioral studies were performed in the Mouse Behavior Core at the University of Minnesota (supported by NIH grant NS062158). We thank core director Dr. Benneyworth for help with the Barnes maze testing. This study has been previously presented at conferences and has been published in abstract form in conference proceedings.
AUTHORs' CONTRIBUTIONS
K.M.P.-P., K.L.: Carried out the experiments and analyzed data. S.S., T.T.N., M.S., A.T.: Carried out experiments. K.K.: Conceptualization. R.S.M.: Conceptualization, direction, planning, and article writing. L.B.: Planning and article writing.
AUTHOR DISCLOSURE
The work described in this article was supported by REGENXBIO. Karen Kozarsky was an employee of REGENXBIO.
FUNDING INFORMATION
This study was funded by grants R41 DK094538 and P01-HD32652 from the National Institutes of Health awarded to REGENXBIO and the University of Minnesota, respectively.