
Abstract
Histone H3 is a nucleosome scaffold protein that is involved in a variety of intracellular processes. Aberrant modification of H3 is important in carcinogenesis. In contrast, free histones in cells can act as stimuli to trigger cellular immune responses and cell death. In this study, we linked cell-penetrating peptide HIV Tat to a histone H3 fragment to achieve intracellular delivery in tumor cells. We found that Tat-conjugated histone polypeptides localized to nuclei of lung and breast cancer cells and caused cell death. A trans-configured Tat sequence displayed dramatically improved peptide half-life and cytotoxicity. Mechanistic studies demonstrated that treatment with the peptides significantly elevated mitogen-activated protein kinase (MAPK) signaling, reactive oxygen species (ROS) production, as well as levels of stress-inducible transcription factor ATF3 (activating transcription factor 3) and AP-1 (activating protein-1). Cytotoxicity of the peptide was significantly reduced by inhibition of AP-1 activity and ROS production. These results suggest the potential of Tat-conjugated H3 peptides as antitumor agents to induce cell death via increased cellular stress response by activating p38-MAPK signaling and intracellular ROS production.
INTRODUCTION
Histones (H3, H4, H2A, H2B)
Additionally, under various stress conditions, free histones can be detected in the cytoplasm and cell membrane. 5 Moreover, together with antimicrobial factors and genomic DNA, free histones have been found in neutrophil extracellular traps that mediate inflammation, apoptosis, and necrosis, 6,7 suggesting that exogenous histones may affect the cellular functions in chromatin-dependent and chromatin-independent manners.
Based on the previous data, manipulating the abundance and location of histones could fine-tune their targeted biological pathways. However, the efficient intracellular delivery of the desired histones is challenging. Cell-penetrating peptides (CPPs) have been demonstrated to be powerful transmembrane vehicles for many types of cargos. 8 The trans-acting activator of transcription (Tat) protein of human immunodeficiency virus 1 (HIV-1) has been identified as an efficient CPP 9 carrying a patch of basic amino acids (RKKRRQRRR) for cell uptake. 10 Because naturally existing cis (L)-type amino acids are easily degraded by proteases in target cells, trans (D)-type Tat with intrinsic cell-penetrating capability was designed to avoid being degraded. 11
Activating transcription factor 3 (ATF3) belongs to the ATF/cAMP-response element binding protein family. 12 It was reported that ATF3 forms homodimers with the activating protein-1 (AP-1) family members, including c-FOS and c-JUN, 13 in response to a variety of stimuli. 14 The ATF3/AP-1 expression was shown to be induced by various stress signals to regulate cell death. 15 Particularly, overexpressed ATF3 initiates apoptosis in ovarian cancer cells as an apoptosis inducer 16 and enhances DNA damage-mediated HeLa cell death via accelerating caspase protease activation. 17
ATF3 acts as an essential transcription factor that regulates the endoplasmic reticulum stress-mediated death receptor 5 (DR5) expression after zerumbone (ZER) and celecoxib (CCB) treatment in human p53-deficient colorectal cancer cells. 18 It was reported that activated c-JUN N-terminal kinases (JNKs) lead to c-JUN phosphorylation and enhance AP-1-dependent genes transcription. 19,20 For instance, JNK/AP-1 signaling mediates Galectin-1 (gal-1)-triggered T-cell death by inducing DNA fragmentation; 21 whereas JNK1 increased AP-1 activity by enhancing phosphorylation of c-JUN in bortezomib-induced apoptosis. 22 The collective findings indicate the involvement of the ATF3/AP-1 axis in stress-related cell death.
In this study, we designed histone H3 peptides in combination with HIV Tat sequence to induce cancer cell death through activating mitogen-activated protein kinase (MAPK)-AP-1 and ATF3 signaling. Moreover, we examined the in vivo tumor killing effect of trans-Tat-H3 peptide in a mouse 4T1 breast cancer model. The findings indicate the therapeutic potential of the CPP-H3 peptide via the activation of stress-responsive ATF3/AP-1 signaling.
MATERIALS AND METHODS
Peptide and protein
H3 peptides were synthesized by Shanghai Top-peptide Biotechnology Co., Ltd. (Shanghai, China). Crude peptides were purified by reverse-phase high-performance liquid chromatography using a C18 preparatory column, with confirmation by mass spectrometry. The purity of the synthesized peptides was >95%. The recombinant H4 protein was a gift from the Li Bing laboratory, Shanghai Jiao Tong University Li Bing.
Chemical regents and antibodies
Z-VAD-FMK (HY-16658B), Necrostatin-1 (Nec) (HY-15760), Ferrostatin-1 (Fer-1) (HY-100579), ARV-825 (HY-16954), Acetylcysteine (NAC; HY-B0215), and L-NAME hydrochloride (HY-18729A) were purchased from MedChemExpress (Shanghai, China).
Antibodies against ATF3 (A13469), c-FOS (A0236), and c-JUN (A0246) were all provided by ABclonal (Shanghai, China). Antibodies for extracellular signal-regulated kinase (ERK; 11257-1-AP), Phospho-ERK (28733-1-AP), JNK (66210-1), Phospho-JNK (80024-1), p38 (66234-1), and Phospho-p38 (28796-1) were purchased from ProteinTech (Wuhan, China). Antibody for cleavage-poly (ADP-ribose) polymerase (PARP; 5625), Bcl-2 (15071), Caspase3 (9662), Cleaved-Caspase3 (9664), LC3Ⅱ (2775), and Cyclin D1 (2922) were purchased from CST (Danvers, MA, USA). Bax (A0207), MLKL (A5579), and Phospho-MLKL (AP1173) were purchased from ABclonal (Wuhan, China).
Cell culture
A549 human lung cancer cells, 4T1 human breast cancer cells, 293T human renal epithelial cells, COS7 African green monkey kidney fibroblast-like cells, and U87 human glioma cells were all purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in high glucose Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (ExCell Bio, Shanghai, China) and 1% penicillin/streptomycin. Tat-conjugated H3/H3ac peptides were dissolved in dimethyl sulfoxide at a storage concentration of 600 μM at −80°C. The peptide was diluted to different concentrations (30, 15, 7.5, and 5 μM) and added to cell culture medium.
Phase-contrast microscopy observation of cell death
Cells were seeded in 96-well plates for treatment with peptides. At the indicated time points, cells were washed three times with phosphate-buffered saline (PBS) and photographed by phase-contrast microscopy (Olympus, Tokyo, Japan). Cells that were darkened and shrunken were defined as dead. Three randomly selected fields of view were photographed using a 20 × lens. The total number of cells and dead cells were counted using the ImageJ software (NIH, Bethesda, MD, USA) to calculate the dead cell rate. Fluorescence of fluorescein isothiocyanate (FITC)-labeled peptide was observed by fluorescence microscopy (Carl Zeiss, Jena, Germany). The localization and distribution of FITC within cells were observed using a 60 × lens. Fluorescence intensity was also quantified using the ImageJ software.
CCK8 assay of cell survival
Cells were plated in 96-well plates at a density of 2.5 × 103 cells per well and cultured overnight. Cells were washed three times with PBS before being assayed. CCK8 solution (Colleagues Association, Tokyo, Japan) was added to cells (100 μL per well) and incubated for 2 h at 37°C. The optical density at 450 nm (OD450) of each well was measured using a microplate reader equipped with a 450 nm filter. The relative cell viability values of each treatment group were calculated by normalizing the average OD450 values with the control group.
RNA extraction and RNA-sequencing analysis
Cells were dispensed in 24-well plates at a density of 1 × 104 cells per well. After being treated with FITC-tat-H3ac or FITC-tat-H3 peptide for 8 h, the total RNA was extracted using TRIzol (Ambion, Dallas, TX, USA) according to the manufacturer's protocol. The extracted RNA samples were sent to Annoroad (Shanghai, China) for subsequent sequencing. Alignment of RNA-sequencing (RNA-Seq) reads with the reference genome (GRCh38) was conducted using STAR, whereas differential expression analysis was performed using DEseq in R.
Dihydroethidium staining
Cells were dispensed in 96-well plates at a density of 2,500 cells per well and incubated overnight. After being treated with peptides, 200 μL of dihydroethidium (DHE) (Keygen Biotech, Nanjing, China) was added at room temperature and incubated for 30 min. 4′,6-Diamidino-2-phenylindole (DAPI) was added to stain nuclei for 5 min. DHE- and DAPI-stained sample images were acquired by fluorescence microscopy.
Western blotting
Protein extracts were prepared using RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS] in PBS). Protein concentrations were determined by the Pierce BCA Protein Assay (23227; Thermo Fisher Scientific, Waltham, MA, USA). Proteins in cell lysates (25–30 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA) and transferred to polyvinylidene difluoride membranes for incubation with the appropriate primary and species-specific secondary antibodies. ECL Prime Western Blotting Detection Reagent (RPN2232; GE Healthcare, Chicago, IL, USA) was used for detection using the Image Lab software (Bio-Rad).
Propidium iodide dye exclusion assay
The propidium iodide (PI) dye exclusion assay was performed as described in previous study. 23 Cells were in Krebs–Ringer–HEPES (KRH) buffer comprising 115 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, and 25 mM HEPES (pH 7.4) containing 30 μM PI. At the end of the experiment, 25 μM digitonin or 5 μM Triton X-100 was added to each well to permeabilize cells and label the nuclei with PI. Fluorescence was measured again to determine a value corresponding to 100% cell death. Percentage viability (V) was calculated as V = 100 (X–A)/(B–A), where A is the initial fluorescence, B is the fluorescence after addition of digitonin or Triton X-100, and X is the fluorescence at any given time with excitation maximum 540 nm and emission maximum 640 nm.
Mouse breast cancer model
Female BABA/c mice 4–6 weeks of age were purchased from Sichuan Dashuo Experimental Animal Company (Chengdu, China). 4T1 cells in passaged culture were prepared and injected at a density of 2 × 106 cells per 100 μL in the abdominal mammary gland of each mouse. Seven days after injection, tumor tissue could be observed, indicating successful subcutaneous tumor loading.
Intratumoral injection of peptide in mice
Intratumoral injection of peptide was performed in 4T1 tumor-bearing mice. Mice were randomly divided into two groups. Mice in the experimental group were injected with the peptide, whereas those in the control group were injected with the same volume of PBS or FITC-1084iTAT-H3ac peptide. Injections were given once every 2 days for a total of four times. Before each intratumoral injection, the tumor size was measured using Vernier calipers and recorded.
Statistical analyses
Statistical calculations of the data were processed using GraphPad Prism 8 Software (Graph Pad, San Diego, CA, USA). Data results are expressed as the mean ± standard deviation. One-way analysis of variance (ANOVA) with t-test was used for comparison between different groups, with * representing p < 0.05, ** representing p < 0.01, *** representing p < 0.001, and **** representing p < 0.0001 as statistically significant, and ns as not statistically significant.
RESULTS
CPPs-H3K14 and H3K14ac peptide fragments induce cell death
We synthesized a series of HIV Tat-conjugated H3 peptides to test their cell-killing effects (Fig. 1A). Amino acids 11 to 19 of histone H3 were chosen (denoted by black capital letters, Fig. 1A). Since residue lysine (K) 14 is a natural site for acetylation, 24 we also synthesized acetylated H3K14 peptide (denoted by “ac,” Fig. 1A). A nuclear localization sequence (NLS; denoted by pink capital letters, Fig. 1A) was designed to allow nuclear localization. 25 Tat was linked to the N-terminus of the NLS to allow transmembrane internalization. 10 In its natural state, HIV Tat consists of L-type amino acids (TAT); therefore, D-type amino acids (tat) were applied to increase the stability of the peptides. 11 We also designed a Tat peptide sequence from type C HIV isolate HIV1084i. To monitor the subcellular localization and kinetics of the peptides, their extreme N-termini were labeled with FITC. Unlike the nucleolus location of classical Tat, 1084i TAT showed uniform nuclear distribution (Supplementary Fig. S1A). 26
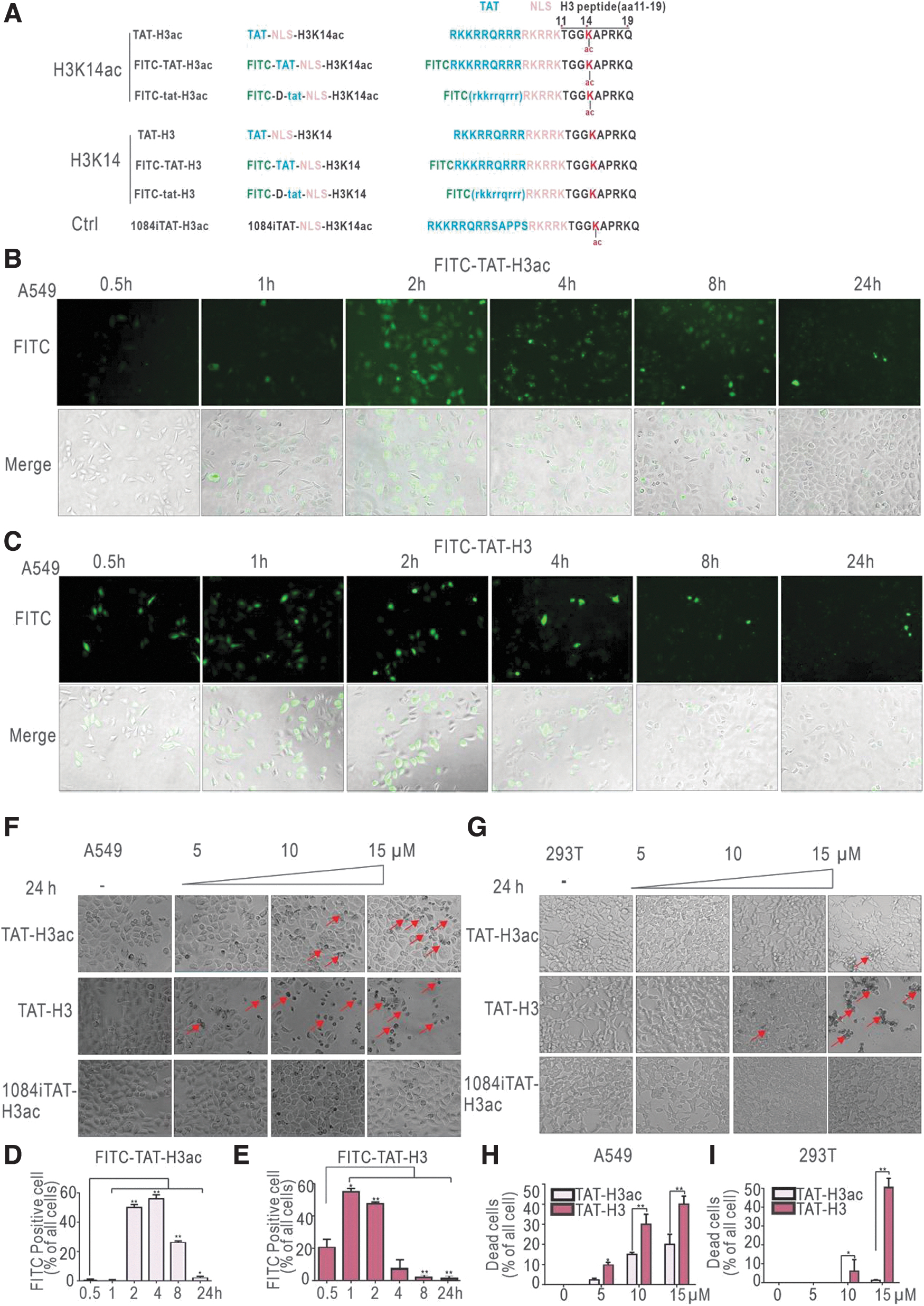
Internalized Tat-conjugated H3 peptide induces cell death.
We treated A549 lung carcinoma cells with 5 μM FITC-TAT-H3 acetylated or non-acetylated peptides and observed the fluorescence intensity and distribution within cells at different time points. As shown in Fig. 1B and 1D, fluorescence of FITC-TAT-H3ac reached maximal level at 2 h after peptide administration and was diminished after 8 h. In contrast, fluorescence of FITC-TAT-H3 could be detected at 30 min and gradually diminished until 4 h after administration (Fig. 1C, E). No significant cell death was evident in either treatment group. These results indicated that the designed Tat-H3 peptides could be delivered into the nucleolus, whereas the acetylated peptide displayed distinct kinetics compared with non-acetylated peptide.
When A549 cells were treated with increasing amounts of peptides for 24 h, darkened and shrunken cells debris were observed, particularly in higher concentration groups (Fig. 1F, red arrows). Interestingly, non-acetylated TAT-H3 displayed more potent cell killing activity than the acetylated peptide (Fig. 1F, H), whereas control 1084i-TAT did not produce any cell damage. A similar trend was observed in 293T cells (Fig. 1G, I), but not in COS7 monkey kidney fibroblasts or U87 human glioma cells (Supplementary Fig. S1B–E). These findings indicate that the peptides we tested possessed cell-type specificities.
Involvement of MAPK signaling pathway in TAT-H3 peptide-mediated cell death
To explore the mechanism of Tat-H3 peptide-induced cell death, RNA-Seq was performed to reveal transcription profile changes in A549 cells treated with FITC-TAT-H3acH3ac/H3 peptide. As shown in Fig. 2A and B, mRNA levels of ATF3, nuclear factor of activated T cells 2 (NFATC2), inhibitor of DNA binding 1 (ID1), and FOS genes were significantly upregulated in both groups. Protein–protein interaction analysis was performed to investigate the physical interaction and functional association among differentially expressed genes. Interestingly, most of the identified targets physically interacted with each other (Fig. 2C). FOS encodes transcription factor c-FOS protein to form a heterodimer of AP-1 with the c-JUN proto-oncogene protein. 27

MAPK and ATF3 are involved in peptide-induced cell death. (
Additionally, c-FOS and c-JUN were found not only bound with ATF3 but also served as ATF3 target gene products. 28 As shown in Fig. 2D, both acetylated and non-acetylated peptides induced ATF3 expression, whereas non-acetylated peptide induced c-FOS expression. MAPK includes the ERK (ERK1/2), JNK (JNK1/2/3), and p38 signaling pathways, which are crucial in cell death. 29 The detection of phosphorylated ERK and phosphorylated JNK indicates activated ERK and JNK signaling, respectively. 30 In Fig. 2E, we observed that both acetylated and non-acetylated TAT-H3 peptides induced phosphorylation of p38, ERK, and JNK, as well as the damage signal-PARP cleavage. 31 On the contrary, in COS7 cells, there was no significant activation of ERK/JNK/P38 signaling pathway after peptide treatment (Supplementary Fig. S2). The collective findings suggest that the TAT-H3 peptides induced obvious lung cancer cell stress, likely via MAPK signaling and the ATF3/AP-1 axis.
D-type Tat-conjugated H3 peptides are more stable and cytotoxic
To increase the cell half-life of peptides, we replaced the natural cis amino acids (TAT-H3ac/H3) in Tat with trans-configuration (tat-H3ac/H3). As shown in Fig. 3A, both D-typed tat-H3 peptides significantly increased intracellular fluorescence and the cell death rate compared with L-type Tat-H3 peptides (red arrows). Moreover, tat-H3 induced more cell death (Fig. 3B), which agreed with CCK8 assay results (Fig. 4A). Consistently, tat-H3 also increased ATF3, c-FOS, and c-JUN expression (Fig. 4B). Using lipopolysaccharide as a positive control 32 we observed that tat-H3 significantly activated the MAPK pathway and PARP cleavage (Fig. 4C). The collective findings suggest that the trans-configuration of the tat sequence increased the stability of H3 peptides in cells, which was accompanied with elevated cytotoxicity. Moreover, D-type Tat H3 peptides caused cell death through the similar pathway as the L-type Tat-H3 peptide.

D-type Tat-conjugated H3 peptide is more stable in cells.
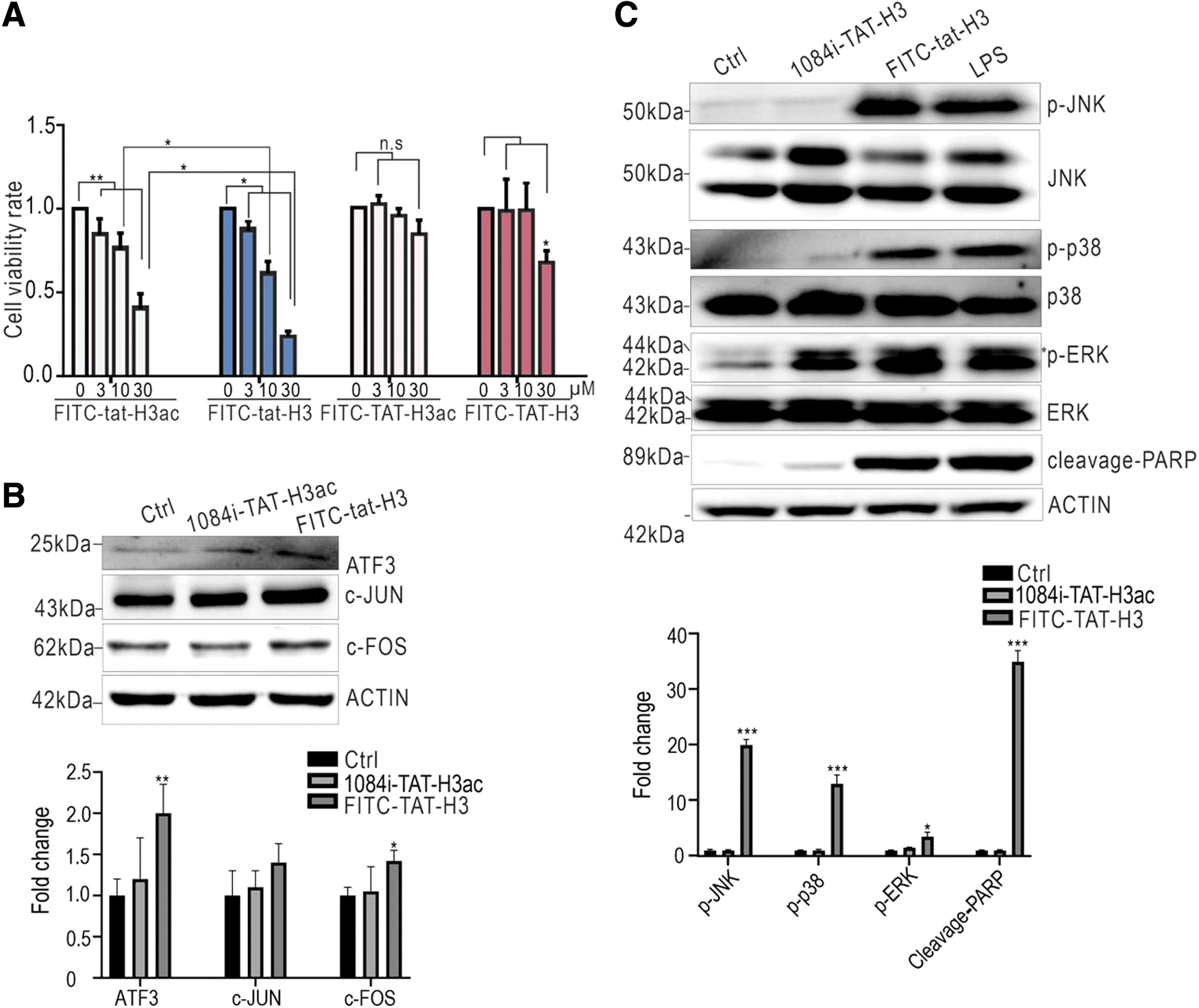
D-type Tat-conjugated H3 peptide is more cytotoxic to cells.
AP-1 inhibition partially rescues cell death due to D-type tat-conjugated H3 peptide
T5224, a c-FOS/AP-1 DNA binding inhibitor, was applied to validate the involvement of AP-1 in cell death. As shown in Fig. 5A, FITC-conjugated H3 peptide carrying the D-type Tat sequence induced significant cell death. Pretreatment with 80 μM T5224 reduced cell death, particularly in cells treated with non-acetylated peptide (Fig. 5A, B) without inhibiting peptide internalization. Intriguingly, cell death was not rescued by the pan-caspase inhibitor Z-VAD-FMK, RIPK1 inhibitor Nec, iron death inhibitor Fer-1, or the acetylated histone H3 binding protein BRD4 inhibitor ARV-825 (Supplementary Fig. S3). However, Bax, Cleavage-caspase 3, and MLKL phosphorylation levels were increased after FITC-TAT-H3/H3ac treatment (Supplementary Fig. S4), suggesting that the mechanisms of cell death may be complex and diverse. This also explains the inability of using apoptosis inhibitor or necrosis inhibitor alone to rescue the cell death induced by the peptide.
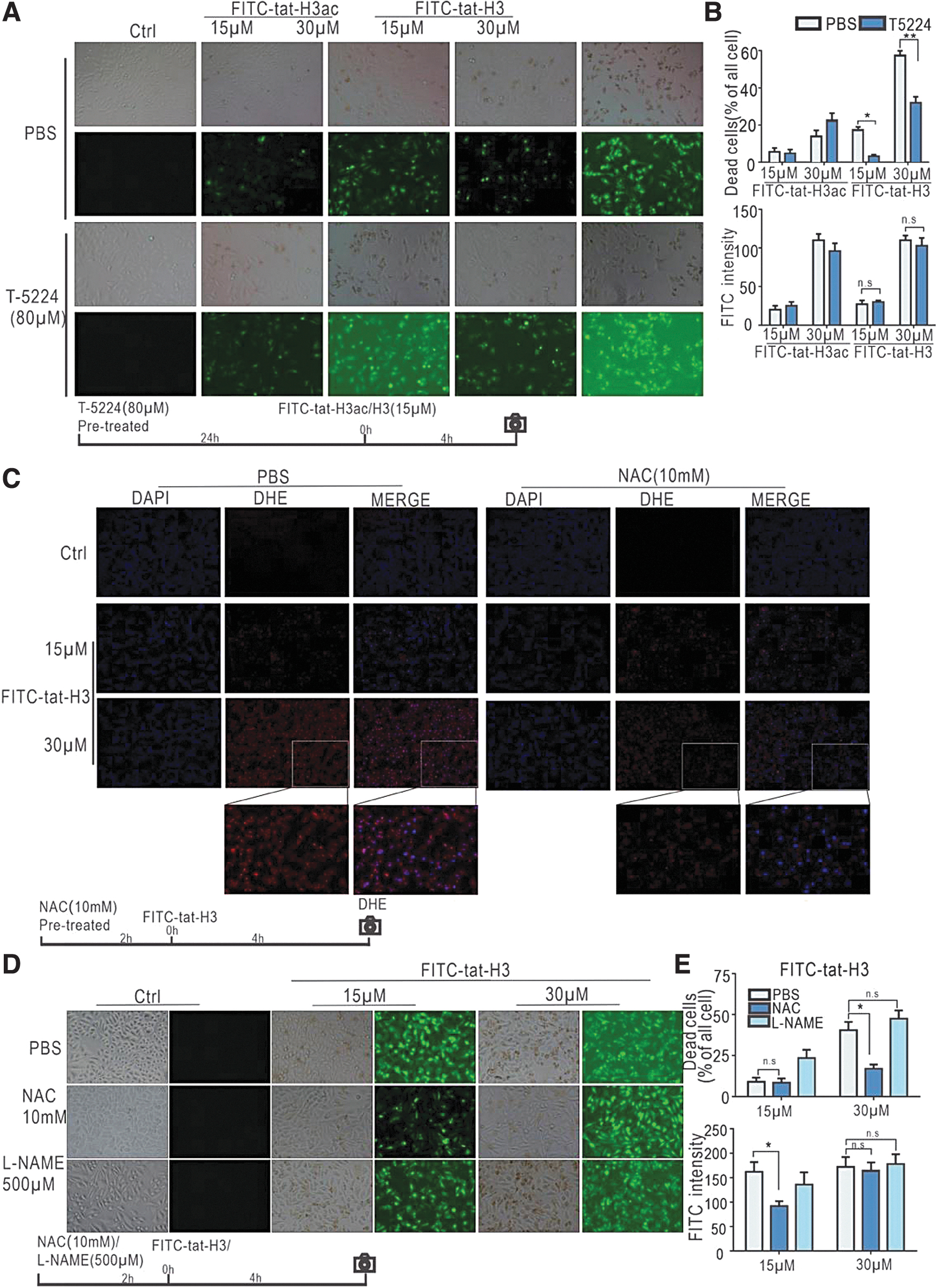
T5224 and NAC partially rescue cells from peptide-induced death.
A previous study described that extracellular histone H4 protein could induce cell death via membrane cleavage. 7 To exclude this possibility in the present study, PI staining was used to detect DNA leakage upon membrane cleavage. Peptide treatment did not affect cell membrane integrity (Supplementary Fig. S5). We further measured the leakage of Golgi matrix component GM130 shown in Golgi fragmentation 33 and cytochrome c in mitochondrial injury 34 (Supplementary Fig. S6). The unchanged levels of the above proteins in whole-cell extracts suggested that peptide treatment did not induce Golgi and mitochondrial damage directly.
D-type Tat-H3 peptide induces production of reactive oxygen species
Reactive oxygen species (ROS) are important in cell death and are related to MAPK pathway activation. 35 To check if oxidative stress was involved in our experimental setup, DHE staining was performed to label the intracellular ROS changes in peptide-treated cells. We observed that FITC-tat-H3 peptide induced ROS production (Supplementary Fig. S7). As shown in Fig. 5C, compared with the PBS (control) group, the ROS scavenger NAC significantly lessened ROS in cells treated with 30 μM FITC-tat-H3, confirming that NAC improved cell viability by diminishing intracellular ROS production. Compared with the PBS group, cells pretreated with 10 mM of NAC showed a significant reduction in cell death upon treatment with 15 or 30 μM FITC-tat-H3 (Fig. 5D, E). The reactive nitrogen species inhibitor L-NAME did not rescue cell death. CCK8 assay results were consistent with the phase-contrast microscopy observations (Supplementary Fig. S8). The collective findings indicate that oxidative stress was induced during cell death via intracellular ROS.
Combined inhibition of AP-1 and ROS production synergistically protects cells from death upon treatment with peptides
Next, we treated cells with T5224 and NAC before exposure to D-type peptides. In the NAC group, the percentage of dead cell upon FITC-tat-H3 administration was significantly lower than that in the PBS group. The combined use of NAC and T52224 further improved cell survival without influencing internalization of the peptides (Fig. 6). These findings indicate that the toxicity of FITC-tat-H3 can be significantly inhibited by simultaneous inhibition of both the AP-1 and ROS pathways.
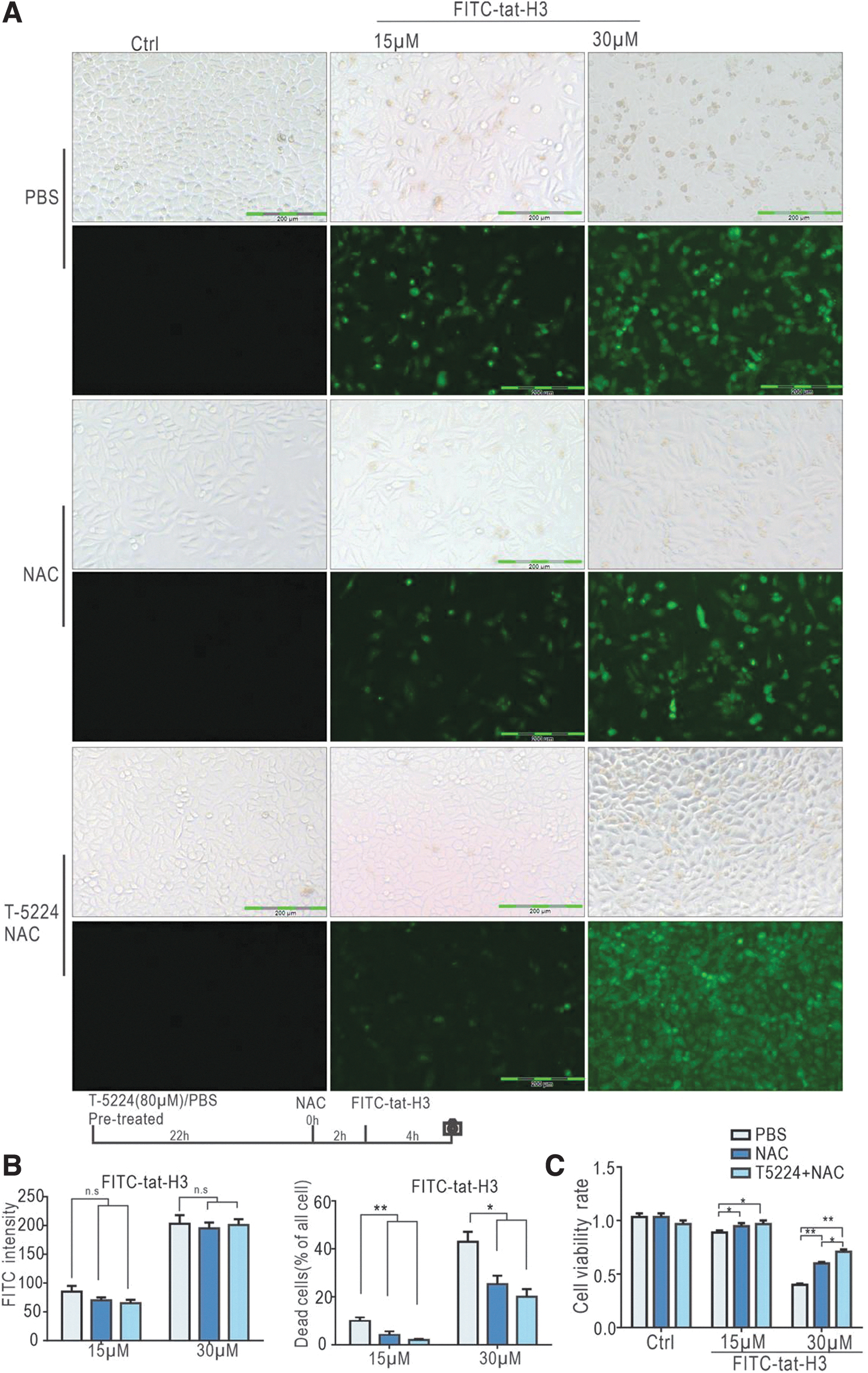
Combined pretreatment of T5224 with NAC further improves cell state after peptide treatment.
D-type Tat-H3 peptide significantly inhibits tumor growth in vivo
To further explore the potential application of our designed peptide in antitumor therapy, FITC-tat-H3/H3ac toxicity was verified in 4T1 mouse breast cancer cells by the observation of substantial cell-killing effects (Supplementary Fig. S9). We then constructed a mouse tumor model by injecting 4T1 cells into the mammary region of BALB/C mice. After 7 days, intratumoral injection of FITC-tat-H3 was initiated for another 8 days. Subsequently, tumor tissue was collected, and tumor volume was measured. The injection of FITC-tat-H3 had little influence on mice body weight (Supplementary Fig. S10) but significantly inhibited tumor growth (Fig. 7A, B). To evaluate apoptosis in tumor cells, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) stain assay was performed. More apoptotic cells were observed in the FITC-tat-H3 group than in the control group (Fig. 7C). The collective findings indicate that D-type H3 peptide inhibited tumor growth in vivo.

Effects of D-type Tat-H3 peptides on tumor growth.
DISCUSSION
Given the critical roles of histone acetylation in the regulation of gene transcription, abnormal abundance and genome location of acetylated histone have been linked with carcinogenesis. 36 Therefore, histone acetylation is considered an attractive target for cancer therapy. 37 We originally hypothesized that acetylated and non-acetylated H3K14 peptide may perform differently because the acetylated lysine residue was shown to affect gene transcription by interacting with histone modification “writer” or “reader” proteins. 38 However, in our experiments, we only observed mild cytotoxicity difference between acetylated and non-acetylated cis-Tat H3 peptides, both of which induced MAPK signal pathway activation and ATF3 expression comparably (Fig. 2D, E). In addition, trans-Tat sequence enhanced the cytotoxic effects of peptides regardless of acetylation (Fig. 3). Therefore, our data suggested that the mechanism of peptide-induced cell death may not be related to the function of histone acetylation itself.
A previous study demonstrated that acetylated H4 peptide disrupted histone methyltransferase G9a and HDAC1 genome occupancy of the p53 promoter, resulting in P53-mediated apoptosis by upregulating the level of DNA damage-induced transcription of p21 and Noxa gene. 3 In another study, the H3K36me2 peptide inhibited diffuse intrinsic pontine glioma cell proliferation through the disassociation of the H3K36me2/3 “Reader” proteins-LEDGF/HDGF2 39 from chromatin. 4 These data suggested that exogenously modified histone peptides could interfere with gene transcription. Our results showed that internalized Tat-conjugated H3 and H3ac peptides induced cell death (Figs. 1F, G and 3) and gene transcription changes in target cells (Fig. 2A, B).
RNA-Seq data revealed upregulated expression of ATF3, NFATC2, ID1, and FOS mRNA in both the acetylated and non-acetylated peptide groups, indicating that cell death may not result from lysine acetylation. A prior study reported that bromodomain-containing protein 4 (Brd4) could recognize and combine with histone acetylation to mediate gene transcription. 40 However, in our pharmacology assay, ARV-825, an inhibitor of Brd4, did not change peptide toxicity in either H3 or H3ac treatment (Supplementary Fig. S3A), implying that Tat-H3 peptide-mediated cell death might not be due to histone H3 reader proteins. Moreover, it has been shown that membrane cleavage mediated by extracellular histone H4 in smooth muscle cells triggers arterial tissue damage. 7
The internalization of Tat peptide mainly depends on membrane association, followed by vesicle-mediated cytosol translocation 41 and lipid-dependent micropinocytosis. 42 Consistent with these findings, the PI dye exclusion assay in our study showed that histone H4 protein, but not Tat-H3 peptide, caused physical damage to the cell membrane (Supplementary Fig. S5). These findings exclude the possibility that the Tat-H3 peptide induced cell death by membrane cleavage.
ATF3 functions as an immediate early gene induced by stress signals such as cAMP, ultraviolet radiation, calcium influx, and cytokines. 13 Together with other immediate early gene encoded proteins, such as c-FOS and c-JUN, ATF3 composes the transcription factor activating protein 1 (AP-1), which regulates important cellular functions. 43 It has also been demonstrated that ATF3 regulates gene transcription by binding to canonical ATF/CRE cis-regulatory element or AP-1 sites, which are distal to transcription start sites and enriched with p300 and H3K27ac. 44 c-JUN forms stable heterodimers with c-FOS or ATF family members to bind with different DNA elements. 45
Our RNA-Seq data revealed a subset of genes whose transcription levels were altered upon peptide stimulation. Among these genes, mRNA levels of ATF3, NFATC2, ID1, and FOS were significantly increased (Fig. 2A, B). Western blot analysis revealed obviously increased ATF3, c-JUN, and c-FOS protein levels in cells treated with D-type Tat H3 peptides (Fig. 4B). Additionally, cell death was rescued by pretreatment with AP-1 inhibitor T-5224 (Fig. 5A). These results indicate that H3 peptides may induce cell death via AP-1 signaling. AP-1 activity is induced by various signals, including cytokines, chemokines, growth factors, and DNA damage. 46 These stimuli activate MAPK cascades that enhance AP-1 activity through the phosphorylation of distinct substrates. 19
The induction of AP-1 by proinflammatory cytokines and genotoxic stress is mostly mediated by the JNK and p38 pathways. Once activated, JNKs phosphorylate c-JUN on Ser 63/73 and Thr 91/93, which potentiates the activation of transcription upon homo- or heterodimerization with c-FOS. 47 ROS induces NF-E2-related factor 2-dependent ATF3 48 and activates AP-1, 49 while causing MAPK activation, such as ERK, 50 JNK, and p38. 51 We observed that upon treatment with Tat-H3 peptides, JNK, p38, and ERK phosphorylation was obviously elevated (Figs. 2E and 4C).
Although a series of cell death inhibitors failed to rescue peptide-induced cell death, Bax, Cleavage-caspase3, and MLKL phosphorylation levels were increased after FITC-TAT-H3/H3ac treatment (Supplementary Fig. S4), whereas ROS production was enhanced (Supplementary Fig. S3 and Fig. 5C) and PARP cleavage was activated (Figs. 2E and 4C). These findings strongly suggest that the mechanisms of peptide-induced cell death may be complex. Meanwhile, these data provide convincing evidence that the internalization of Tat-H3 peptides initiates potent cellular stress, which activates ATF3/AP-1 signaling to induce cell death via ROS. However, whether ATF3 acts cooperatively with or functions upstream of AP-1 remains to be investigated.
Tat enters cells by micropinocytosis, a specialized form of fluid-phase endocytosis that occurs in all cells. 42 The natural composition of the L-type Tat peptide is readily degraded by proteases present in cells, whereas the trans-form of Tat (D-type Tat) peptide exists for a much longer time in cells. 11 We applied D-type Tat to extend the cellular half-life in peptide-sensitive tumor cells. We observed prolonged existence of peptides accompanied by elevated cytotoxicity (Fig. 3A). By using different cell lines, we observed potential cell specificity of Tat-H3 peptides. Moreover, in vivo, peptides induced obvious cell death in mouse tumor tissue and inhibited tumor growth (Fig. 7).
Finally, our collective findings suggest that D-type Tat-H3 peptides could potentially be applied in antitumor treatment. Because of their tremendous potential to deliver biologically active cargoes into intracellular spaces, CPP have impressively broad versatility for antitumor therapy. Besides optimizing the cargo transduction capacity, a tricyclic Tat construct was recently used to deliver functional immunoglobulin-G antibodies and Fab fragments that bind intracellular targets in the cytosol and nuclei of live cells into cancer cells to promote treatment efficiency. 52 Additionally, selective organ-targeting nanoparticles have been systematically engineered to exclusively edit extrahepatic tissues. 53 In the future, engineered Tat-conjugated H3 peptides could be combined with selective organ targeting nanoparticles, monoclonal antibodies, and other advanced drug delivery systems to achieve target selectivity and efficiency.
CONCLUSIONS
In conclusion, this study indicated that Tat-conjugated H3 peptides can cause cancer cell death and inhibit mouse breast tumor growth via cellular stress response. Mechanistically, our data suggested that Tat-H3 peptides could induce ROS production, activate p38-MAPK pathway, and increase AP-1 expression, which may mediate peptide-induced cell death.
Footnotes
AUTHORs' CONTRIBUTIONS
Q.X.: Investigation, data curation, and writing—original draft. K.L.: Investigation and data curation. F.Z.: Investigation. Y.P.: Data curation. Q.X. and Y.R.: Methodology, and writing—original draft. N.H.: Supervision. Y.W.: Conceptualization, supervision, and writing—original draft.
ETHICS STATEMENT ANIMAL EXPERIMENTATION
Our animal experiments have been approved by the ethics committee of West China Hospital of Sichuan University under the approval number 20220525002.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
Financial support was provided by the National Natural Science Foundation of China (NSFC) (31701098 to K.L., 31860287 to Y.W.) and the Science and Technology Bureau Fund of Sichuan Province (2021YFS0051 to Y.W.).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.