
Abstract
Duchenne muscular dystrophy (DMD) is caused by the lack of dystrophin, but many patients have rare revertant fibers that express dystrophin. The skeletal muscle pathology of DMD patients includes immune cell infiltration and inflammatory cascades. There are several strategies to restore dystrophin in skeletal muscles of patients, including exon skipping and gene therapy. There is some evidence that dystrophin restoration leads to a reduction in immune cells, but dystrophin epitopes expressed in revertant fibers or following genome editing, cell therapy, or microdystrophin delivery after adeno-associated viral gene therapy may elicit T cell production in patients. This may affect the efficacy of the therapeutic intervention, and potentially lead to serious adverse events. To confirm and extend previous studies, we performed annual enzyme- linked immunospot interferon-gamma assays on peripheral blood mononuclear cells from 77 pediatric boys with DMD recruited into a natural history study, 69 of whom (89.6%) were treated with corticosteroids. T cell responses to dystrophin were quantified using a total of 368 peptides spanning the entire dystrophin protein, organized into nine peptide pools. Peptide mapping pools were used to further localize the immune response in one positive patient. Six (7.8%) patients had a T cell-mediated immune response to dystrophin at at least one time point. All patients who had a positive result had been treated with corticosteroids, either prednisolone or prednisone. Our results show that ∼8% of DMD individuals in our cohort have a pre-existing T cell-mediated immune response to dystrophin, despite steroid treatment. Although these responses are relatively low level, this information should be considered a useful immunological baseline before undertaking clinical trials and future DMD studies. We further highlight the importance for a robust, reproducible standard operating procedure for collecting, storing, and shipping samples from multiple centers to minimize the number of inconclusive data.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive, progressive neuromuscular condition affecting 1:5,000 male births. 1 It is caused by mutations (mainly deletions) in the DMD gene, which codes for the protein dystrophin. 2 The lack of dystrophin in skeletal muscle fibers causes them to degenerate; this is followed by cycles of regeneration/degeneration, ultimately resulting in the progressive loss of skeletal muscle (reviewed 3 ). Individuals with DMD lose their ability to walk by their early teens, but corticosteroids can postpone the age at which ambulation is lost by 3–4 years. 4
Many approaches to restore dystrophin have been tested preclinically and four antisense drugs (eteplirsen, golodirsen, viltolarsen, and casimersen) have been approved in the USA and a small molecule (ataluren) in Europe; these drugs restore a small amount of functional dystrophin. In addition, adeno-associated viral (AAV) gene therapy is a promising approach that is currently in clinical trials, for example,
There are several different dystrophin isoforms, but only the full-length dp427 is expressed in skeletal muscle fibers (reviewed 1,2 ). Dystrophin is not always completely absent in skeletal muscles of individuals with DMD: depending on the location of the mutation in the DMD gene, shorter, partially functional or nonfunctional dystrophin protein may be produced. 3 Many patients and mouse models have a small percentage of “revertant” muscle fibers that express truncated dystrophin protein 5 (reviewed 6 ).
These revertant fibers arise from aberrant, stochastic splicing events that allow the production of small amounts of protein and the resulting dystrophin epitopes expressed in them 7 might elicit T cell production. The latter may accelerate an immune response to the restored dystrophin in treated patients 8,9 (reviewed 10 ). Alternatively, dystrophin expressed in revertant fibers may reduce the immune response to myofibers expressing restored dystrophin. 11 However, the timing of the appearance of these revertant fibers is likely to be crucial. They are already present in newborn mdx mouse muscles 7 and in DMD fetal muscle, 4 and their prenatal onset is likely to induce tolerance to the expressed epitopes.
It has been shown that increasing age correlates with an increased risk for a T cell-mediated immune response to dystrophin and in a previous cross-sectional study on 70 individuals with DMD, ∼50% of the steroid-naive and 20% of the steroid-treated population were reported to have circulating dystrophin primed T cells. 9 To confirm and extend these observations, we performed a multicenter, longitudinal natural history study to determine whether patients with DMD had a pre-existing T cell-mediated immune response to dystrophin, and whether this changed over time.
We performed enzyme- linked immunospot (ELISPOT) interferon (IFN)-gamma assays on individuals recruited into this 4-year DMD natural history study that recruited 50 ambulant and 27 nonambulant boys with DMD from four clinical centers. ELISPOT assays on all patients were performed with a full-length dystrophin peptide set; we also studied one individual who had an exon-skippable deletion with peptides corresponding to unique epitopes generated by the potential exon skipping event. We correlated our data to factors such as age, ambulation status, steroid regime, and DMD deletion.
METHODS
Subjects
Blood samples from DMD subjects belonging to a cohort of boys enrolled in the Association Francaise contre les Myopathies (AFM)-funded iMDEX multicenter natural history study were used for our experiments. Specimens from subjects recruited in London (Center 1), Newcastle (Center 2), Paris (Center 3), and Leiden (Center 4) were analyzed. All samples used for this project are listed in Table 1.
Summary of Elispot results
DMD, Duchenne muscular dystrophy.
This study was approved in United Kngdom by the Bromley Research Ethics committee (REC 12/LO/0442), and the ethics committee of all other institutions. All subjects and their legal representatives provided written informed consent for the study. This study is registered with the Clinical Trial Gov website with the number NCT02780492.
Boys with DMD were assessed annually over up to 4 years with an ELISPOT IFN-γ assay performed with a full-length dystrophin peptide set as previously described. 8,9 A patient with an exon-skippable deletion was additionally assessed with peptides corresponding to unique epitopes that would have been generated in the case of a single exon skipping intervention to restore the reading frame. We also assessed four healthy adult controls as well as six neuromuscular disease controls from female children with nondystrophinopathies. These consisted of one individual with muscle-eye-brain disease (6 years of age), four with limb girdle muscular dystrophy (9–16 years of age), and one with Ullrich congenital muscular dystrophy (14 years of age).
Sample collection and preparation
Around 5–20 mL (ideally, at least 10 mL) of blood was collected from individuals into either heparin tubes or Vacutainer® CPT™ Cell Preparation Tubes with sodium citrate. All samples were stored at room temperature for a maximum of 24 h before processing. Samples from the two UK sites were delivered to UCL as blood within 24 h and processed. Samples from France and the Netherlands were processed locally, and frozen peripheral blood mononuclear cells (PBMCs) shipped on dry ice to minimize loss of cell viability during shipment. A standardized procedure was used by all blood processing sites. Briefly, an equal volume of phosphate-buffered saline (PBS) was added and a maximum of 20 mL of diluted blood was carefully layered on top of 15 mL Ficoll. The tubes were centrifuged at 400 g for 30 min at room temperature with slow acceleration and no brake.
Plasma was removed, aliquoted, and stored at −80°C; some of these samples were used for miRNA assays. 12 The PBMC layer was extracted and washed three times with 30 mL PBS and centrifuged at 100 g for 10 min at room temperature, with the brake on and high acceleration. The cell pellet was resuspended in chilled freezing medium (10% dimethyl sulfoxide [DMSO], 90% fetal calf serum) in 1 mL aliquots (∼10–20 million PBMCs/mL) and frozen in a Mr Frosty at −80°C overnight. When required, cells were thawed rapidly at 37°C and resuspended in 5 mL warmed AIM-V medium (AIM-V: Invitrogen; 12055-091) containing 2% human serum (Human AB serum, Gemini Bio Products 100–512, heat inactivated for 30 min at 56°C), and a count of viable cells was performed.
Peptides
Peptides (20mer) overlapping by 10 amino acids were used, collectively these span the entire dystrophin protein (Proimmune Ltd, Oxford, UK). There was a total of 368 peptides, organized into 9 peptide pools 8 (Fig. 1). Stock vials of individual peptides at 5 mg/mL were made up in 10% DMSO, 90% sterile water, and stored at −80°C. Peptide pools were made up at 40 μg/mL (of each peptide), diluted in sterile water, and kept at −80°C in small aliquots. Peptide mapping pools were used to further localize the immune response in a subset of positive individuals. In the mapping pools, each peptide is present in two subpools.
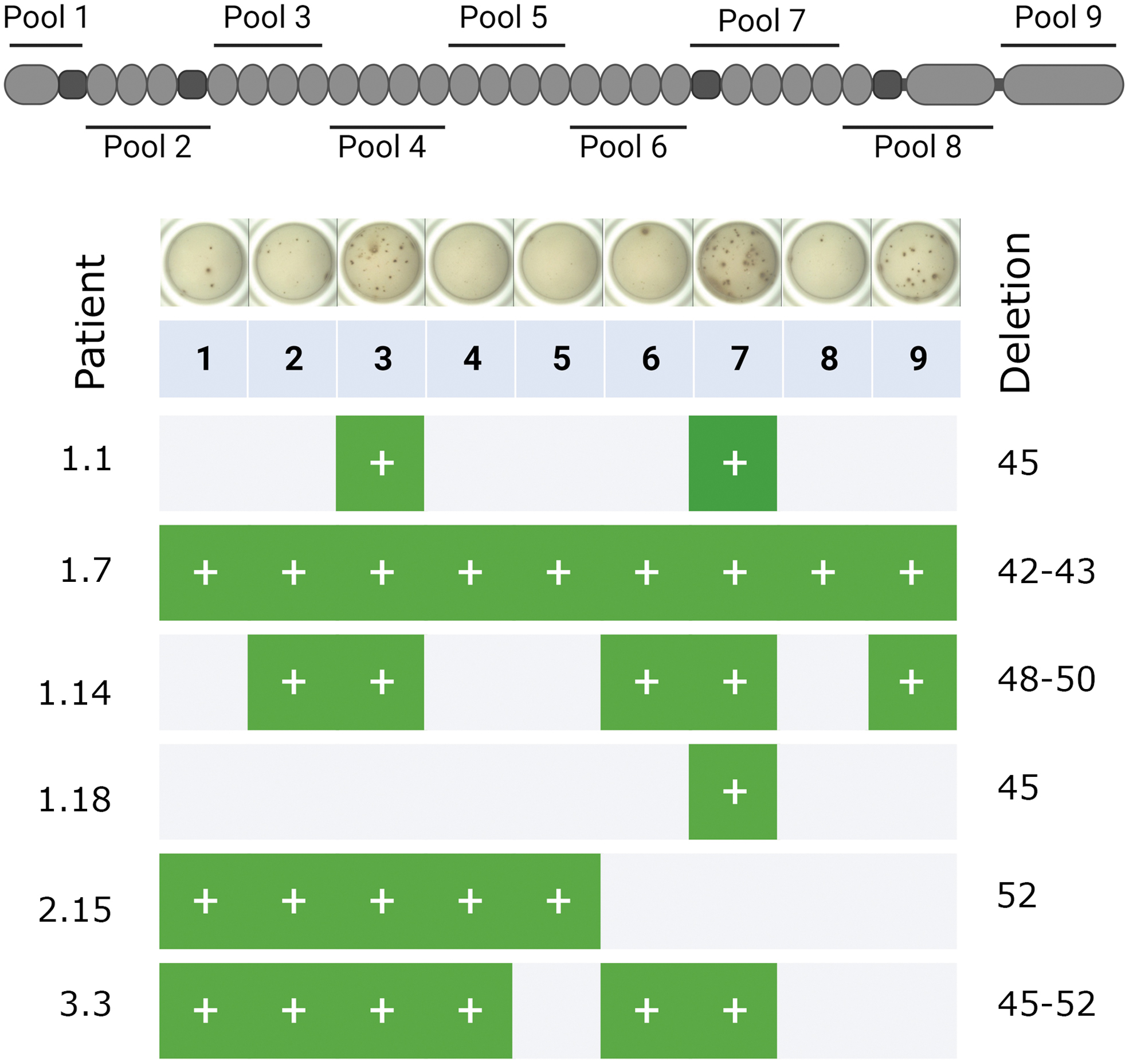
The location of the nine dystrophin peptide pools is illustrated in relation to the structural features of the dystrophin protein. An example of ELISPOT wells from one patient (Center 1, patient 14; (Supplementary Table S1) at the 0-year time point is provided. The table summarizes the results of the six positive patients at the 0-year time point showing which peptide pools returned a positive result. ELISPOT, Enzyme-Linked Immunospot.
ELISPOT assay
Peripheral blood T cell responses to dystrophin were quantified using the ELISPOT assay. 8 This was performed using the Human IFN-gamma Elispot kit (U-CyTech; CT230-PB5) and Millipore IP filter plates (Millipore; S2EM004M99) according to the manufacturers' instructions.
Around 3 × 105 cells were plated/well when screening for dystrophin responses and 75,000 cells/well were plated for the positive control or polyclonal stimulation. Concanavalin A (Sigma C0412) at final concentration of 2.5 μg/mL in PBS was used as a stimulus positive control. For peptide stimulations, the peptide pools were used at 1–2 μg/mL final concentration for each peptide. Cells were plated in duplicate wells, with a total volume of 200 μL (100 μL cells, 95 μL medium, and 5 μL antigen). The ELISPOT plate was covered with a lid and incubated at 37°C, 5–7% CO2, and 100% humidity for 24–36 h.
Spots were imaged and counted using an automated AID reader. The same camera and count settings were used for all samples. Each well was manually assessed to remove any debris mistaken as spots. Over 50% saturation was considered too numerous to count. The intensity was set to a minimum of 20 brightness units and the spot size set as 40–500 pixels with a minimum gradient of 5°. In line with Flanigan et al, a result was considered positive only when both duplicates were >15 spot forming units (SFC)/106 PBMCs. 8 We used this low threshold after discussion with Dr. Mendell's group, whose reported responses to dystrophin are typically very low level. 8 For further confidence, to record a positive result, the positive control (patient's PBMC reaction to Concanavalin A) must also be >15 SFC/106 PBMCs and the negative control (patient's PBMCs without stimulation) must be <5 SFC/106 PBMCs.
RESULTS
Boys with DMD were assessed annually (or semiannually for patient 1.1) over three (and in one case four) years with an ELISPOT IFN-γ assay performed with a full-length dystrophin peptide set. 9 The results are summarized in Fig. 1 and Table 1 (full data in Supplementary Table S1). Twenty three patients from Center 1, 20 patients from Center 2, 23 patients from Center 3, and 11 patients from Center 3 were included, ranging from 5 to 18 years of age. Details of the different steroid regimes are summarized in Supplementary Table S2. Thirty six were on prednisolone, 16 were on deflazacort, 15 were on prednisone, and 2 were on prednisone followed by deflazacort. Of the participants who remained on the same treatment throughout, 39 were on a daily regime (14 prednisolone, 14 deflazacort, and 11 prednisone) and 27 were on an intermittent regime (22 prednisolone, 2 deflazacort, and 3 prednisone).
A total of 6 (8%) individuals were positive at the first baseline visit (Fig. 2). The positive epitopes are located before and/or after the patient's deletion with no apparent association (Supplementary Table S3). All individuals who had a positive result had been treated with corticosteroids (five with prednisolone and one with prednisone); two were ambulant and four were nonambulant and ranged from 6 to 16 years of age (Supplementary Table S1). None of the 16 deflazacort-treated patients had a positive result. We compared the rates of positive results between individuals who had been treated with prednisone, prednisolone, deflazacort, and prednisone followed by deflazacort with those on no/discontinued treatment and between individuals who had been treated with prednisolone compared to deflazacort, using a Fishers Exact Test.
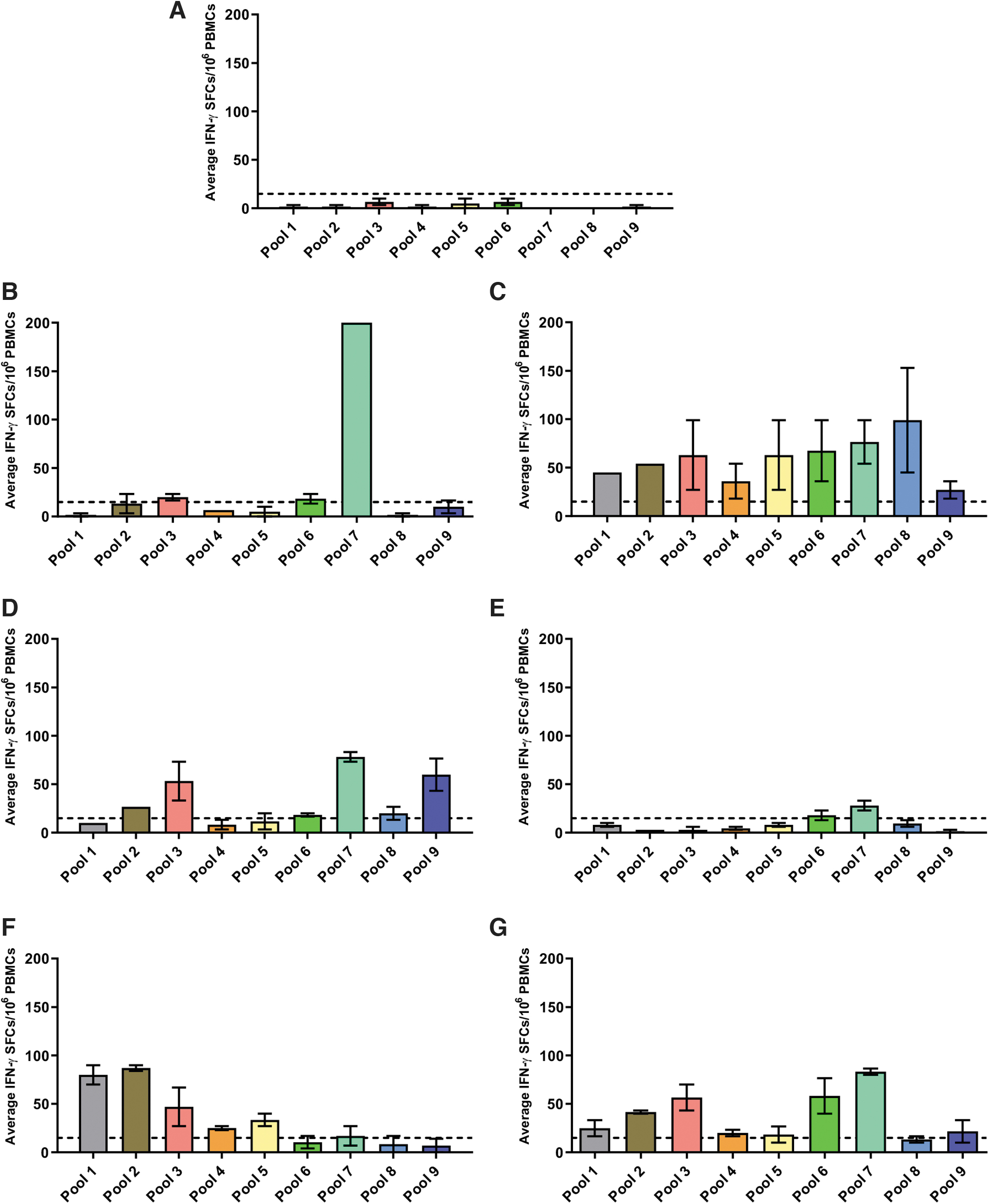
Graphs plotting the average (±SEM) IFN-γ SFCs/106 PBMC values across each peptide pool for:
We found no significant difference between the any of the groups (deflazacort vs. no/discontinued treatment—p = 1; prednisone vs. no/discontinued treatment—p = 1; prednisolone vs. no/discontinued treatment—p = 0.5661; prednisone followed by deflazacort vs. no/discontinued treatment—p = 0.2; and prednisolone vs. deflazacort p = 0.3077).
The positive individuals had deletions in exons 45 (2 patients), 42–43, 48–50, 52, and 45–52 (Supplementary Tables S1 and S3). These mutations are expected to lead to the lack of full-length dystrophin (Dp427), but all should have been able to produce the shorter dystrophin proteins Dp116, Dp71, and Dp40, and one patient would be expected to produce Dp140 (Supplementary Table S3). None of these shorter dystrophin proteins, with the exception of Dp71, 13 is expressed in skeletal muscle. 14
Due to the frequent occurrence of inconclusive results (defined below) at subsequent time points, we were unable to capture full longitudinal data on all individuals. Longitudinal data for two participants are presented in Fig. 3. Patient 1.1 (patient 1 from Center 1), carrying an exon 45 deletion, had an extremely strong response to peptide pool 7 (peptides encoded by exons 50–59) at baseline, which remained the strongest response among all pools at all subsequent visits (Fig. 3). In contrast, patient 1.18, also deleted for exon 45, was only weakly positive for peptide pool 7 at baseline and 1-year follow-up, but then showed a strong response to several peptide pools (including pool 7) at the 3-year visit (Fig. 3).
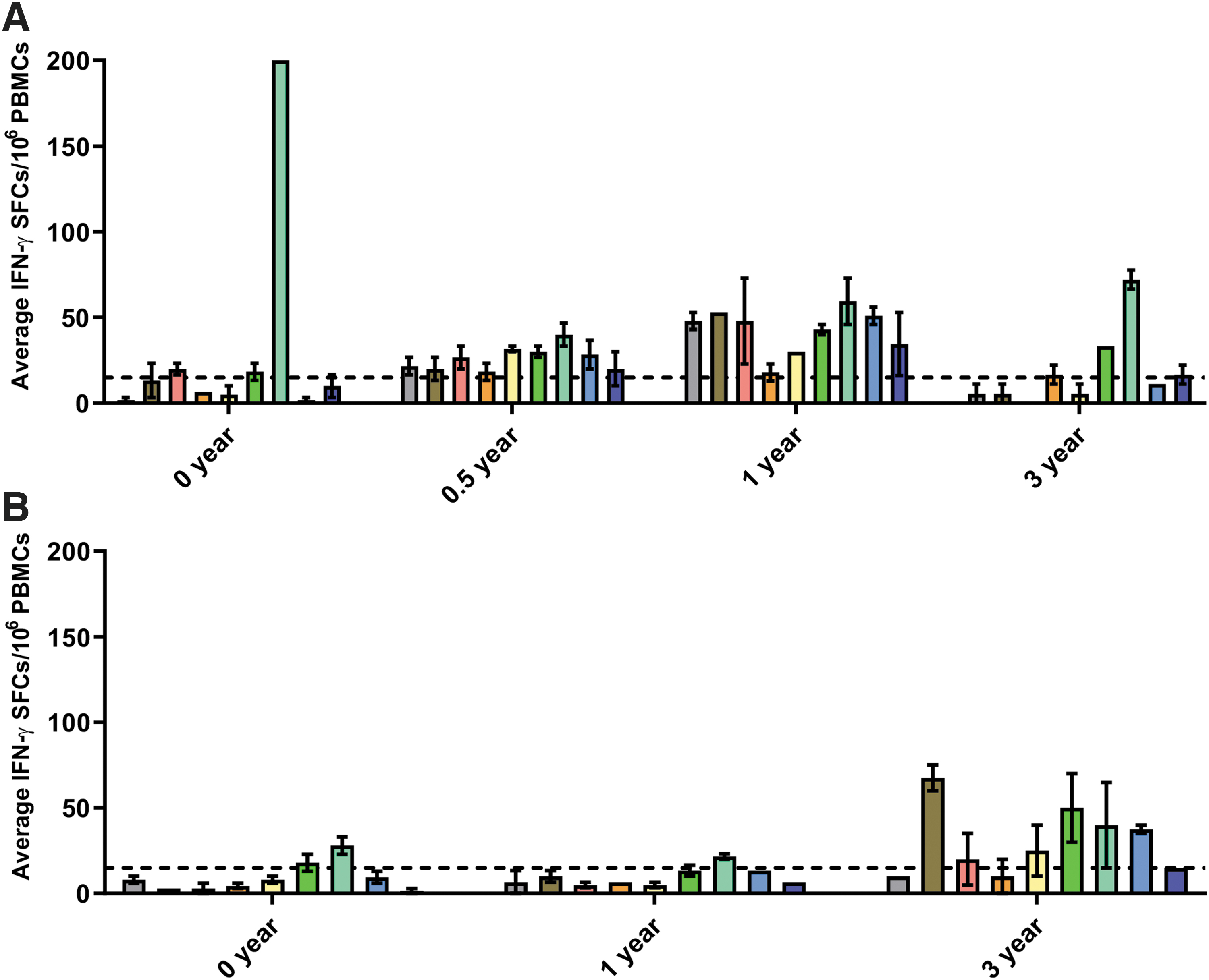
ELISPOT results for patients 1.1
Since patient 1.1 had a consistently strong response to peptide pool 7 at all time points, we performed additional ELISPOT assays using the mapping pool for peptide pool 7 at the 0- and 6-month time points (Fig. 4). At the 0-year time point, this patient was positive for pools 7C, 7L, 7M, and 7N, which together map to peptides encoded for by exon 54. The patient was also positive for pools 7A, 7L, 7M, and 7N, which maps to peptides encoded for by exon 51.
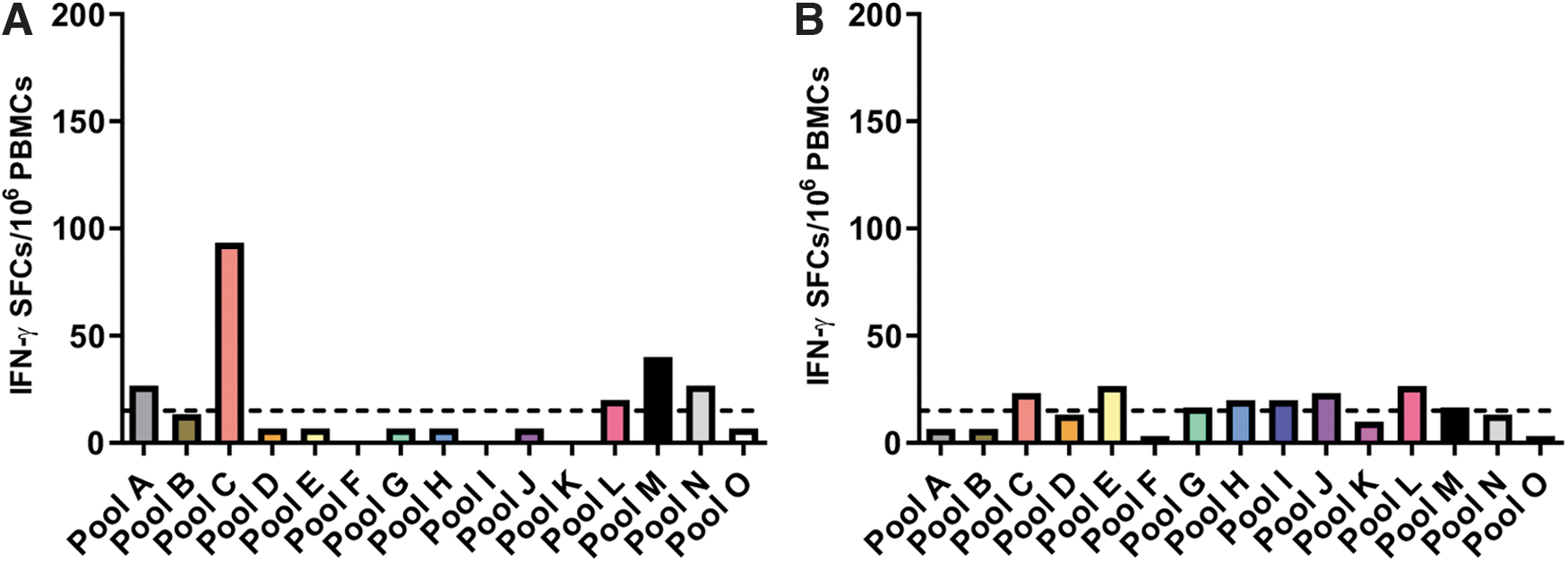
Results from mapping pool 7 at 0-
In summary, 4% of ambulant and 14.8% of nonambulant patients had a T cell response to dystrophin. The mean ages of ambulant and nonambulant patients at the start of the study were 8 and 14 years, respectively. All the positive patients were taking prednisolone (five patients) or prednisone (one patient). Of the patients in our study taking prednisolone, 13.9% returned a positive ELISPOT result. Only eight patients had not been treated with corticosteroids and these either had a negative or an inconclusive result.
One patient (1.14, Supplementary Table S1) with an exon 48–50 deletion, theoretically skippable for exon 51, was additionally assessed with peptides corresponding to unique junctional epitopes that would be generated by exon skipping. In this patient, the unique junctional epitope did not have a positive response, despite being positive to the full-length dystrophin peptide set (Fig. 5). We also analyzed four healthy adult controls and six disease controls, who all returned a negative result (data not shown).

Graph plotting the average (±SEM) IFN-γ SFCs/106 PBMC values for a peptide pool corresponding to the unique epitopes that would be generated by exon 51 skipping for patient 1.14 (48–50 deletion). A peptide pool for the unskipped scenario was also tested. The dotted line represents the positive cutoff value of 15 SFCs/106 PBMCs; note both duplicates must be >15 to be considered positive.
On occasions when the positive control was not positive, the negative control was positive, and/or there were not enough viable cells to perform the assay, samples were scored as inconclusive (Supplementary Table S1, marked with *). Sampling issues contributing to insufficient viable cells included the patient not attending the clinic, the appointment being cancelled, failure to take a blood sample, insufficient blood taken (a relatively high volume of blood (at least 10 mL) is required), or poor cell count and/or viability. There were (with rare exceptions) not enough cells remaining to perform repeat assays in cases where the original results were inconclusive. The assay was repeated on patient 3.1 at the first time point: the repeated assay was also inconclusive, as the negative control (patient's PBMCs without stimulation) was >5 SFC/106 PBMCs on both occasions.
CONCLUSIONS/DISCUSSION
When designing clinical trials to restore dystrophin, it is important to identify patients who have a pre-existing T cell-mediated immune response to dystrophin. It is also important to establish a natural history baseline and to try to understand factors that might affect this immune response and how it might be attenuated so that it does not interfere with treatment. This is especially important when considering AAV-mediated gene therapy approaches, which in pre-clinical work elicit significant dystrophin restoration (reviewed 15 –17 ), although similar considerations apply to any experimental therapy employed to restore dystrophin, from genome editing to cell therapy.
The fact that none of the neuromuscular disease controls had a T cell response to dystrophin suggests that a pathological muscle environment, including inflammation (reviewed 18 ), does not on its own play an obvious role in the process. The muscle fibers of patients with other types of neuromuscular disease contain dystrophin, so they would have been tolerized to the protein. Our findings indicate that this tolerance was not broken by the immune cells, which would include T cells that are present within pathological muscle. However, our small control group size is a limitation and a larger set of disease control individuals should be studied to conclusively address this question.
The ELISPOT assay is a highly sensitive and widely used immunoassay that measures the frequency of cytokine-secreting cells at the single-cell level (reviewed 19 ). It is in theory easy to perform and provides both qualitative and quantitative information. Pre-existing cellular immune responses to dystrophin have already been reported and quantified using the ELISPOT assay. 8,9 The assay has also been used to examine T lymphocyte responses to dystrophin in a clinical trial of AAV-minidystrophin, showing that some patients had an immune response either before the start of treatment (2/6 patients) or after treatment (4/6 patients). 9 These patients were given prednisolone 4 h before treatment. In contrast, none of the six dogs included in a preclinical study of AAV-microdystrophin, which were transiently immunosuppressed with cyclosporine and mycophenolate mofetil, had a post-treatment T cell response to dystrophin, but pretreatment response was not quantified. 20
T cell-mediated immune responses to dystrophin in patients enrolled in our study were relatively rare and occurred at a fairly low level. Approximately 9% (6/69) of steroid-treated DMD individuals had a pre-existing T cell-mediated immune response to dystrophin. A lower percentage of patients in our cohort had a response to dystrophin than in a previous study, 8 which reported that 20/70 (29%) of patients had T cell immunity against dystrophin. In this study, 91 subjects were enrolled, including 70 patients with DMD and 21 age-matched normal control subjects.
Among the patients with DMD, 29 were treated with deflazacort, 24 were treated with prednisone, and 17 were untreated. This may be due to the fact that the majority of our patients (69/77) were on corticosteroids, which would reduce the inflammatory response that occurs as part of the pathological process in dystrophin-deficient skeletal muscle (reviewed 21,22 ) and which may exacerbate any T cell response. In addition, the ages of patients in the two studies were slightly different—from 4 to 18 years of age at the start of our study, and from 3 to 25 years of age in Flanigan et al's study. 8 As older subjects were shown to have an increased probability of having an immune response to dystrophin, 8 it is possible that the different ages of subjects in the two studies may have contributed to the different findings.
However, we cannot determine whether, as previously suggested, 8 a smaller percentage of patients who had been steroid treated compared to nontreated have an immune response to dystrophin, as we had so few patients (5/77) who were steroid naive (and none of these had a T cell response). The fact that all our positive patients were on prednisolone or prednisone is in accordance with Flanigan et al, who found a lower incidence of T cell response in patients treated with deflazacort than prednisolone. 8 Hoever, we found no significant difference in the percentage of individuals who had a T cell response between those treated with either deflazacort or prednisolone. However, we only had 16 deflazacort-treated patients in our study, which is too low to draw any firm conclusion.
Interestingly, the shorter dystrophin protein products not affected by the genomic deletions, which are therefore expected to be produced by the patients studied (Supplementary Table S2), did not appear to give any protection by tolerizing against epitopes that they share with full-length dystrophin (Dp427). Revertant fibers, which are present in ∼50% of individuals with DMD, might either tolerize the individuals or induce an immune response to dystrophin. The fact that the number of revertant fibers does not change much with time 5 and our findings that individuals often have a T cell response to dystrophin at one time point, but not at others, argues against the idea that dystrophin in revertant fibers is eliciting the response.
Pre-existing immunity may be more of an issue in patients treated by gene therapy, which induces considerably higher levels of dystrophin production than exon skipping. In support of this, out of the 12 patients who had AON (Eteplirsen)-mediated restored dystrophin, there was no T cell response to dystrophin after 6 months of treatment. 8 Nevertheless, it is still important to identify, and if possible control, any immune response to dystrophin in patients both before they embark on any treatment intended to restore dystrophin and at time points after the onset of treatment.
Unfortunately, we encountered some issues that gave rise to inconclusive results in a high percentage of our assays (Supplementary Table S1). These included high background levels in negative controls that may be due to difficulties in processing blood within 24 h. Twenty-three patients had an inconclusive result at their first time point. If these patients are removed from the analyses, then 11% of all patients had a positive result at at least one time point. Of all 185 assays performed, 74 (40%) had an inconclusive result.
Eleven patients had either an inconclusive result or sample problems at every time point. These problems may well have skewed our findings. To overcome such problems, we suggest that center(s) collecting blood samples also isolate and freeze the PBMCs and send these, rather than the entire blood sample, to the laboratory doing the analysis. Obtaining a sufficient volume of blood (at least 10 mL to achieve enough duplicate wells of 3 × 105 PBMCs/well) to isolate PBMCs can be challenging, especially for younger DMD patients and those with neurobehavioral difficulties.
Despite the missing data indicated above, our longitudinal study clearly identified the T cell -mediated immune response to dystrophin in two DMD patients who were simply followed using standards of care. We complement and extend previous studies and show for the first time that having another type of muscular dystrophy, in which dystrophin is present, does not appear in itself to elicit an immune response to dystrophin.
In line with the fact that an individual's immunological memory response can vary over time, we show that an individual's overall natural immune response to dystrophin and response frequency can vary, as two patients who were positive at early time points were negative at the year 3 time point. Earlier work has suggested that the likelihood of an immune response to dystrophin increases with age. 5 In our cohort, four out of six patients who have a positive response were above the mean age (9 years) at the start of the study.
Overall, it is likely that the responses we observed are a result of low avidity T cells that have not quite escaped tolerance mechanisms. We cannot rule out cross-reactive responses from peptides that might be present in other proteins; it is also important to consider that each patient, even if they have the same deletion, will likely have different HLA types, which might govern different responses. Our findings highlight the need for a robust, reproducible standard operating procedure for collecting, storing, and shipping samples and for performing assay, so that different intralaboratory and interlaboratory operators achieve comparable results. Such a protocol could be used to routinely monitor patients' T cell response to dystrophin, especially in gene therapy clinical trials for DMD.
Further investigations of the T cell response might include use of the FluoroSpot assay, which utilizes fluorochrome-conjugated detection antibodies, thereby allowing the simultaneous detection of several individual cytokines and subsequent analysis of T cell subpopulations. 23,24 It would also be of interest to determine whether there is an antidystrophin humoral response in ELISPOT-positive patients.
In conclusion, our results show that pre-existing T cell responses to dystrophin are uncommon (8%), inconsistent, and low level. While this does provide some confidence for dystrophin restorative treatments, the fact that some patients are responsive warrants that baseline T cell response should be considered before interpreting any data from dystrophin restoration.
Footnotes
ACKNOWLEDGMENTS
We acknowledge Dr. Katie Campbell, Prof. Kevin Flanigan, Prof. Christopher Walker, and Prof. Jerry Mendell for sharing their ELISPOT expertise. We would like to thank Dr. Valentina Sardone for her help in preparing cells and Dr. Petra Disterer for her assistance. We thank Georgia Stimpson for statistical advice. The support of the MRC Centre for Neuromuscular Diseases Biobank is gratefully acknowledged. Je.M. was supported by Great Ormond Street Hospital Children's Charity. This research was supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
AUTHORs' CONTRIBUTIONS
K.A.: methodology (equal), formal analysis (equal), writing—original draft (supporting), writing—review and editing (equal), and performed experiments (equal). P.A.: methodology (equal), formal analysis (equal), writing—review and editing (equal), and performed experiments (equal). F.C.: methodology (equal), formal analysis (equal), writing—review and editing (equal), and performed experiments (equal). J.M.: methodology (equal), formal analysis (equal), writing—review and editing (equal), and performed experiments (equal). J.D.: methodology (equal) and formal analysis (equal); M.P.: methodology (equal), formal analysis (equal), writing—review and editing (equal), and performed experiments (equal). V.R.: methodology (equal), formal analysis (equal); and writing—review and editing (equal).
K.M.: writing—review and editing (equal). L.C.P.: writing—review and editing (equal). L.S.: writing—review and editing (equal). A.M.S.: writing—review and editing (equal). S.D.L.: writing—review and editing (equal). I.G.: writing—review and editing (equal). Y.D.K.: writing—review and editing (equal). J.G.M.V.: writing—review and editing (equal). E.H.N.: writing–—review and editing (equal). V.S.: writing—review and editing (equal). M.G.: writing—review and editing (equal). T.V.: writing—review and editing (equal). Je.M.: writing—original draft (lead) and writing—review and editing (equal). F.M.: conceptualization (lead), methodology (lead), and writing—review and editing (equal).
AUTHOR DISCLOSURE
L.S. reports consultancy for Pfizer, Sarepta, Santhera, Biophytis, and Regenx Bio., and is on the data and safety monitoring board of Fibrogen. I.d.G. was in the past principal investigator of several drug trials (Italfarmaco, NSPharma, and ReveraGen). E.H.N. report grants from Spieren voor Spieren, Duchenne Parent Project, ZonMW, AFM, and PPMD. He has been site principal investigator for clinical trials conducted by BioMarin, GSK, Eli Lilly, Santhera Pharmaceuticals, Italfarmaco SpA, Roche Pharma, Reveragen, NS Pharma, Fibrogen, Sarepta, Alexion, Janssen, and Argnx outside the submitted work. He also reports ad hoc consultancies for PTC therapeutics, WAVE Life Sciences, Edgewise, Epirium Bio, Janssen, Sarepta, and Regenxbio. All reimbursements were received by the LUMC. No personal financial benefit was received.
V.S. is or has been on advisory boards for Astellas Gene Therapies, Biogen, Edgewise Therapeutics, Kate Therapeutics, ML Bio Solutions, Novartis Gene Therapies, Roche, Sanofi Genzyme, Sarepta Therapeutics, Vertex Pharmaceuticals, and Wave Therapeutics. He received speaker honoraria from Sanofi Genzyme and Sarepta Therapeutics and has research collaborations with the two companies. M.G. is Study Chair for VBP15-004 study sponsored by ReveraGen, Chief Medical Coordinator for DMD study funded by NIH, Chief Investigator/Principal Investigator for clinical trials of Pfizer, Italfarmaco, Santhera, Roche, ReveraGen, Dynacure, and Edgewise. M.G. is a member of Advisory boards for Pfizer, NS Pharma, and Dyne (honoraria through Newcastle University), and has been given speaker honoraria from Sarepta and Novartis (2019, 2021, 2022).
T.V. has received consulting fees from BioMarin, Debiopharm, Dynacure, Italfarmaco, PTC Therapeutics, Sarepta Therapeutics, Santhera Antisense Therapeutics ,and Biophytis; he is an SAB member and holds equity in Constant Pharmaceuticals; he is a co-founder and holds equity in Dinaqor AG.; and is supported by the National Institute of Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust, and University College London.
FUNDING INFORMATION
M.G. received research funding from Sarepta, PTC (through TREAT NMD Enterprise or Newcastle University). J.M. has received research funding from Sarepta Therapeutics. F.M. has received research funding from Sarepta Therapeutics and personal fees from Roche, Pfizer, Dyne Therapeutics, and Sarepta, outside the submitted work.
SUPPLEMENTARY MATERIAL
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.