
Abstract
Duchenne muscular dystrophy (DMD) is a fatal muscle disease caused by dystrophin deficiency. Dystrophin consists of the amino terminus, central rod domain with 24 spectrin-like repeats and four hinges (H), cysteine-rich domain, and carboxyl terminus. Several highly abbreviated micro-dystrophins (μDys) are currently in clinical trials. They all carry H1 and H4. In this study, we investigated whether these two hinges are essential for μDy function in murine DMD models. Three otherwise identical μDys were engineered to contain H1 and/or H4 and were named H1/H4 (with both H1 and H4), ΔH1 (without H1), and ΔH4 (without H4). These constructs were packaged in adeno-associated virus serotype-9 and delivered to the tibialis anterior muscle of 3-month-old male mdx4cv mice (1E12 vector genome particles/muscle). Three months later, we detected equivalent μDys expression in total muscle lysate. However, only H1/H4 and ΔH1 showed correct sarcolemmal localization. ΔH4 mainly existed as sarcoplasmic aggregates. H1/H4 and ΔH1, but not ΔH4, fully restored the dystrophin-associated protein complex and significantly improved the specific muscle force. Eccentric contraction-induced force decline was best protected by H1/H4, followed by ΔH1, but not by ΔH4. Next, we compared H1/H4 and ΔH1 in 6-week-old male mdx mice by intravenous injection (1E13 vector genome particles/mouse). Four months postinjection, H1/H4 significantly outperformed ΔH1 in extensor digitorum longus muscle force measurements but two constructs yielded comparable electrocardiography improvements. We conclude that H4 is essential for μDys function and H1 facilitates force production. Our findings will help develop next-generation μDys gene therapy.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscle-wasting disease. 1 Affected boys show delayed development of motor function and lose the ability to walk in their early teenage. Patients also develop dilated cardiomyopathy at the late stage of life. The lifespan of DMD patients is reduced to about a quarter of the normal population. The primary causes of death are respiratory failure and cardiac failure.
Null mutations in the DMD gene are the genetic root of DMD. 2 The DMD gene encodes dystrophin, a 427 kDa subsarcolemmal cytoskeleton protein. 3,4 The full-length dystrophin protein contains four major functional domains, including the N-terminal (NT) domain, a rod domain composed of 24 spectrin-like repeats and 4 interspersed hinges, the cysteine-rich (CR) domain, and the C-terminal domain. Dystrophin and its associated proteins (dystroglycan, sarcoglycan, syntrophin, dystrobrevin, and neuronal nitric oxide synthase [nNOS]) form the dystrophin-associated glycoprotein complex (DGC). Dystrophin maintains the sarcolemma integrity primarily by linking the cytoskeleton with the extracellular matrix.
Adeno-associated virus (AAV)-mediated systemic gene delivery holds promise to treat all muscles in the body. 5 However, the full-length dystrophin expression cassette greatly exceeds the packaging capacity of a single AAV vector. To overcome this hurdle, investigators have developed highly abbreviated micro-dystrophin (μDys). 6 –9 Synthetic μDys is only one-third the size of the full-length protein. It carries the NT domain, a truncated rod domain, and the CR domain. Despite this dramatic size reduction, μDys was found to provide basic muscle protection in preclinical studies. 10 Several μDys AAV vectors have entered clinical trials in human patients. 11 Although the exact configuration of the μDys constructs used in each trial is different, they all carry hinge 1 (H1) and hinge 4 (H4). Hinges are proline-rich regions and are thought to provide structure flexibility. 12 The full-length dystrophin protein contains four hinges. H1 is located between the NT domain and spectrin-like repeat 1 (R1). H2 is located between R3 and R4. H3 is located between R19 and R20. H4 is located between R24 and the CR domain.
The two central hinges seem dispensable because the clinical trial μDys constructs are engineered to contain no central hinges (Solid Biosciences trial, SGT-001), 8,9 only H2 (Sarepta trial, SRP-9001; Genethon trial, GNT0004), 6 or only H3 (Pfizer trial, PF-06939926). 7 It is currently unclear whether H1 and H4 are essential for μDys-mediated muscle protection.
To understand the physiological implications of H1 and H4 in the context of μDys, we engineered three constructs that differ in hinge configurations (Fig. 1A). The starting μDys construct contained the NT domain, H1, R1, R16, R17, R24, H4, and the CR domain and was named H1/H4 μDys because it carried both H1 and H4 (Fig. 1A). This construct has been shown to effectively protect dystrophic muscle in murine and canine models of DMD following local and systemic delivery. 13 –16 Two new constructs were made by deleting H1 (ΔH1 μDys) or H4 (ΔH4 μDys) from the parental construct.
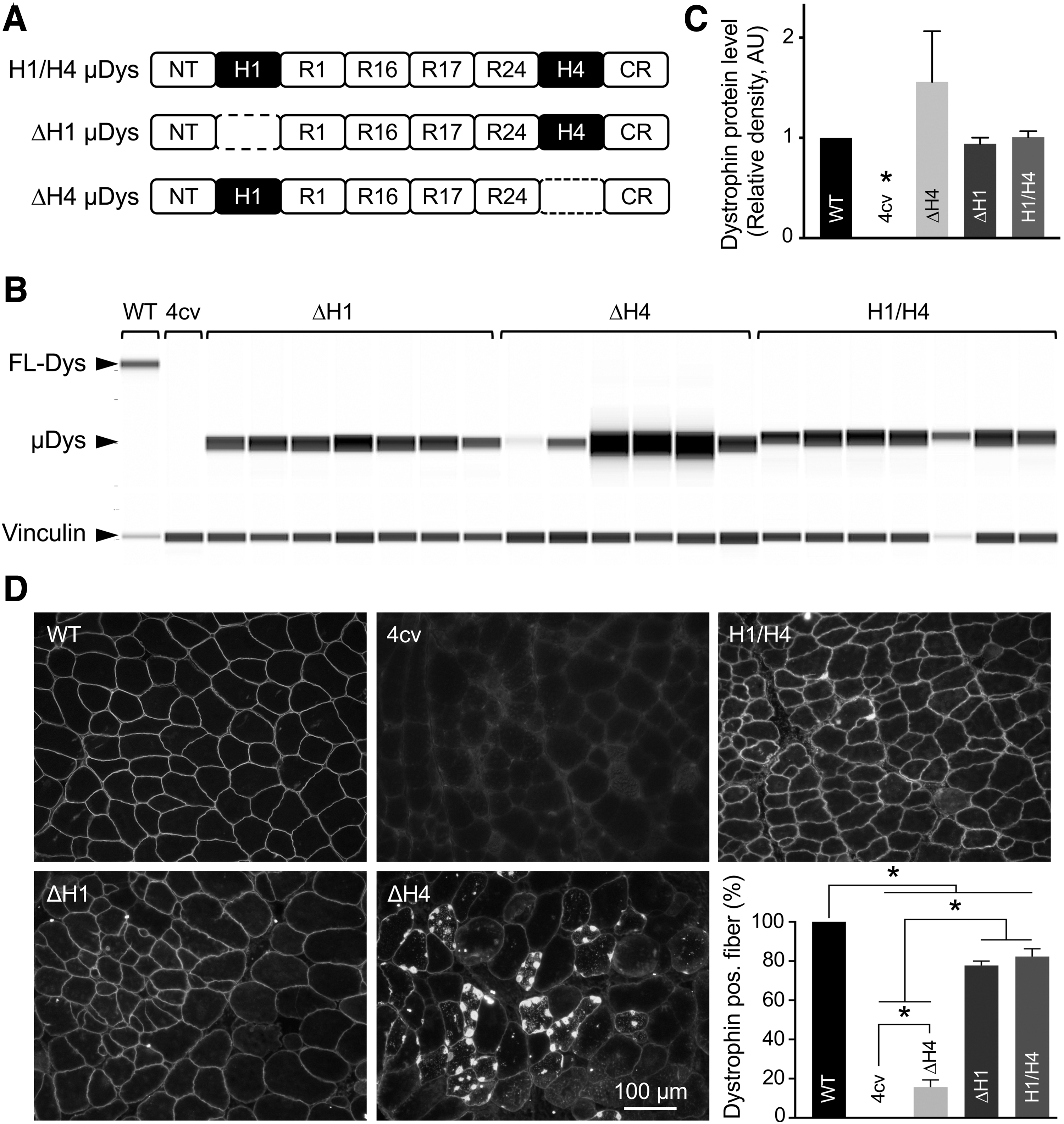
Absence of H4 but not H1 affected μDys localization.
We packaged these constructs in AAV serotype-9 (AAV9) and examined μDys expression, DGC reconstitution, and muscle contractility following local injection to the tibialis anterior (TA) muscle of mdx4cv mice. H1 deletion did not affect subcellular localization and DGC reconstitution but reduced protection against eccentric contraction-induced force drop. Deletion of H4 drastically altered subcellular localization of μDys, compromised DGC restoration, and impaired muscle force rescue.
To further compare H1/H4 and ΔH1 constructs, we performed systemic delivery in mdx mice. Consistent with local injection results, contractility of H1/H4 μDys outperformed ΔH1 μDys in the extensor digitorum longus (EDL) muscle. Nevertheless, two constructs resulted in comparable improvements in the electrocardiography (ECG) assay. Our results suggest that H4 is an essential component of μDys, and H1 may help force generation. These findings should be considered in the development of next-generation μDys gene therapy.
MATERIALS AND METHODS
Experimental animals
All animal experiments were approved by the Institutional Animal Care and Use Committee and were in accordance with NIH guidelines. Four strains of mice were used in the study, including dystrophin-deficient mdx mice and the corresponding wild-type control C57/BL10 mice, and dystrophin-deficient mdx4cv mice and the corresponding wild-type control C57/BL6 mice. All experimental mice were generated in a barrier facility using breeders purchased from The Jackson Laboratory (Bar Harbor, ME) (mdx, stock no. 001801; C57/BL10, stock no. 000476; mdx4cv, stock no. 002378; and C57/BL6, stock no. 000664). Only male mice were used in the study. The sample size is listed in Tables 1 and 2. All mice were maintained in a specific pathogen-free animal care facility on a 12-h light (25 lux)/12-h dark cycle with access to food and water ad libitum.
Bodyweight and anatomical properties of the tibialis anterior muscle in the local injection study
Statistically different from all other groups.
Statistically different from all other groups, except for the WT group.
μDy, micro-dystrophin; CSA, cross-sectional area; WT, wild-type.
Bodyweight and anatomical properties of the extensor digitorum longus muscle in the systemic injection study
Statistically different from the WT and H1/H4 μDys groups. p < 0.05.
EDL, extensor digitorum longus.
Construction of H1- and H4-deleted μDys expression cassette
Three different human μDys vectors were used in the study. The parent H1/H4 μDys construct (also called YL90) was published before under the name of ΔR2–15/ΔR18–23/ΔC
13,15,16
and R16–17/ΔC.
14
It contained the NT domain and CR domain, four spectrin-like repeats (R1, 16, 17, 24), and two hinges (H1, H4) (Fig. 1A). The expression of H1/H4 μDys was regulated by the cytomegalovirus promoter and SV40 polyadenylation signal. The entire expression cassette was flanked by the inverted terminal repeats from AAV2. The ΔH1 (also called pLW1) and ΔH4 (also called pLW2) vectors were generated by selective deletion of H1 and H4, respectively, using standard molecular cloning techniques (Fig. 1A). H1 is located between amino acids 253 and 327 (amino acids) and H4 is located between amino acids 3,041 and 3,112 in full-length human dystrophin according to the human dystrophin protein sequence in the Leiden muscular dystrophy database (
AAV production and delivery
All vectors were packaged in AAV9 using our previously published protocol. 17 For local delivery, AAV was injected into the TA muscle of 3-month-old male mdx4cv mice at the dose of 1E12 vector genome particles (vg)/muscle, and treated mice were harvested 3 months postinjection. For systemic delivery, AAV was injected through the tail vein to 6-week-old male mdx mice at the dose of 1E13 vg/mouse, and treated mice were harvested 4 months postinjection.
TA muscle function assay
TA muscle contractile properties were assessed in situ using our published protocols. 18,19 Briefly, the mouse was anesthetized through intraperitoneal injection of a cocktail containing 25 mg/mL ketamine and 2.5 mg/mL xylazine at 2.5 μL/g body weight. The TA muscle and the sciatic nerve were exposed. The mouse was transferred to a custom-designed thermocontrolled platform of the footplate apparatus. The sciatic nerve was stimulated using a custom-made 25G platinum electrode. The force was measured with a 305C-LR dual-mode servomotor transducer under the control of LabView-based Dynamic Muscle Control software (Aurora Scientific, Inc., Aurora, ON, Canada). Data were analyzed using the LabView-based Dynamic Muscle Analysis software (Version 3.12, Aurora Scientific).
The absolute tetanic force was measured by stimulating the muscle at 200 Hz for 300 ms. The eccentric contraction was performed by stimulating the muscle for a total of 350 ms at 200 Hz. During the last 200 ms, the muscle was stretched by 10% of the optimal length at the speed of 0.5 optimal length per second. The absolute tetanic force was converted to the specific muscle force by dividing it with the muscle cross-sectional area and reported as mN/mm2. TA muscle cross-sectional area was calculated according to the published protocol. 18
EDL muscle function assay
EDL muscle function was assessed ex vivo using our published protocols. 18,19 Briefly, the mouse was anesthetized as described above. The EDL muscle was gently dissected and mounted on an organ bath muscle test system (Aurora Scientific, Inc.) containing oxygenated (95% O2 and 5% CO2 at 30°C) Ringer's buffer. Muscle force was evaluated with a 305B dual-mode servomotor transducer (Aurora Scientific, Inc.). Data acquisition and analysis were performed with the Dynamic Muscle Control and Analysis software (Aurora Scientific, Inc.). The absolute tetanic force was measured by stimulating the muscle at 200 Hz for 300 ms. The eccentric contraction was performed by stimulating the muscle for a total of 700 ms at 200 Hz. During the last 200 ms, the muscle was stretched by 10% of the optimal length at the speed of 0.5 optimal length per second. The absolute tetanic force was converted to the specific muscle force by dividing it with the muscle cross-sectional area and reported as mN/mm2. EDL muscle cross-sectional area was calculated according to the published protocol. 18
ECG assay
Cardiac electrophysiology was evaluated using our published protocols as described in the standard operating protocol in the Cardiac Protocols for Duchenne Animal Models (
Morphological studies
Tissues were harvested at the end of the study. Harvested tissues were immediately frozen by covering in Tissue-Plus® optimal cutting temperature compound (Scigen Scientific, Gardena, CA) and freezing in liquid nitrogen-cooled 2-methylbutane. Ten-micron cryosections were used for staining. General muscle histology was evaluated by Hematoxylin and Eosin (H&E) staining.
Immunostaining of a single protein was performed as described in Kodippili et al. 22 Dystrophin was detected using the MANEX44A monoclonal antibody, which recognizes human dystrophin R17 (1:500, a gift from Dr. Glenn Morris at Wolfson Center for Inherited Neuromuscular Disease, RJAH Orthopedic Hospital, Oswestry, United Kingdom). β-Dystroglycan was detected with a mouse monoclonal antibody against the β-dystroglycan C-terminus (NCL-b-DG, 1:100; clone 43DAG1/8D5, IgG2a; Novocastra, Newcastle upon Tyne, United Kingdom). α-Sarcoglycan was detected with a mouse monoclonal antibody against α-sarcoglycan amino acid residues 217–289 (VP-A105; 1:1,000; clone Ad1/20A6, IgG1; Vector Laboratories, Newark, CA).
Syntrophin was detected with a pan-syntrophin mouse IgG1 monoclonal antibody that recognized the syntrophin PSD-95/Dlg/ZO-1 domain (ab11425, 1:2,000; clone 1351, IgG1; Abcam, Cambridge, MA). Dystrobrevin was detected with a mouse monoclonal antibody against dystrobrevin amino acid residues 249–403 (no. 610766, 1:200; clone 23, IgG1; BD Biosciences, San Diego, CA). nNOS was detected with a rabbit polyclonal antibody (N7280, 1:2,000; Sigma-Aldrich, St. Louise, MO).
Triple-immunostaining was used to codetect μDys, syntrophin, and nNOS in the same tissue section. Briefly, the tissue section was blocked using 20% normal goat serum (Thermo Fisher Scientific, Waltham, MA). A cocktail of antibodies against μDys, syntrophin, and nNOS was applied to the slide. μDys was recognized with MANDYS106, a mouse IgG2a monoclonal antibody against dystrophin R16 (1:100, a gift from Dr. Glenn Morris at Wolfson Center for Inherited Neuromuscular Disease, RJAH Orthopedic Hospital, Oswestry, United Kingdom). Syntrophin and nNOS were detected with the pan-syntrophin antibody and nNOS polyclonal antibody, respectively, as described above.
To visualize the target protein, a secondary antibody (Thermo Fisher Scientific) cocktail was applied to the slide. The secondary antibody mixture contains Alexa Fluor™ 647-conjugated goat anti-mouse IgG2a (1:100, to visualize μDys), Alexa Fluor 594-conjugated goat anti-mouse IgG1 (1:100, to visualize syntrophin), and Alexa Fluor 488-conjugated F(ab’)2-goat anti-rabbit IgG antibodies (1:100, to visualize nNOS). Slides were coverslipped using ProLong™ Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole. Slides were viewed at the identical exposure settings for each antibody for all animal groups using a Nikon E800 fluorescence microscope. Photomicrographs were taken with a QImage Retiga 1300 camera. Centrally nucleated myofibers were determined from H&E-stained images using the Fiji imaging software (
Protein Lysate Preparation
Proteins were extracted in homogenization buffer (10% sodium dodecyl sulfate, 5 mM ethylenediaminetetraacetic acid, 62.5mM Tris-HCl pH 6.8) supplemented with a protease inhibitor cocktail (Roche diagnostics, Indianapolis, IN). Samples were homogenized in Bullet Blender Storm 24 (Next Advance, Averill Park, NY) at the speed 12 for 8 min at 4°C followed by centrifugation at 14,000 rpm for 3 min (Eppendorf centrifuge, model 5417C; Eppendorf-Netheler-Hinz GmbH, Hamburg, Germany). The supernatant was used to measure the total protein concentration using the DC Assay Kit (Bio-Rad, Hercules, CA). Protein lysate concentrations were adjusted to 3 μg/μL and diluted to 1:1 using 2 × Laemmli buffer (Bio-Rad) supplemented with 20% 2-mercaptoethanol and denatured at 95°C for 5 min, chilled on ice for 2 min before loading.
Capillary Western (Wes) Blot
Protein samples were mixed with the Wes™ sample buffer and analyzed with a Simple Western assay designed for the Wes instrument by ProteinSimple (San Jose, CA). For the assay, reagents of the Wes (ProteinSimple) were used, and all steps of the assay were performed according to the manufacturer's protocol. Protein separation modules were preloaded with the separation matrix. The module plate was loaded with samples, primary antibodies, antibody diluent, and wash buffer. The plate was placed in the Wes instrument. Wes instrument processed all assay steps automatically using default settings. Using Compass V. 4.0.0 software (ProteinSimple, San Jose, CA), electropherograms were generated for each sample and each protein. The area under the curve, which represents the signal intensity of the chemiluminescent reaction was analyzed for each detected protein. Values given for each protein expression were normalized to vinculin followed by normalization to the wild-type data.
Dystrophin was detected by MANDYS103(8E11) monoclonal antibody, which recognizes human dystrophin R16 (a gift from Dr. Glenn Morris). Vinculin was detected with a polyclonal antibody (cat. no. 155120; Abcam).
Western Blot Evaluation of DGC Components
Western blot was performed as we described before with 21 μg of total protein lysate loaded per lane.
23
β-Dystroglycan was detected with a mouse monoclonal antibody against the β-dystroglycan C-terminus (NCL-b-DG, 1:100; clone 43DAG1/8D5, IgG2a; Novocastra). Syntrophin was detected with a pan-syntrophin mouse monoclonal antibody that recognizes the syntrophin PDZ domain (ab11425, 1:2,000; clone 1351, IgG1; Abcam). Dystrobrevin was detected with a mouse monoclonal antibody against dystrobrevin amino acid residues 249 to 403 (no. 610766, 1:1,000; clone 23, IgG1; BD Biosciences). For the loading control, we used an antibody against Vinculin (ab155120, 1:2,000; Abcam). Western blot image quantification was performed using LI-COR Image Studio Version 5.0.21 (
Statistical Analysis
Data are presented as mean ± stand error of mean. One-way analysis of variance with Tukey's multiple comparison analysis was performed using GraphPad PRISM software version 7.0 for Mac OSX (GraphPad Software, La Jolla, CA). A p < 0.05 was considered statistically significant.
RESULTS
Subcellular Localization of μDys Was Affected by H4 But Not H1 Deletion
To study the function of H1 and H4 in the context of μDys, we generated three AAV vectors. The parental H1/H2 μDys vector carries both H1 and H4, the ΔH1 μDys vector carries H4 but not H1, and the ΔH4 μDys vector carries H1 but not H4 (Fig. 1A). AAV vectors were delivered to the TA muscle of mdx4cv mice by intramuscular injection (1E12 vg/muscle). μDys expression was examined in whole muscle lysate and muscle cross-sections by capillary western blot and immunofluorescence staining, respectively (Fig. 1B–D). On capillary western blot, the expression of ΔH4 μDys appeared higher compared with H1/H4 μDys and ΔH1 μDys. However, the difference did not reach statistical significance (Fig. 1B). On average, the expression of all three μDys reached the level of full-length dystrophin in wild-type mice (Fig. 1C). On immunostaining, H1/H4 μDys and ΔH1 μDys showed the expected sarcolemmal expression. Despite some sarcolemmal localization, ΔH4 μDys was mainly presented as dense aggregates in the sarcoplasm (Figs. 1D and 2A and Supplementary Fig. S1).
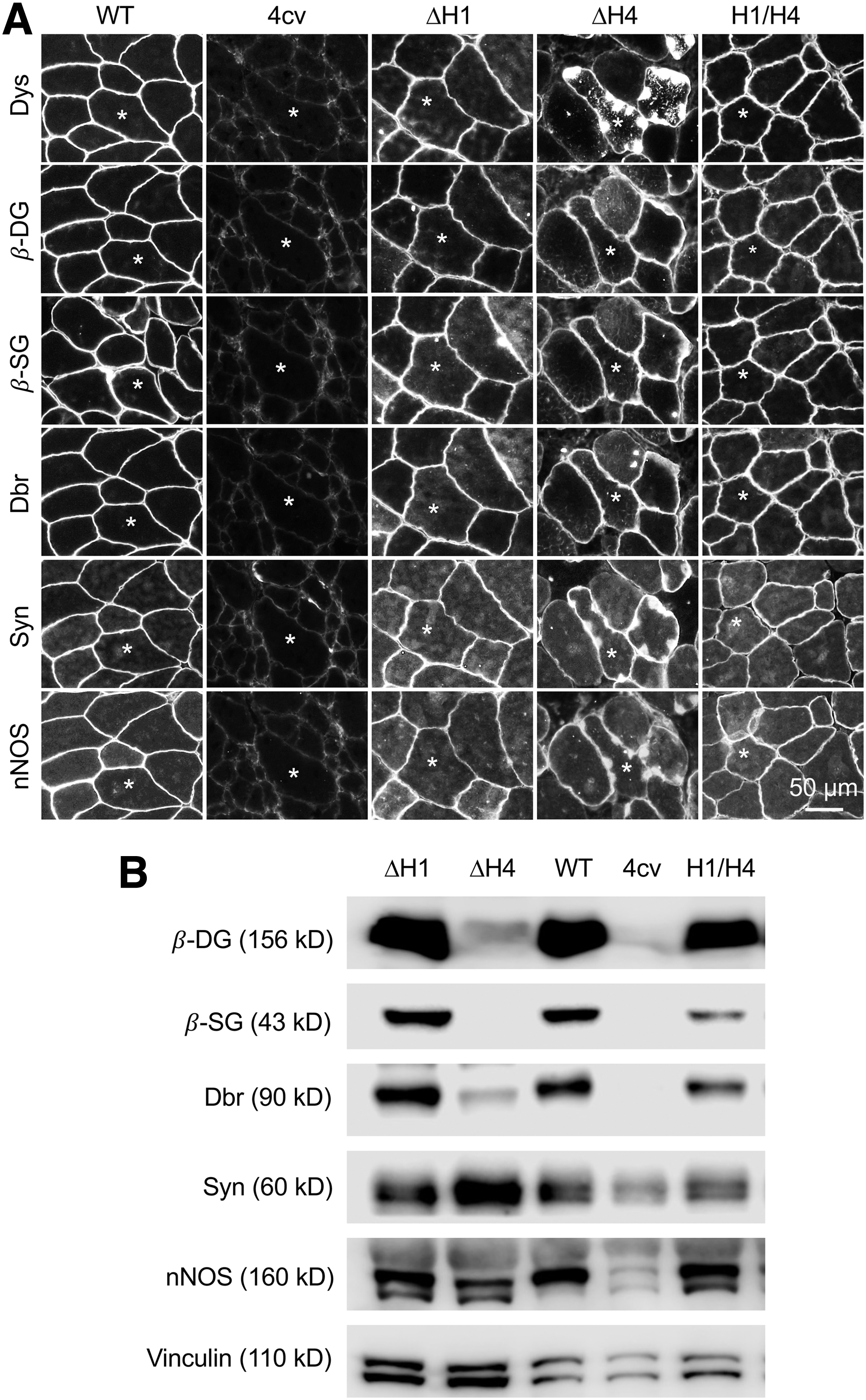
H4 was essential for dystrophin-associated glycoprotein complex assembly.
Interestingly, some aggregates accumulated around myonuclei (Supplementary Fig. S1A). On quantification, more than 70% of myofibers were dystrophin positive in the H1/H4 μDys and ΔH1 μDys groups. However, only 15.6% of myofibers were dystrophin positive in the ΔH4 μDys group (Fig. 1D and Supplementary Fig. S2).
H4 Deletion Compromised DGC Reconstitution
The biological function of dystrophin is achieved by the DGC. We examined DGC components by immunofluorescence staining and western blot (Fig. 2 and Supplementary Figs. S1 and S3). On immunostaining, all three types of μDys were able to recruit β-dystroglycan, β-sarcoglycan, dystrobrevin, syntrophin, and nNOS to the sarcolemma (Fig. 2A and Supplementary Fig. S1). Interestingly, the subcellular localization of syntrophin and nNOS, but not β-dystroglycan, β-sarcoglycan, and dystrobrevin, colocalized with dystrophin in myofibers that expressed ΔH4 μDys (Fig. 2A and Supplementary Fig. S1). On western blot, the levels of β-dystroglycan, β-sarcoglycan, dystrobrevin, syntrophin, and nNOS in the H1/H4 μDys and ΔH1 μDys groups were comparable to those of normal muscle (Fig. 2B). However, in the ΔH4 μDys group, the expression of β-dystroglycan and dystrobrevin was substantially reduced, and β-sarcoglycan was barely detected (although β-sarcoglycan was detected on the sarcolemma by immunostaining) (Fig. 2 and Supplementary Fig. S3).
ΔH4 μdys failed to improve muscle contraction
Next, we examined the histological and physiological consequences of H1 and H4 deletion (Fig. 3). On H&E staining, μDys-treated muscles were indistinguishable from untreated mdx4cv muscle. The percentage of centrally nucleated myofibers, a marker for muscle degeneration and regeneration, was similar in untreated and μDys-treated mdx4cv muscles (Fig. 3A).
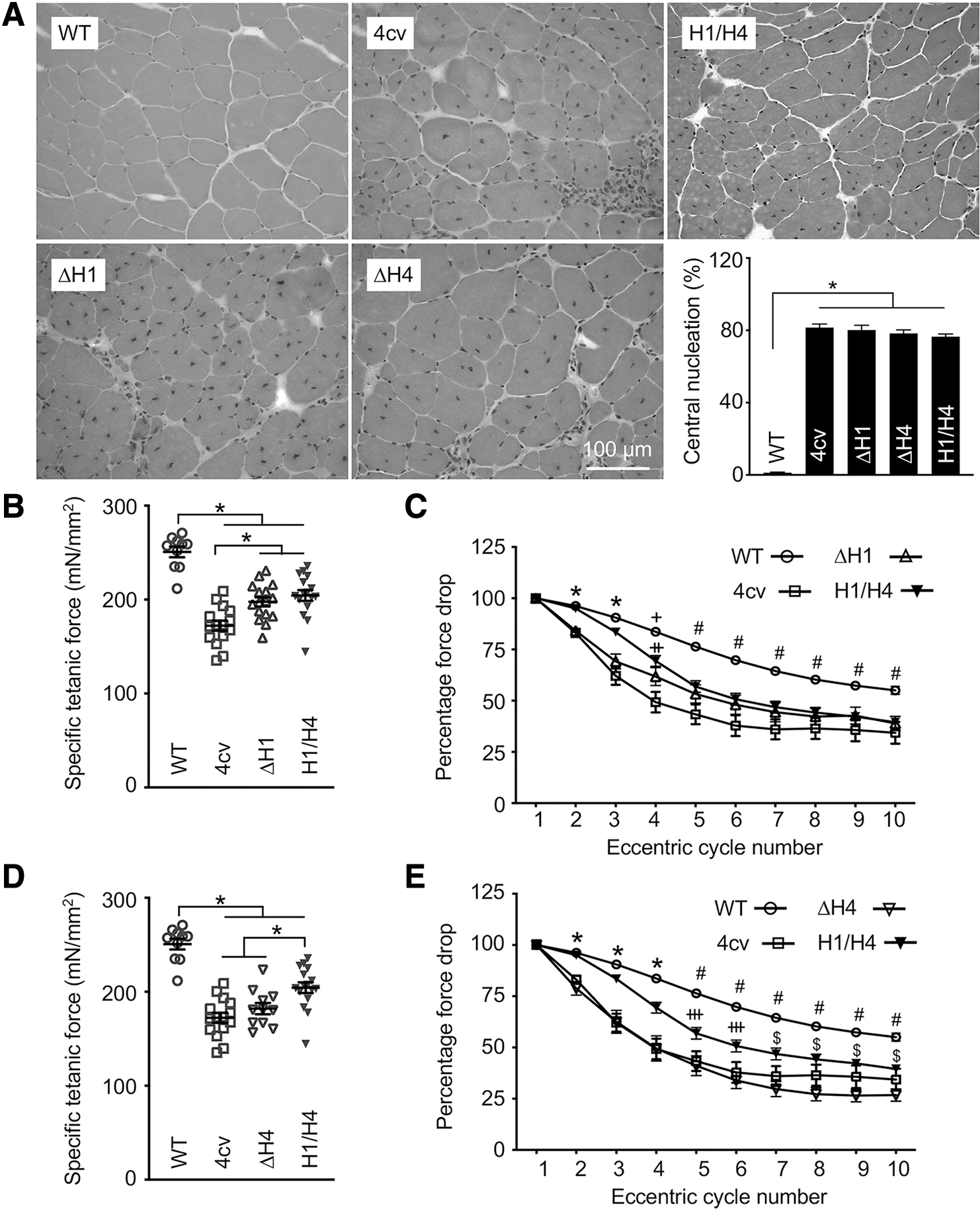
TA muscle function was differently impacted by H1 deletion and H4 deletion.
We evaluated the contractile properties of the TA muscles in situ (Fig. 3B–E). H1/H4 μDys and ΔH1 μDys treatment significantly increased but did not normalize the specific tetanic force (Fig. 3B). H1/H4 μDys completely prevented force drop induced by one round of eccentric contraction (Fig. 3C and Supplementary Fig. S4A). However, it only partially prevented force drops in subsequent rounds of eccentric contraction. ΔH1 μDys failed to prevent force drop induced by the first two rounds of eccentric contraction. In subsequent rounds, H1/H4 μDys and ΔH1 μDys showed a similar pattern. In contrast, ΔH4 μDys did not improve the specific muscle force (Fig. 3D). On eccentric contraction assay, ΔH4 μDys behaved exactly as mdx4cv in the first five cycles and appeared worse than mdx4cv in subsequent cycles (Fig. 3E and Supplementary Fig. S4B).
We also examined the TA muscle anatomy (Table 1). In untreated mdx4cv mice, we detected the well-described muscle hypertrophy. 24 This was partially corrected by H1/H4 μDys but not by ΔH1 μDys and ΔH4 μDys (Table 1).
EDL muscle function was improved by H1/H4 μDys in the systemic gene therapy study
H1/H4 μDys and ΔH1 μDys were further compared following systemic injection in mdx mice (Figs. 4 and 5, Table 2 and Supplementary Figs. S5 and S6). To study skeletal muscle, we examined μDys expression and force generation in the EDL muscle. On immunostaining, ∼65% of myofibers were dystrophin positive in the H1/H4 μDys group, whereas ∼40% of myofibers were dystrophin positive in the ΔH1 μDys group (Fig. 4A and Supplementary Fig. S5). Capillary western blot showed a similar pattern (Fig. 4B, C). The μDys level reached 16.8% of the normal in wild-type mice muscle in the H1/H4 μDys group but only 12.6% in the ΔH1 μDys group (Fig. 4C). H1/H4 μDys treatment significantly prevented the EDL muscle hypertrophy (Table 2), significantly improved the specific muscle force (Fig. 4D), and significantly limited eccentric contraction-induced force decline (Fig. 4E).
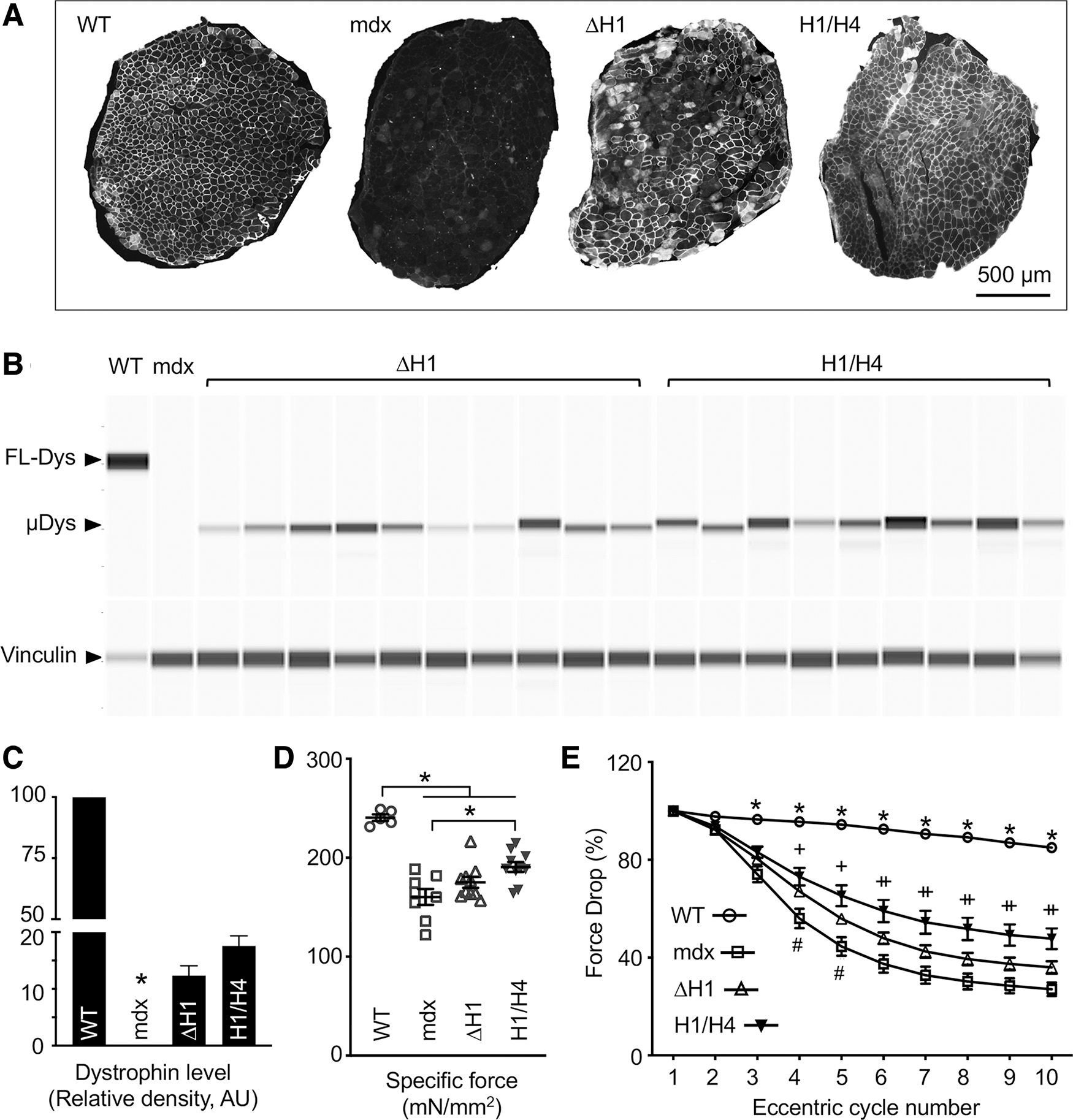
H1 deletion compromised force recovery in the EDL muscle.
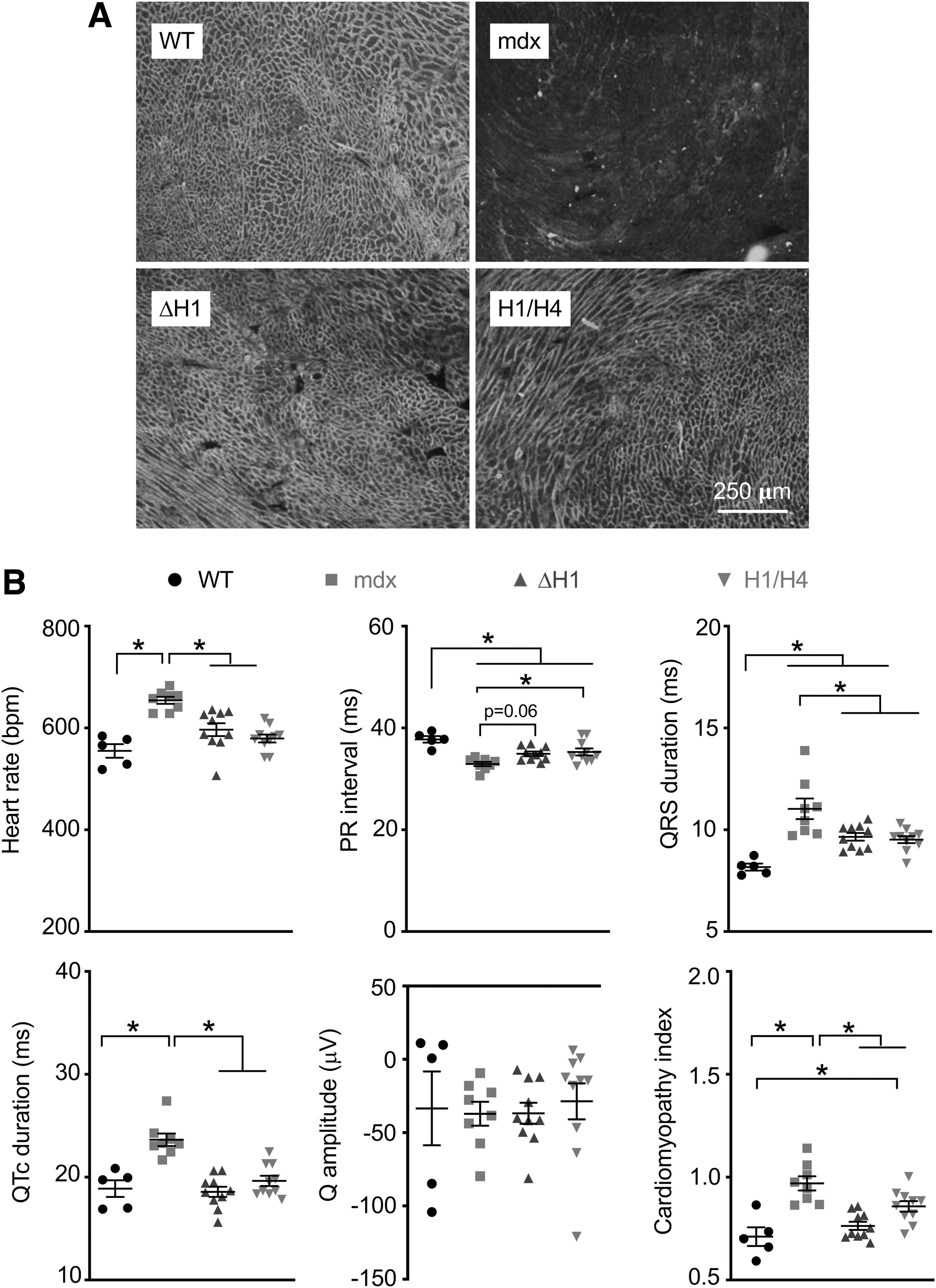
ΔH1 μDys and H1/H4 μDys were equally effective in improving ECG.
In contrast, ΔH1 μDys neither prevented muscle hypertrophy nor did it increase the specific muscle force (although there was a trend of improvement) (Fig. 4D and Table 2). Nevertheless, ΔH1 μDys-treated muscle showed a slightly better profile than untreated mdx muscle in the eccentric contraction assay (Fig. 4E and Supplementary Fig. S6).
ECG Was Similarly Improved by H1/H4 and ΔH1 μDys
Mdx mice do not develop cardiomyopathy until later in life. 25 However, ECG abnormalities can be detected in young adult mdx mice. 26 Hence, we examined heart μDys expression and ECG (Fig. 5). Consistent with our previous publications, 27 –29 we observed saturated μDys expression on immunostaining (Fig. 5A). On ECG assay, several parameters (heart rate, QRS duration, QTc duration, and the cardiomyopathy index) showed comparable levels of improvement in H1/H4 μDys- and ΔH1 μDys-treated mice. H1/H4 μDys treatment resulted in significant improvement in PR interval. ΔH1 μDys also improved PR interval but was marginal on statistical analysis (p = 0.06) (Fig. 5B).
DISCUSSION
In this study, we investigated the importance of H1 and H4 to μDys function. We showed that H4 is absolutely required. The absence of H4 distorted subcellular localization of μDys and impaired DGC restoration (Figs. 1 and 2 and Supplementary Figs. S1 and S3). Importantly, H4-deleted μDys did not improve muscle force (Fig. 3 and Supplementary Fig. S4B), suggesting H4 is essential for achieving the therapeutic effect. In contrast, H1 deletion did not interfere with the subcellular localization of μDys nor did it affect DGC restoration (Figs. 1 and 2). Furthermore, H1-deleted μDys ameliorated ECG abnormalities (Fig. 5). Nevertheless, it did not prevent muscle hypertrophy and was less effective in improving muscle function (Figs. 3 and 4, Tables 1 and 2, and Supplementary Figs. S4A and S6). Collectively, our data suggest that the inclusion of H1 enhances μDys function.
Hinges are enriched with proline. Unlike spectrin-like repeats, hinges do not have a defined helical structure. Full-length dystrophin has four hinges. These hinges are dispersed in the rod domain, one at the beginning, one at the end, and two in the middle. They are named H1 (75 amino acids, encoded by exons 8–10, and located between the NT domain and R1), H2 (50 amino acids, encoded by exon 17, and located between R3 and R4), H3 (47 amino acids, encoded by exons 50–51, and located between R19 and R20), and H4 (72 amino acids, encoded by exons 61–64, and located between R24 and the CR domain, and partially overlaps with the CR domain). Hinges were initially thought to act as flexible linkers between blocks of highly structured spectrin-like repeats to provide elasticity to the protein. 12 Subsequent animal studies and clinical investigations on H2, H3, and H4 suggest that hinges may have an important function.
Unlike other hinges, H2 contains a stretch of five consecutive proline residues. 12 Banks et al. found that this unique polyproline configuration profoundly influenced skeletal muscle maintenance, maturation, and structure when H2 was flanked by R3 and R24 in μDys. 30,31 Specifically, the authors observed chronic myotendinous strain injury and ring fiber formation in mdx muscles that expressed ΔR4–R23 or ΔR4–R23/ΔC μDys, and abnormal neuromuscular synapses in mdx muscles that expressed ΔR4–R23/ΔC μDys. The authors concluded that the rigid polyproline structure may compromise the integration of muscles into nerves and tendons. Interestingly, these structural abnormalities were eliminated by deleting the polyproline stretch or substituting H2 with H3. 31
Becker muscular dystrophy (BMD) patients carry in-frame deletions in the DMD gene. Interestingly, clinical studies in BMD patients suggest that the absence of H3 could be beneficial. Carsana et al. studied clinical presentations of BMD patients that have deletions between exons 45 and 55. 32 Muscle disease onset was not affected by H3 deletion. However, patients without H3 showed much slower disease progression. In a separate study, Kaspar et al. found that the onset of dilated cardiomyopathy was significantly delayed in patients without H3. 33 Harper et al. compared skeletal muscle disease between two lines of μDys transgenic mice. 6 Structure-wise, two μDys proteins were identical, except for the inclusion of H3 in the rod domain of one μDys protein. Interestingly, the one without H3 showed better histology and force, despite being expressed at a lower level. 6
H4 contains the WW domain, a protein-binding module characterized by two highly conserved tryptophan residues that are spaced 20–22 amino acids apart. 34 –36 The WW domain has been well characterized for its role in multiprotein complex assembly. Biochemical and crystal structure studies suggest that the WW domain in H4 and the first EF-hand in the neighboring CR domain bind the cytoplasmic tail of β-dystroglycan. 37 –40 This interaction forms the basis for dystrophin to interact with the extracellular matrix. Our ΔH4 μDys vector results reinforced the importance of H4 for dystrophin function. Specifically, the μDys protein was mislocalized, the DGC assembly was compromised, and protection against contraction-induced stress was lost in the absence of H4 (Figs. 1–3).
The immunostaining results of ΔH4 μDys AAV injected muscle were intriguing (Figs. 1D and 2A and Supplementary Fig. S1). The percentage of dystrophin-positive myofibers was only approximately one-fourth of what was detected in H1/H4 μDys- and ΔH1 μDys AAV-injected muscles. All three μDys genes were delivered by the same AAV capsid and their expression cassettes were regulated by the same promoter and pA signal; it is thus unlikely that the dramatical difference in the number of dystrophin-positive cells is due to differences in AAV transduction (uptake, intracellular trafficking, and single-strand to double-strand genome conversion) or μDys transcription. Future studies are needed to determine (1) whether the vector genome and μDys transcripts can be detected in dystrophin-negative cells in ΔH4 μDys AAV-injected muscles using the newly developed in situ RNAscope technique, 41 and (2) whether the difference is caused by translation and/or stability of ΔH4 μDys. Another intriguing observation is the peculiar pattern of ΔH4 μDys localization in dystrophin-positive cells.
In addition to the sarcolemma, most ΔH4 μDys proteins formed large inclusions in the sarcoplasm (Fig. 2A and Supplementary Fig. S1). Some aggregates appeared as ring-shaped structures around myonuclei (Supplementary Fig. S1). We suspect that sarcolemmal expression is likely due to dystroglycan-independent membrane-anchoring mechanism(s). 42 Sarcoplasmic aggregates have only been reported in transgenic mice that express excessive amounts of dystrophin. 6,43 This seems unlikely the case here. One hypothesis is the potential involvement of β-dystroglycan in assisting the intracellular trafficking of dystrophin. Substantial future investigations are warranted to test this hypothesis. One more interesting finding in ΔH4 μDys vector-injected muscle is the differential expression of the DGC components. On immunostaining, all DGC components were detected at the sarcolemma (Fig. 2A). Surprisingly, on western blot, only syntrophin and nNOS levels were comparable to those of normal muscle and other two μDys vector-injected muscles (Fig. 2B).
β-Dystroglycan and dystrobrevin were greatly reduced and β-sarcoglycan was barely detected. We do not have a biological explanation for this unexpected finding. If this is indeed a true finding, it may imply that the assembly of the DGC is more complex than what we know. Alternatively, our findings could be an artifact of antibody sensitivity. We have previously shown that the same antibody may show different detection sensitivity for different assays (e.g., immunostaining versus western blot). 22
The role H1 plays in dystrophin has never been appreciated until recently. It was suggested that dystrophin H1 may carry a dominant immunogenic epitope. 44,45 Importantly, the presence of H1 in μDys may underlie severe adverse events seen in clinical trials. 44 While the removal of H1 from μDys may attenuate the transgene-triggered immune response, it is unclear whether such modification can compromise μDys function.
We found that the loss of H1 did not affect subcellular localization of μDys, DGC restoration, and ECG improvement (Figs. 1D, 2, 4A, and 5). However, it resulted in functional deficiencies in muscle force assays (Figs. 3 and 4 and Supplementary Figs. S4A and S6). In both local and systemic injection studies, ΔH1 μDys was less effective than H1/H4 μDys in preventing eccentric contraction-induced damage (Figs. 3C and 4E and Supplementary Figs. S4A and S6). Interestingly, when μDys levels were comparable to that of wild type (WT) muscle, ΔH1 μDys and H1/H4 μDys were equally effective in improving the tetanic muscle force (Fig. 3B). When μDys levels were about one-fifth of the WT muscle, only H1/H4 μDys significantly increased tetanic force (Fig. 4C).
Our results suggest that H1 is important for force production, especially when μDys expression is low. It is currently unclear how the loss of H1 compromises muscle function rescue. Interestingly, we found that ΔH1 μDys did not prevent pathological muscle hypertrophy in mdx mice (Tables 1 and 2). It has been shown that hypertrophy in mdx mice is caused by myofiber branching. 24 Branched myofibers are more susceptible to mechanical stress damage. 46 –48 Failure to rescue muscle hypertrophy may thus result in more branched myofibers, hence, poorer function.
The limited packaging capacity of AAV has prevented the use of a full-length dystrophin expression cassette for DMD gene therapy. The need to abbreviate the dystrophin coding sequence for AAV delivery has stimulated the development of synthetic μDys genes. Although several highly functional μDys AAV vectors have entered clinical trials, there is a need to further improve μDys design. Removal of nonessential components in μDys would allow the inclusion of additional regulatory elements (in the expression cassette) that may improve the safety of the AAV μDys vector. In this study, we examined the importance of H1 and H4. Our results suggest that H4 is fundamental to μDys and H1 facilitates muscle contraction in the context of μDys. These findings provide novel insights for the engineering of next-generation μDys AAV vectors in the future.
Footnotes
ACKNOWLEDGMENTS
The authors thank Sean Duan and Thomas McDonald for their help in quantifying revertant fibers in mdx4cv and mdx mice.
AUTHORs' CONTRIBUTIONS
Conceived and designed experiments: L.P.W., Y.L., and D.D. Performed the experiments: L.P.W., T.B.W., N.B.W., M.J.B., and Y.Y. Analyzed data: L.P.W., T.B.L., N.B.W., Y.L., G.Y., and D.D. Wrote the article and made figures: L.P.W., G.Y., and D.D. All authors edited the article, and approved submission.
AUTHOR DISCLOSURE
D.D. is a member of the Scientific Advisory Board for Solid Biosciences and equity holders of Solid Biosciences. D.D. is a member of the Scientific Advisory Board for Sardocor Corp. The Duan Laboratory received research support unrelated to this project from Solid Biosciences in the last 3 years. The Duan Laboratory has received research support unrelated to this project from Edgewise Therapeutics in the last 3 years. Other authors have no disclosure.
FUNDING INFORMATION
This work was supported by grants from the National Institutes of Health (NS-90634), Jackson Freel DMD Research Fund, and Jett Foundation.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.