
Abstract
Solid cancers remain a major health challenge in terms of research, not only due to their structure and organization but also in the molecular and genetic variations present between tumors as well as within the same tumor. When adding on the tumor microenvironment with cancer-associated cells, vasculature, and the body's immune response (or lack of), the weapons used to tackle this disease must also be diverse and intricate. Developing gene-based therapies against tumors contributes to the diverse lines of attack already established for cancers and can potentially overcome certain obstacles encountered with these strategies, the lack of tumor selectivity with chemotherapies, for example. Given the high mortality and relapse rate associated with pancreatic cancer, novel treatments, including gene therapy, are actively being investigated. Even though no gene therapy for pancreatic cancer is currently on the market, a significant amount of clinical trials are underway, especially in active and recruiting or recently completed phases.
PANCREATIC CANCER
Pancreatic cancer is the 12th leading malignancy worldwide with worldwide incidence and mortality rates predicted to nearly double by 2040. The most common and dire form of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC) accounting for 90% of cancers of the pancreas. Symptoms of PDAC are nonspecific 1,2 and diagnostic is often late since clinical signs generally appear at later stages of disease progression, with merely 15–20% of patients diagnosed when the tumor is operable. In addition, no effective biomarker of PDAC exists, further hindering early diagnostic. 3 PDAC is the digestive cancer with the worst prognosis, having a 5-year overall survival rate of <9%, all stages considered.
The mortality rate being close to the incidence rate can be partly explained by the lack of early diagnosis, an ineffective treatment plan, and the high relapse rate. The best therapeutic option remains surgery, which is only possible in less than a fifth of diagnosed patients, accompanied by high morbidity and relapse rates. Several chemotherapies are available for treatment of PDAC depending on disease progression, results of tumor biopsy, and patients' characteristics (age, well-being, co-morbidities, etc.), including gemcitabine (Gemzar), 5-fluorouracil (5-FU), nab-paclitaxel (Abraxane), oxaliplatin, and irinotecan. To this day, pancreatic cancer still remains incurable. These chemotherapy regimens are accompanied by numerous side effects and risk of innate or acquired resistance. In addition, these treatment protocols are inadequate when faced with the cellular and molecular complexity of pancreatic cancers and their tumor microenvironment (TME). 4 Due to these shortcomings, gene therapy has a promising future where current treatments are lacking.
GENE THERAPY
Gene Therapy Medicinal Products (GTMPs) are innovative therapies developed from tissues, cells, or genes currently being studied as medicine in a wide range of diseases. They are biological medicinal products with an active substance containing/consisting of a recombinant nucleic acid for human use or administration with the aim of regulating, repairing, replacing, adding, or deleting a genetic sequence with a direct relation between transgene sequence or expression and the biological effect induced. 5 GTMPs include both gene therapy and cell-based gene therapy (tumor vaccines and chimeric antigen receptor [CAR]-T cells) products, but exclude vaccines against infectious diseases.
Development of GTMPs implicates both the therapeutic transgene in itself, which can take various genetic forms, and of its transport and delivery, either by administration procedures or in vector design. GTMPs can have both a direct effect on tumor cells through induction of cell lysis (oncogene inhibitors and tumor suppressors) and potentiating antitumoral efficacy of chemotherapies (chemosensitizers), or indirectly through immunostimulating/modulating signaling (vaccines, CAR cells) and oncolytic virotherapy (Supplementary Fig. S1). Due to the complexities and increased ethical and safety considerations involved, regulations and guidelines have been set up by health administrations to ensure the best efficacy and safety during development, testing, and use of these medicines.
There has been debate on whether oncolytic viruses (OVs) can be considered GTMPs. Considering the safety implications when using viruses in therapy, specific control and monitoring of viral gene activities are essential, similar to other gene-based compounds. OVs have therefore been included in the accounts of gene therapy techniques and applications. In addition, recombinant OVs are being conceptualized to express transgenes of tumor suppressor genes or immune stimulation on top of their oncolytic capacities. Currently, 23 approved clinically used human gene therapy or human cell-based gene therapy products exist, six of which are used in oncology (Supplementary Table S1). 6
ONGOING CLINICAL TRIALS
Many sources and databases exist that register clinical trials. The increase in transparency of studies at both national and international levels, adding to the increase in the number of multicountry trials, has allowed for a compilation of global completed and ongoing clinical trials. The clinical trials studied henceforth were obtained from multiple references so as to have an utmost complete index. 7 –11 According to the Wiley “Gene Therapy Clinical Trials Worldwide” online library, nearly 2,600 completed or ongoing trials are of gene therapy. 12 Other than monogenic diseases, oncology research has had a significant impact with the use of gene-based therapies, especially within immunotherapies with the development of CAR-T cells.
The majority of pancreatic cancer-related trials is in phases I to II (Fig. 1). This could be explained by the fact that gene therapy is still an innovative field and many therapeutics are only just arriving at the clinical trial stage or have failed in early phases. Another reason could be that certain aspects of gene-based products are unforeseeable, particularly when transitioning to in vivo administrations. This is especially the case when considering viral or bacterial vectors. To this regard, it would be interesting to know how many research studies do not make it to clinical trials or to the next phase of trials. Nonetheless, a notable increase in the number of trials are entering later phases of clinical trials (Supplementary Table S2). 13
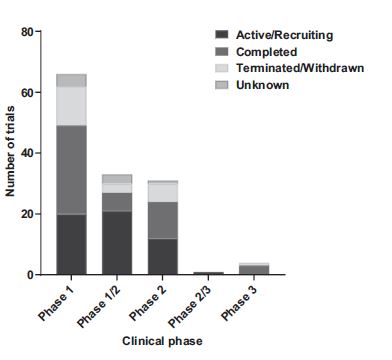
GTMP clinical trials for pancreatic cancer depending on phase and trial status. Source: Clinical
PDAC is an indication for a wide variety of GTMPs currently in clinical trials. After compilation of the current gene therapy trials enrolling patients with all forms of pancreatic cancer, 53 trials are in an active or recruiting status, 51 trials have been completed, and 23 have been prematurely terminated or withdrawn (Fig. 1). Nine trials under the category “unknown” have passed completion date without verification of their status, but with last known status as either active or recruiting. Finally, a study research in the United States (BLESSED, NCT04091295) based on a replication-incompetent retrovector with a cytocidal cyclin G1 construct (DeltaRex-G) is currently in expanded access. Meaning that, although still being under investigation, it is available for patients who are not currently in the clinical trial.
It would be interesting to investigate the reasons behind the prematurely terminated and withdrawn trials and the status of those completed, but have not moved on to the next phase. As such, there are clinical trials (CTs) that have completed a phase III trial, but results have not yet been transmitted. One of these concluded in 2016 in the United States is for a HyperAcute vaccine, which is now in a long-term follow-up study over the next 15 years (NCT03165188). 14 The HyperAcute vaccine consists of intradermally administered irradiated allogeneic pancreatic cancer cells transfected to express the murine gene encoding Alpha-(1,3) galactosyltransferase. Murine alpha-gal epitopes induce a HyperAcute rejection of the cancer cell allograft resulting in the rapid activation of antibody-dependent cell-mediated cytotoxicity (ADCC) toward allograft cells and, in consequence, endogenous pancreatic cancer cells.
The murine alpha-gal gene is transfected in allogeneic pancreatic cancer cells by means of a retroviral vector. Another completed phase III trial is the use of TNFerade with 5-FU and radiation therapy for the treatment of unresectable locally advanced PDAC (NCT00051467). TNFerade consists of a second-generation replication-deficient adenoviral vector transporting tumor necrosis factor-α cDNA as transgene and injected directly into the tumor.
Following a well-tolerated response and dose-dependent increase in the stabilization of treated tumors, phase III results, however, failed to be effective in prolonging survival in patients, despite being safe. 15 It is important to note that certain clinical trials are the succession of those “completed.” For example, when looking at CTs in France, one can see a phase I trial completed and an ongoing phase II trial, both on the study TherGAP 16 ; after positive results from the phase I completed in 2013, a phase II was launched in 2017. To decrypt the potential future drugs for pancreatic cancer and to avoid multiple counts of the same trial in different phases, the following reviews ongoing (active and recruiting) clinical trials and those completed in the last 3 years (since 2018), totaling up to 82 trials (Fig. 2).

Ongoing or recently completed pancreatic cancer GTMP trials depending on gene-therapy technique. Source: Clinical
In these 82 trials, a variety of gene therapy techniques are studied (Fig. 2), the majority being CAR-T/natural killer (NK)/M cells, followed by vaccines and OVs. The latter can be further divided depending on whether utilized for oncolytic effect only or whether also used as a vector to transport antitumoral transgenes. The “suicide gene” category encompasses both chemosensitizing agents and tumor suppressors.
These GTMP compounds are either delivered alone or associated to chemotherapies, targeted therapies, immunotherapies, and other treatments such as Stereotactic Body Radiation Therapy; the benefits of these associations are often an objective of the trials. As a whole, biological (viral, bacterial, and yeast) vectors are the more popular choice (Supplementary Fig. S2), also having the largest variety of vectors. Taken individually, “naked” DNA such as plasmids are the most used vectors, being the simplest gene delivery method and mainly used in the generation of vaccines through genetic incorporation of either tumoral or immunological cells ex vivo before being administered to patients.
VACCINES
Tumor vaccines are engineered cells that present tumor-associated antigens (TAAs) or epitopes to the immune system, prompting an immune response toward the tumor cells presenting these TAAs or oncoproteins. Gene-based tumor vaccines incorporate genes coding for proteins that are overexpressed in cancer, differentiation antigens or tumor-specific epitopes or neoepitopes. 17 These genetically engineered cells can either be PDAC cells or dendritic cells (DCs) responsible for the presentation of these antigens to cytotoxic immune cells directly.
As mentioned, the majority of vaccines is composed of naked or plasmid DNA (Fig. 3). An example of a DNA vaccine is composed of autologous pancreatic cancer cells genetically modified to secrete the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) and irradiated to prevent further cell division (GVAX). 14,18 GM-CSF stimulates the immune system by promoting the activation of DCs and increasing antigen presentation to B and T cells. In addition, GM-CSF enhances ADCC and interleukin-2 (IL-2)-mediated lymphokine-activated killer cell function. Six trials from early phase I to phase I/II and II in the United States are currently underway, studying intradermal administrations of GVAX. Two other phase II trials involving GVAX vaccines are accompanied by a second vaccine composed of live, attenuated Listeria monocytogenes expressing mesothelin (CRS-207) delivered intravenously (intravenous+intradermal).
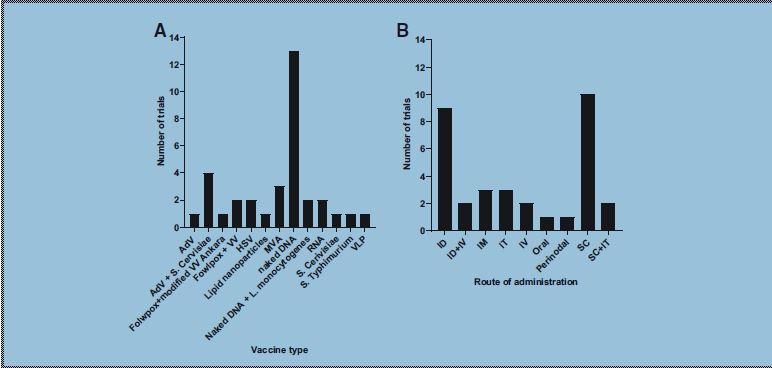
Vaccine trials repartition according to the vector used
Mesothelin is a TAA overproduced by certain tumors, including pancreatic. 19 Latest results show that GVAX is safe and feasible to use as both adjuvant and neoadjuvant therapy 20 with a better overall survival when combining GVAX to CRS-207, 21 despite showing no improvement on patient survival when compared to standard chemotherapy. 19 Furthermore, no improvement of combining GVAX to immune checkpoint inhibitors (ICIs) has yet been shown, 22,23 but other combinations with immunotherapies are underway after signs of immunostimulation. Another version of the GVAX vaccine uses, this time, allogeneic irradiated pancreatic cells transfected with a plasmid encoding GM-CSF (PANC 10.05 pcDNA-1/GM-Neo and PANC 6.03 pcDNA-1/GM-Neo) currently studied in four phase II (one non available [N/A]) studies.
The results of a study showed, here again, a lack of impact of the vaccine associated with ICI on overall survival, although signs of clinical responses and effects on immune cells are worth a further study. 23 The human telomerase reverse transcriptase (hTERT) is also being investigated as a DNA vaccine (hTERT/INO-1400, phase I). 14,24 The enzyme hTERT prolongs cells' lifespan by maintaining telomere length, playing an important role in tumor cell immortality. No clinical result is available yet. A phase I study debuted in 2018 involves the use of B cells and monocytes transfected with a recombinant HER2/neu (receptor tyrosine-protein kinase erbB-2, CD340) gene and loaded with alpha-galactosyl ceramide, a natural killer T (NKT) cell ligand.
The HER2 protein is part of the epidermal growth factor receptor family overexpressed in multiple solid tumors, and so this study is aimed at patients with HER2/neu-positive gastric tumors (BVAC-B). The results show this vaccine to have an acceptable toxicity profile with doses deemed acceptable for a phase II study. 25 A personalized neoantigen DNA vaccine encompassing polyepitope inserts of multiple prioritized neoantigens and mesothelin epitopes, administered by intramuscular injection using an electroporation device (TDS-IM Electrode Array System), is currently in a phase I study. Very recently, a study was presented during American Society of Clinical Oncology Meeting showing positive phase 1 data from mRNAa-based individualized neoantigen-specific immunotherapy in patients with resected PDAC (NCT04161755).
Three phase I tumor vaccine trials use RNA transgenes. The first is a lipid nanoparticle-formulated mRNA-derived vaccine targeting the four most commonly occurring KRAS oncogenic mutations: G12D, G12V, G13D, and G12C (V941). 14,26 After intramuscular vaccination, the mRNA is taken up by antigen-presenting cells (APCs) and the epitopes presented at the surface by the major histocompatibility complex. This induces cytotoxic and memory T lymphocytes directed toward tumor cells harboring these mutations.
Preclinical studies by Moderna show enhanced CD8+ T cell stimulation following mutated KRAS vaccination in mice models. The second is a recently started (2020) trial involving perinodal delivery of a DC vaccine loaded with PDAC lysate and mRNA as adjuvant therapy following completion of standard chemotherapy (DECIST). 27 Finally, RO7198457 is a personalized mRNA vaccine developed on a patient-to-patient basis after sequencing tumor and blood samples and identifying tumor-specific antigens. This vaccine strategy is also in play in phase II colorectal cancer and malignant melanoma trials.
A popular route of administration of these vaccines is by subcutaneous injection (Fig. 3), especially for vaccines involving biological vectors. Four phase I/II U.S. trials involve a cocktail of drugs administered subcutaneously, the majority comprising two gene-based therapies, one of which is an OV (QUILT-3.070, QUILT-3.039, QUILT-3.080, and QUILT-3.060 NANT vaccines). 28 The first is an adenoviral serotype 5 vector with deleted E1 and E2b genes—potentially bypassing an anti-adenovirus immunity—encoding the human carcinoembryonic antigen (CEA), a TAA overexpressed in various tumors (Ad5-CEA(6D), ETBX-011). 29 The second is a heat-killed recombinant of brewer yeast Saccharomyces cerevisiae transfected with genes encoding mutated forms of RAS oncoproteins (GI-4000). 14,30 One of these cocktails of drugs involves only S. cerevisiae transfected with the YE-NEO-001 neoepitope (QUILT-2.025 NANT vaccine) 31 and approved by the FDA for a phase I trial in 2018.
Other subcutaneous or intravenous vaccines involve attenuated viruses, so as to act solely as a vector. One such vaccine is the replication-deficient modified vaccinia Ankara (MVA) virus—an attenuated strain of vaccinia virus. MVA encoding the p53 gene (p53MVA) 32 is studied in mutant p53-overexpressing cancers in a phase I trial. The p53 gene is a tumor suppressor gene with a key role in cell division and cell death, commonly mutated in cancer cells. The trial resulted in a safe and feasible treatment, despite a fatal cardiac toxicity—leading to enhanced cardiac monitoring—and clinical responses in less than a third of patients. 32 Recently, a phase I/II trial involving an even more attenuated strain, MVA-BN, was launched with an intravenous viral-vector vaccine targeting brachyury and HER2 proteins (TAEK-VAC-HerBy).
Another phase I MVA trial (MVA Brachyury-TRICOM) 33 encodes the brachyury gene, a member of the T-box family of transcription factors overexpressed in numerous cancer cell types and correlated with increased epithelial-mesenchymal transition, and cancer resistance and progression. This same vaccine also associates a triad of T cell co-stimulatory molecules (TRICOM): B7.1, ICAM-1, and LFA-3. 34 The trial results in safety of the virus vaccine with activation of brachyury-specific T cells, further studies in combination therapies are planned. 33 The MVA TRICOM vaccine is also investigated in a phase I/II trial combination with a fowlpox virus encoding CEA and mucin-1 (MUC-1) TAAs as booster (CV301). 34,35 The fowlpox virus is a nonreplicative and nononcolytic viral vector.
A bacterial vector used in a phase II trial involves the oral administration of Salmonella typhimurium expressing IL-2 (Saltikva), a cytokine involved in T cell differentiation. Although there were no survival advantages in the phase I study, there was a significant increase in circulating NK and NK/T cells, conveying an immunological response. 36 CMP-001 comprises an oligonucleotide, which, when taken up by antigen-presenting cells, will activate toll-like receptor 9 to stimulate both an innate and adaptive immune response. This oligonucleotide is encompassed in virus-like proteins that trigger the APCs to release the oligonucleotide. 37 CMP-001 is currently in a phase I/II trial in combination with a human IgG1 monoclonal antibody (INCAGN01949) in multiple neoplasms, including metastatic PDAC.
CAR-T/NK/M CELLS
The CAR-T cell therapy (Supplementary Fig. S3) involves obtaining T cells from a patient and genetically engineering them ex vivo to produce antigens against neoepitopes. In an interesting technique, especially in hematological neoplasms, tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta) are approved therapies against B cell acute lymphoblastic leukemia and large B cell lymphoma, respectively. Limitations of CAR-T cells include the associated cost of production and the risks of understimulation or overstimulation of the immune response depending on the neoepitopes expressed.
Other immune cells are also being investigated to express genetically engineered CARs, 38 including gamma-delta (γδ) T cells, NK cellas, NKT cells, and macrophages. 39,40 γδ T cells are a subset of T lymphocytes that also possess an NK receptor (NKG2D) and cytotoxic activities in cancer cells, which are not yet completely elucidated. These cells could be engineered to express CAR-T cell products all the while, maintaining their unique tumor infiltrating and killing capacities. Similar to T lymphocytes, arming cytotoxic NK cells to target specific antigens (CAR-NK) has shown promising results in preclinical and numerous CTs, targeting both hematological and solid cancers. Moreover, bispecific and trispecific killer engagers contain a single variable portion of an antibody linked to one or two variable portions, respectively, from other antibodies of different specificities. 40 These increase cell potency and persistence of CAR-NK cells, while maintaining their specificity.
Although vector information is incomplete on a portion of these trials, the only two vectors used to transduce CAR-T cells are retrovirus and lentivirus vectors (Fig. 4). Four trials in the United States—two phase I active, one phase II/III recruiting, and one phase I completed—involve intrahepatic artery delivery of retroviral (one N/A)-transduced CEA-CAR T. 27 All four trials are aimed to combat liver metastases of pancreatic cancer (PC). Similarly, a phase I trial targeting peritoneal metastases or malignant ascites uses the intraperitoneal route to deliver anti-CEA CAR-T cells.
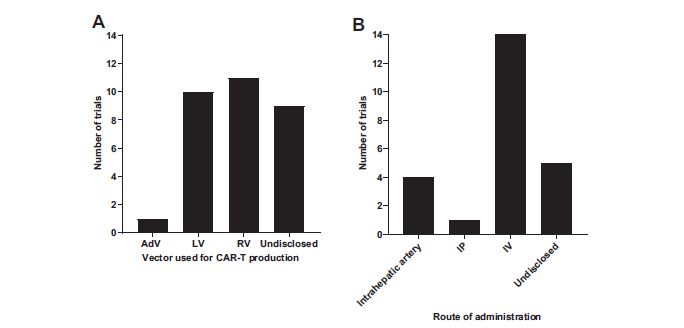
CAR-T/NK cell trial repartition according to the vector used
A phase I study in patients with CEA+ liver metastases showed safe hepatic artery infusion delivery of anti-CEA CAR-T cells as well as encouraging clinical results with increased necrosis and fibrosis in liver metastases. 41 Further, a single-subject phase Ib trial displayed complete metabolic response in the liver, an increased overall survival, and a less immunosuppressive milieu in the TME. 42 Three other trials of anti-CEA CAR-T cells in China in phases I and I/II involve lentiviral transduction and intravenous administration (one N/A).
The rest of the trials involving CAR-T cells are administered by IV and transduced to express a variety of TAAs (Fig. 5). A popular choice is mesothelin (meso-CAR T) 27 investigated in seven trials between early phase I and phase I/II, one of which was terminated before reaching phase II (results not published). Other antigens studied in trials spread between phases I and I/II in the United States or China include: MUC-1, KRASG12V, KRASG12D, Claudin-18 (CLD18), CD133, receptor tyrosine kinase-like orphan receptor 2 (ROR2), epithelial cell adhesion molecule (EpCAM), CD70, and prostate stem cell antigen (PSCA) (Fig. 5). 43 An early phase I trial involves concomitant delivery of T cells, both armed with a murine-derived single chain antibody fragment (scFv), cluster of differentiation antigen 137 co-stimulatory domain (41BB), and cluster of differentiation antigen 3 zeta chain (CD3ζ) signaling domain transduced by lentivirus, targeting either mesothelin or CD19.
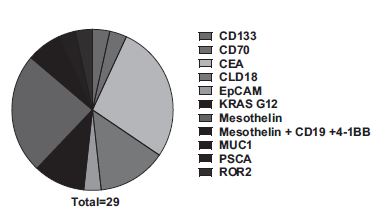
Chimeric antigens using in CAR-T cell-based strategies for PDAC patients' management. Source: Clinical
On top of a mesothelin-targeted immune response (CARTmeso), this trial aims to deplete B cells, involved in the negative regulation of antitumor T cell immunity, with CD19-CAR T cells (CART19). 44 Very recently, CAR-T cells targeting mutant KRAS G12D mediated objective regression of metastatic PDAC for more than 6 months. 45 Importantly, the patient received 600 mg intravenous tocilizumab before infusion, to prevent cytokine release syndrome followed by intravenous cyclophosphamide. Following therapy, nausea and myelosuppression were noted and associated with cyclophosphamide preconditioning therapy, but no toxic effect of the engineered T cell therapy was observed. This warrants prospective clinical trials for patients with PDAC and KRASG12D mutation.
Only two clinical trials involve CAR-NK cells, both in phase I/II in China. The first is a bichimeric antigen receptor-NK cell produced with a lentivirus vector to express the Roundabout homolog 1 (ROBO-1) receptor, overexpressed in pancreatic tumors. 46,47 ROBO-1 is a member of the axon guidance receptor family with a reported role in T cell chemotaxis modulation and tumor angiogenesis. The other involves intravenous injection of peripheral NK cells expressing a chimeric antigen specific for MUC-1 in patients with MUC-1-positive relapsed or regressive tumors. A recent phase I trial has begun involving CAR macrophages for the treatment of tumors overexpressing the HER2 receptor (CT-0508). The autologous macrophages in question are beforehand transduced using an adenoviral vector engineered to contain an anti-HER2 chimeric antigen and administered intravenously.
Preclinical studies show the generation of a proinflammatory environment, an increase in antitumor T cell activity, and recruitment of bystander immunosuppressive M2 macrophages into proinflammatory M1 macrophages. 39 Finally, one can speculate that the recent development of CAR γδ T cells in melanoma 48 may translate soon for the management of patients with PDAC. On one hand, this strategy should be immune to potential deleterious graft versus host disease, cytokine toxicity, antigen escape, and secondary inflammation of normal tissue that may occur with αβ T cells. On the other hand, the role of γδ T cells in PDAC is still controversial, 49 and these cells suffer in general from limited tumor tropism. While promising, more preclinical investigations are needed to fully capture the potential of CAR γδ T cells in PDAC management.
SUICIDE GENES
Suicide gene is the introduction of a transgene into a target cell that, through various mechanisms, will induce cell death. 50 For instance, by metabolizing a previously indolent chemical, such as the cytosine deaminase gene from Escherichia coli that converts 5-fluorocytosine (5-FC) into 5-FU, which is then taken up by cellular enzymes and transformed into cytotoxic antimetabolites. Another is the herpes simplex virus' thymidine kinase (HSV-TK) gene, which converts ganciclovir to ganciclovir monophosphate, further converted to ganciclovir triphosphate by the cancer cells' enzymes.
Ganciclovir triphosphate is found to delay proliferation processes in cancer cells, provoking apoptosis. Suicide gene products can be made from sequences transcribed by particular promoters, like the H19 RNA oncofetal or the hTERT genes, 51 which can be found abnormally expressed in certain tumors. Blocking the promoters by suicide genes leads to the death of cancer cells that overexpress them. Other suicide gene approaches include monoclonal antibodies or toxins such as the Corynebacterium diphtheria toxin-A chain (DTA-H19). 52
As mentioned, a phase II trial in France combines the CYL-02 DNA plasmid and gemcitabine (TherGAP) by intratumoral injection. CYL-02 plasmid encodes the mouse somatostatin receptor subtype 2 (sst2) and the fusion protein of human deoxycytidine kinase (DCK) and uridine monophosphate kinase (UMK) complexed to a synthetic polyethylenimine carrier. Expression of the DCK::UMK fusion protein converts gemcitabine into its toxic metabolite. Expression of the sst2 protein—whose gene expression is often lost in pancreatic and colorectal cancers and negatively regulating multiple processes such as epithelial cell proliferation—is believed to induce both antioncogenic and local antitumor bystander effects. Combining the two allows for a lower dose of gemcitabine to cause tumor cell lysis. Results of phase I showed safety and feasibility with disease stability in patients and identification of biomarkers predictive of response to treatment. 53
A DNA-based tumor suppressor used in a phase II trial in the United States and Taiwan involves a liposome-based complex targeting the transferrin receptor, found to be overexpressed in pancreatic tumors. 54 This liposomal vector carries a plasmid containing wild-type p53 gene (SGT-53) and is injected intravenously. Restoration of wild-type p53 has been linked to antineoplastic activity and enhancing chemotherapeutic activity. 55 This complex was found to target pancreatic tumor cells and sensitize them to gemcitabine in both in vitro and in vivo models. 56 Other tumor suppressor or chemosensitizing-based CTs involve a viral vector such as retrovirus (Fig. 6). One of these is a replication-incompetent retrovector with a cytocidal cyclin G1 construct (DeltaRex-G). Cyclin G1 is one of the target genes of p53 and induces a cell cycle arrest between the G2 and M phases.
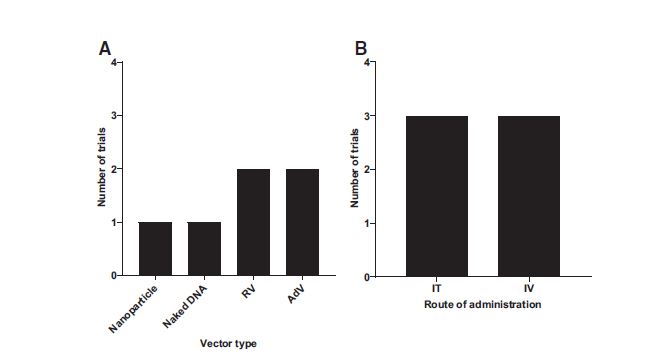
Suicide gene trial repartition according to the vector used
A phase I/II study demonstrated the safety and antitumor activity of Rexin-G in gemcitabine-refractory PDAC with a 10-year survivor patient. 57 The other is a replication-competent retrovirus transporting a modified yeast cytosine deaminase (yCD) gene, currently in a phase II/III trial for glioma (Toca 511). Toca 511/5 treatment was found to be an efficient prodrug to 5-FC with tumor-targeted replication, low systemic biodistribution, and a significant inhibition of tumor growth in murine models, leading to the trial at hand. 58 Another vector used in these clinical trials is a replication-deficient adenovector engineered to bear the HSV-TK gene (aglatimagene besadenovec), currently in phase II. 59 Phase I supported this gene-mediated cytotoxic immunotherapy with standard of care treatment. 60
A phase I investigation with replication-competent adenoviral vector involves double suicide fusion genes: yCD and a mutant form of HSV-1 TK (HSV-1 TKSR39). This OV turned vector also incorporates the adenovirus death protein (ADP) gene with potential oncolytic activity (Ad5-yCD/mutTK(SR39)rep-ADP). 61 The recently completed South Korean trial demonstrated a good tolerance of the treatment in patients, 61 phase II of the trial having started in February 2021 (phase I completed in 2019, not included).
ONCOLYTIC VIRUSES
The idea of treating cancer patients with replicating viruses comes from observed clinical tumor regression after natural virus infections. 62 To be considered an oncolytic agent, a virus should have a tumor-selective infection, replication, and propagation. Properties of cancer cells are ultimately beneficial to viral replication, including sustained proliferation, resistance to apoptosis, and immune evasion. In addition, certain malignant cells readily present or upregulate membrane receptors necessary in viral entry, as well as certain signaling pathways that are appropriated by the OV (e.g., wnt/β-catenin pathway), including downregulation of antiviral (Interferon) and antiproliferative signaling.
In most neoplasms, the TME is characteristically immunosuppressed and therefore a compelling niche for viral replication. Viral infection of the tumor and lysis of tumoral cells cause local inflammation, innate immune activation, and danger signal (damage associated molecular pattern and pathogen associated molecular pattern) liberation, ultimately turning “on” an immune-deficient tumor. Furthermore, the release of tumor neoantigens could potentially act as an in situ tumor vaccine (Supplementary Fig. S4). For more information, excellent reviews have been recently produced on the topic. 63
OVs are sought out for their natural ability to replicate preferentially in tumors, but can be engineered to increase their therapeutic potential, for example, by modifying receptor tropism, by including site-specific targets such as through a tumor-specific promoter, or by inserting certain transgenes, whereas the OV acts as a vector, in addition to its oncolytic maneuver. The first OV used in the United States was talimogene laherparepvec (Imlygic), a modified herpes virus encoding GM-CSF, approved by the FDA in 2015 for melanoma treatment.
In these OV studies, the adenovirus and HSV are administered intratumorally, whereas the reovirus and Vaccinia virus are delivered intravenously, and the H-1PV uses both routes of administration (Fig. 7). Two recently completed phase I trials in Spain studied a PH20 hyaluronidase-expressing adenovirus (VCN-01). 64 Hyaluronic acid is a glycosaminoglycan found in the TME frequently overproduced by tumor cells, contributing to tumor cell growth, metastatic capacities, and resistance to chemotherapeutics. The hyaluronidase expressed by the OV degrades hyaluronic acid, decreasing the interstitial space viscosity and the tumor interstitial fluid pressure, resulting in increased viral spread, and facilitating the access of other drugs. Preclinical results show safety and selectivity, as well as delayed tumor growth. 64 Clinical trial results indicate that VCN-01 has an acceptable safety profile following injection of more than 1013 viral particles. 65 The authors report indirect evidences of tumor replication and immune response.
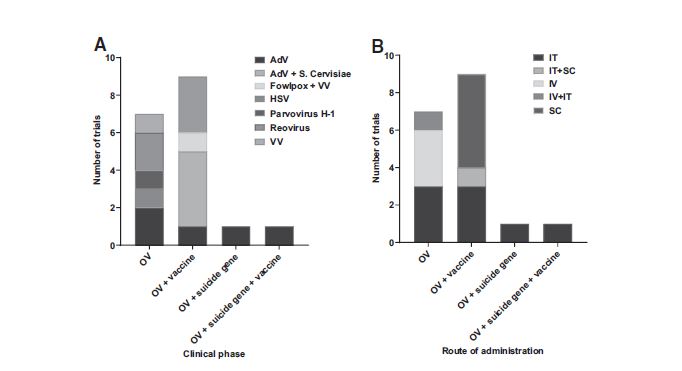
Oncolytic virus trial repartition according to the virus used
Wild-type serotype 3 dearing strain reovirus (pelareorep) 66 was tested as an OV against PC in combination with standard chemotherapy and pembrolizumab in a recently completed phase I trial (REOLYSIN) and is now studied with pembrolizumab alone in an active phase II trial in the United States. 67 This combination is in accordance with previously performed phase II trials of pelareorep in PDAC patients, observing an increase in ICIs CTLA-4 and PD-L1. 68,69 Results of phase I show the treatment cocktail is well tolerated in patients with advanced, pretreated PDAC, encouraging efficacy and viral immunomodulation as a potential dynamic predictive biomarker. 70
A spontaneously attenuated replication-competent strain of HSV-1 (TBI-1401(HF10), canerpaturev) 66 injected orthotopically is currently being investigated in phase I in Japan, combined with standard chemotherapy for patients with grades III and IV PC after failed gemcitabine-based first-line chemotherapy. Genetically modified Vaccinia virus (GL-ONC1, olvimulogene nanivacirepvec) is investigated through intravenous administration in a phase I trial for solid organ tumors, including PDAC. Finally, a phase I/II German study has recently finished exploring parvovirus H1 (ParvOryx), 66 administered both intravenously and intratumorally in liver metastases, as an OV treatment for patients with metastatic inoperable PC. Results of this trial are pending. 71,72
A phase I trial combines the previously described fowlpox virus encoding CEA and MUC-1 (Falimarev/PANVAC-F), and a vaccinia virus encoding the same TAAs, in addition to TRICOM (Inalimarev/PANVAC-V) in combination with GM-CSF. 34,73 A phase III study of this viral vaccine in patients with advanced pancreatic cancer concluded in 2006 without meeting its primary endpoint of increasing progression-free survival compared to standard chemotherapy.
The third adenovirus-centered research is an ongoing phase I/II U.S. and Sweden trial of a modified immunostimulatory adenovirus encoding TMZ-CD40L and 4-1BBL (delolimogene mupadenorepvec, LOAd7). 66 Preclinical studies show reduced tumor size, additional effect when combined with gemcitabine and signs of immunostimulation. 74 Talimogene laherparepvec (OncoVEXGM-CSF/T-vec) approved for melanoma (Imylgic) is now in a phase I study in the United States for PC. Another herpes-derived virus used is the HG52 strain, a less virulent HSV-2, engineered to express GM-CSF (OH2). The genetic modifications brought to this phase I/II therapy allow for better cancer-selective replication of the virus and attenuated pathogenicity.
These genetic mutations and the antitumor activity associated were found to be stable throughout viral passaging in preclinical trials. 75 The final study involving OVs (phase I, United States) involves a replication-competent adenoviral vector and double suicide genes, as seen above. This time, instead of the ADP, this virus also serves as a vector to recombinant human interleukin 12 (Ad5-yCD/mutTKSR39rep-hIL12). IL-12 plays a role in T cell differentiation as well as enhances cytotoxic activity of NK and CD8+ T cells, categorizing this oncolytic adenovirus in both the suicide gene and vaccine categories as well.
DISCUSSION
The majority of GTMP trials for pancreatic cancer involves immune-stimulating techniques such as vaccines and CAR-T/NK cells. Pancreatic cancer is considered a “cold” tumor in that the tumoral environment is lacking and suppressed of immune activity. Vaccines and CAR-T/NK cells could help activate a specific immune response toward tumor cells, thereby turning them “hot.” In addition, many of these trials investigate the combination with other immunomodulatory molecules such as ICIs to further the antitumor immune response. These strategies have functioned for other cancers and although showing promising results in PDAC, no significant improvement in patient survival has yet been observed. 76
Other techniques investigated include gene silencing and suicide gene approaches, which also display potential in PDAC. In addition to suppressing oncogenes or inducing tumor suppressor genes, these strategies have the potential of triggering bystander ramifications through the diffusion of therapeutic effect, that is, proapoptotic signaling on environing tumor cells. Problems with these techniques include poor tumor cell uptake efficiency and degradation and clearance, limits that are being tackled by optimizing delivery vectors. 77
Oncology is a field where treatment strategies come from combining multiple therapies. This is also the case in clinical trials where new treatments are associated with conventional chemotherapies, radiotherapy, immunotherapies, and so on, and by combining different GTMPs. Such is the combination of adenoviral and S. cerevisiae vaccines. Another example is found with OVs being genetically modified to include chemosensitizing, tumor suppressor and immunomodulatory transgenes, further increasing the targets of tumor cell lysis, while maintaining oncoselectivity.
Vectors and administration routes are adapted according to the intended target and transgene approach. This is seen by favoring the intradermal, subcutaneous, and intramuscular route for vaccine administrations (Fig. 3), optimizing access to antigen-presenting cells such as DCs, an essential step in inducing an immune response. 78 Another example is intrahepatic arterial delivery of CAR-T cells targeting liver metastasis and intraperitoneal administration when aiming at metastases and malignant ascites (Fig. 4).
CONCLUSION
Breaking the code of pancreatic tumors rather than using brute force will help in defeating this currently incurable disease. Beyond p53 and KRAS, genetic exploration of cancer cells may reveal novel vulnerabilities as targets, and biomarkers to stratify patients so as to best benefit from available and future treatments. This is of particular importance considering OV that so far present with the strongest antitumoral potential for PDAC management.
Indeed, the replicative life cycle of these viruses engages and hijacks key molecular components, resources, and pathways from the infected cells that will eventually die from infection. Thus, better understanding the dynamic molecular interactions between a virus and its host may help determine the host factors and pathways that are essential to support viral infection and oncolysis. In the era of molecular medicine, this will be instrumental not only to determine what patient may benefit the most from virotherapy, but also which therapeutic combination may improve therapeutic efficacy.
The next problem to tackle will be to better consider the stromal compartment in gene therapy program design. For example, cancer-associated fibroblasts, which usually form a tumor protecting shell, were found to promote OV replication in preclinical models. 79 On the other hand, however, the same cells can trigger genomic stress in cancer cells to block OV efficacy. 80 Finally, certain GTMPs can induce antitumoral immune responses that may sensitize PDAC tumors to immunotherapies such as by remodeling the sparse immune microenvironment. Although no ground-breaking treatment for PDAC has yet emerged, it is tempting to speculate that moving toward an era of precision medicine with GTMPs will definitely give hope to uncovering more effective and safe treatment protocols than those in play today.
Footnotes
ACKNOWLEDGMENTS
This research was conducted in partial fulfillment of Dr. Quillien's doctoral degree requirements (
AUTHORs' CONTRIBUTIONS
Conceptualization: L.Q. and P.C. PubMed investigation: L.Q. Writing—original draft preparation: L.Q. Writing—review and editing: L.B. and P.C. Supervision and funding acquisition: P.C. All authors have read and agreed to the published version of the article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This research was funded by Centre Hospitalier Universitaire (CHU) of Toulouse (doctoral contract).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.