
Abstract
Mucopolysaccharidosis type II (MPSII) is a rare pediatric X-linked lysosomal storage disease, caused by heterogeneous mutations in the iduronate-2-sulfatase (IDS) gene, which result in accumulation of heparan sulfate (HS) and dermatan sulfate within cells. This leads to severe skeletal abnormalities, hepatosplenomegaly, and cognitive deterioration. The progressive nature of the disease is a huge obstacle to achieve full neurological correction. Although current therapies can only treat somatic symptoms, a lentivirus-based hematopoietic stem cell gene therapy (HSCGT) approach has recently achieved improved central nervous system (CNS) neuropathology in the MPSII mouse model following transplant at 2 months of age. In this study, we evaluate neuropathology progression in 2-, 4- and 9-month-old MPSII mice, and using the same HSCGT strategy, we investigated somatic and neurological disease attenuation following treatment at 4 months of age. Our results showed gradual accumulation of HS between 2 and 4 months of age, but full manifestation of microgliosis/astrogliosis as early as 2 months. Late HSCGT fully reversed the somatic symptoms, thus achieving the same degree of peripheral correction as early therapy. However, late treatment resulted in slightly decreased efficacy in the CNS, with poorer brain enzymatic activity, together with reduced normalization of HS oversulfation. Overall, our findings confirm significant lysosomal burden and neuropathology in 2-month-old MPSII mice. Peripheral disease is readily reversible by
INTRODUCTION
Lysosomal storage diseases (LSDs) are a group of about 70 disorders that arise mainly from inherited mutations in the genes encoding the proteins responsible for lysosomal catabolism. One group of LSDs is the mucopolysaccharidosis (MPS) family, which has a frequency of about 1 in 7,500 live-births, 1 whose sufferers carry defects in the genes involved in the degradation of glycosaminoglycans (GAGs). MPS type II, known as Hunter disease, is caused by defects in the X-linked gene iduronate-2-sulfatase (IDS), encoding the hydrolase enzyme iduronate-2-sulfatase (I2S), which is essential for the degradation of both heparan sulfate (HS) and dermatan sulfate (DS). 2
As a result, I2S deficiency leads to progressive accumulation of these unmetabolized molecules in cells and tissues, eventually resulting in a chronic multisystemic disease. Mucopolysaccharidosis type II (MPSII) is generally classified into an attenuated and severe form, although disease symptoms manifest as a continuum between the two extremes. 3 Patients usually experience a wide range of somatic symptoms, including skeletal abnormalities, limited joint motility, airway and pulmonary restriction, cardiomyopathy, and hepatosplenomegaly. In addition to these, the severe disease form is also marked by central nervous system (CNS) involvement, characterized by progressive cognitive deterioration, hyperactivity, aggression, and sleep disturbances, with life expectancy often not exceeding the first decade of life. 4
Currently, enzyme replacement therapy (ERT) is the only approved therapeutic approach for MPSII. This consists of weekly intravenous administrations of idursulfase or idursulfase beta, a formulation containing recombinant I2S enzyme, which is internalized by cells through mannose-6-phosphate (M6P) receptor-mediated endocytosis. 5 ERT is particularly effective in ameliorating the somatic symptoms of MPSII, especially at the musculoskeletal and visceral organs level, increasing the quality of life for many patients. However, this strategy fails to treat the neurological symptoms associated with severe MPSII, due to the inability of the enzyme to cross the blood–brain barrier (BBB). 6
Hematopoietic stem cell therapy (HSCT) is able to provide correction of the brain in related disease MPSI, although it has limited efficacy in other MPS subtypes. 7 This therapy involves the infusion of hematopoietic stem cells (HSC) derived from matched healthy donors into patients who have received full myeloablative conditioning. Following engraftment in the bone marrow, a small proportion of donor-derived myeloid cells seems to be able to migrate, cross the BBB, and engraft as microglia-like cells, eventually secreting enzyme for cross-correction in the brain. 8 –12 A recent study has showed that HSCT can partially ameliorate both motor and speech skills in MPSII patients up to 7 years of age post-transplant 13 ; however, results are poor compared to the efficacy achieved in MPSI patients, presumably due to HSCT failing to achieve the required threshold level of I2S enzyme in the brain.
Although there is evidence suggesting that HSCT may stabilize cognition in MPSII patients if transplant is given at early disease stages,
14
the levels of enzyme provided by a healthy donor remain a limiting factor to achieve complete neurological correction.
15
In this respect, HSC gene therapy (HSCGT) represents a promising alternative for MPSII treatment, as the patient's HSCs are genetically modified to overexpress the missing enzyme before delivery, eventually overcoming the threshold limit of enzyme required in the brain.
16,17
Encouraging results were achieved following lentivirus-mediated HSCGT in mouse models of MPSII, IIIA, and IIIB.
10
–12,18
With regard to MPSII, the ability of somatic IDS enzyme to cross the BBB was enhanced by fusing the codon-optimized IDS to the receptor-binding domain of human apolipoprotein E (ApoE) cloned into a third-generation lentiviral vector containing the myeloid-specific CD11b promoter (
Two-month-old MPSII mice were transplanted with HSC transduced with either
Despite these positive outcomes, the efficacy of any HSCGT approach to reverse neuropathology is highly dependent on the level of existing damage at the time of treatment. As a result, determining the time course of neurological deterioration becomes crucial for understanding the right time of therapeutic intervention for severe MPSII patients. Using the MPSII mouse model, we set out to determine the degree of brain HS storage and neuropathology in 2- (young), 4- (middle aged), and 9-month (old) animals. Having established background levels of neuroinflammation, we investigated the efficacy of HSCGT using
MATERIALS AND METHODS
LV.IDS production
A third-generation lentiviral vector containing codon-optimized human IDS
Mice, HSC isolation, lentiviral transduction, and transplantation
Mice on a C57BL/6 (CD45.2) or Pep3 CD45.1 congenic background were bred as described 11 and housed under 12-h light/12-h dark cycles with food and water provided ad libitum in individually ventilated cages in groups of 3–5 per cage. Only male wild-type (WT) or MPSII animals were used for experiments. All in vivo procedures were performed in accordance with the Animal (Scientific Procedures) Act, 1986 (United Kingdom), under home office license P0C3AEEB0 and approved by the University of Manchester Animal Welfare and Ethical Review Body committee. Control age = matched 2-, 4-, and 9-month animals (n = 4–8 per group) were used for comparison studies.
Before transplantation, bone marrow was isolated from CD45.1-MPSII mice, lineage depleted, and HSCs transduced with a multiplicity of infection of 60, as described previously.
19
Four-month-old male MPSII mice were myeloablated using 125 mg/kg busulfan divided into five daily doses through intraperitoneal injection. On day 6, mice received 3 × 105 lineage-depleted
Behavioral analyses
At 8 months of age, motor coordination and balance were measured using the accelerating rotarod. Three trials were performed for a maximum of 300 s per test, with an average of all three trials recorded. Latency to fall was recorded as a percentage of total trial time, as described. 20 The Y-maze was used to assess spatial working memory. Mice at 8 months of age were placed in a Y-maze consisting of three identical arms for 10 min. Entry into each arm with all four paws was noted and analyzed as previously described. 20
Harvesting animals and tissue analysis
At harvest, mice were anesthetized and transcardially perfused with PBS. One brain hemisphere was fixed in 4% paraformaldehyde for 24 h, transferred to 30% sucrose/2 mM MgCl2/PBS for 48 h before freezing at −80°C. Samples of brain, spleen, liver, heart, lung, and kidney were snap frozen on dry ice. Bone marrow samples were collected by flushing the tibia and femur with 2% FBS/PBS followed by red blood cell lysis and storage of the cell pellet at −80°C. Blood samples were taken into citrate buffer on ice and spun at 200 g to isolate plasma. Red blood cells were lysed, and remaining cells pelleted for storage at −80°C.
For enzyme assay samples of organs were ground in homogenization buffer (0.5 M NaCL, 0.02 M Tris, and 0.1% Triton X-100, pH7) and centrifuged at 5,000 g to isolate the supernatant. Starting material was standardized to 40 μg of total protein for liver, heart, lung, spleen, and bone marrow, and 60 μg for brain using Pierce BCA assay kit (ThermoFisher). IDS enzyme levels were determined as previously described 11 and the assay was standardized to WT values to normalize the variability between each assay run. Genomic DNA was isolated from samples of organs using the GenElute Mammalian Genomic DNA miniprep kit (Sigma) and vector copy number (VCN) determined using quantitative PCR as described. 19
HS composition and amounts were determined in frozen tissues as described. 12 Immunohistochemisty for isolectin-B4 (ISB4), lysosomal associated membrane protein-2 (LAMP2), and glial fibrillary associated protein (GFAP) was performed on free-floating coronal brain sections and imaged as described. 12 Quantification of percentage area stained was performed by thresholding in Image J using the following regions: For cortical regions, images from layers IV/V/VI at 0.14, −0.82, and −1.7 relative to Bregma were quantified and averaged; for striatal quantification, images from 0.14 to −0.82 relative to Bregma were quantified and averaged; hippocampus CA3 regions at −1.7 relative to Bregma was used; amygdala regions from −1.7 relative to Bregma were quantified.
Experimental design
For time-course analysis, independent 2-, 4-, and 9-month-old animals were harvested at the indicated time point (n = 4/group for 2- and 4-month groups, and n = 14 for 9-month animals). For transplant analysis data, HSCGT was performed at 4 months of age in n = 8 animals and directly compared to a previous study performed at 2 months of age. 11 Control animals (n = 8 per group, WT and MPSII) were taken independently during this study and combined with controls (n-6 per group) used in the previous study. 11
Blinding in vivo treatment groups was impossible due to the nature of the transplant procedure performed and the severity in phenotype of the mouse model, with transplants staggered over several months due to recipient/donor availability. However, all analysis where possible was performed in a blinded manner to avoid bias.
Statistical analysis
GraphPad Prism was used for one-way or two-way analysis of variance and Tukey post hoc test for analysis. Significance was taken at p < 0.05%.
RESULTS
Significant central pathology is present by 2 months of age with marked HS storage
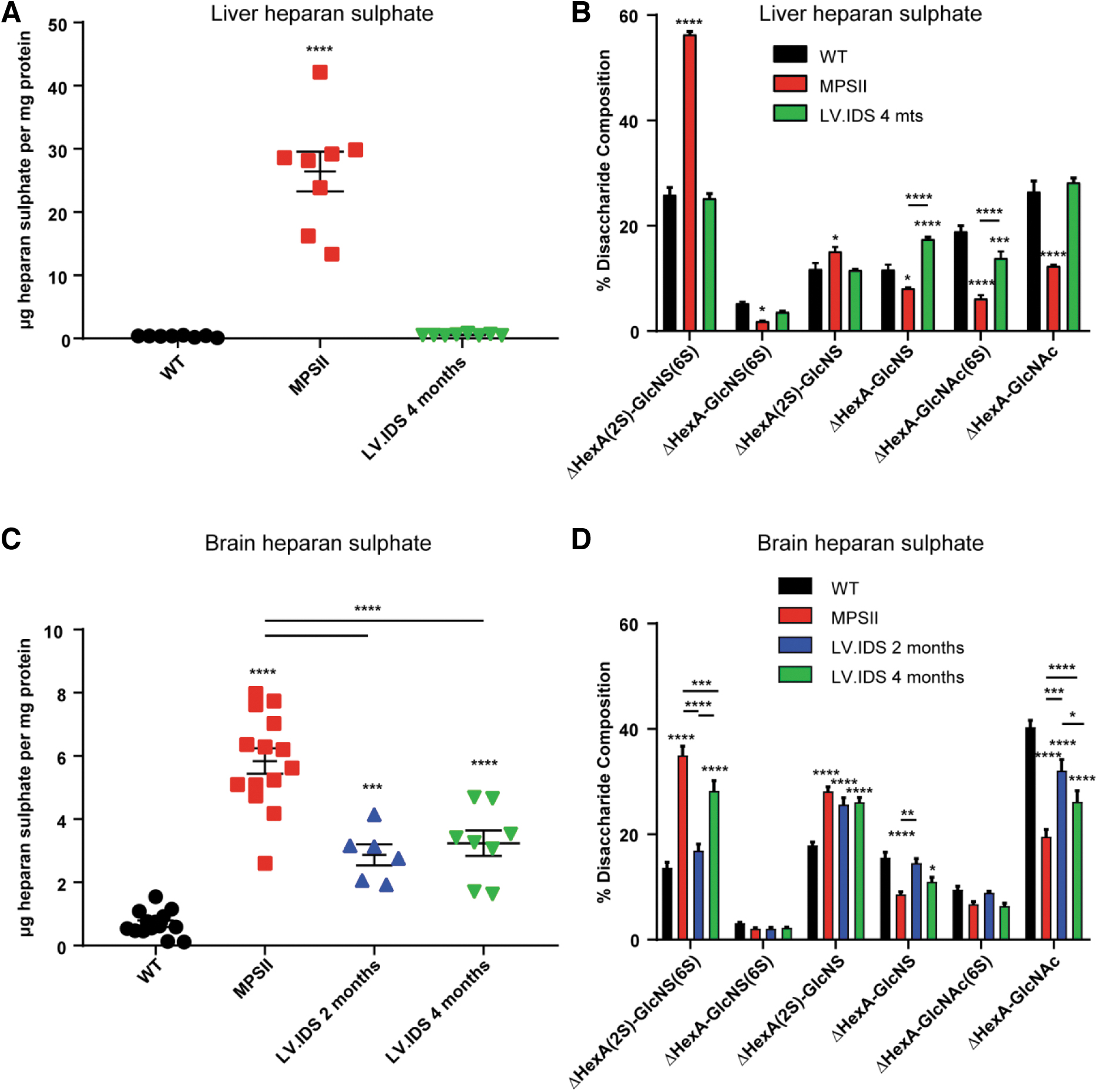
To determine the degree of neuropathology and disease progression in MPSII, brain HS accumulation and pathology were investigated in WT and MPSII mice at 2, 4, and 9 months of age (Fig. 1).

Significant pathological accumulation of HS and neuropathology is evident in 2-month-old MPSII mice with progression seen with age.
Significant HS accumulation was observed over WT by 2 months of age with fivefold more HS in the brain of MPSII mice. Between 2 and 4 months, a significant doubling in HS was observed in MPSII mice, with 10-fold increases in HS over WT by 4 months of age. A plateau in maximum accumulation was recorded between 4 and 9 months of age, with no further increase in HS levels (Fig. 1A). Analysis of the HS composition in MPSII groups revealed that greater than 30% of brain HS was represented by fully sulfated ΔHexA(2S)-GlcNS(6S), compared to 14% in WT mice. While a trend toward further increases in more sulfated disaccharides versus decreases in nonsulfated/low-sulfated disaccharide species was seen between 2- and 9-month MPSII animals, these were not significant (Fig. 1B). This suggests that the pathological alterations in HS patterning occur before 2 months of age in MPSII animals.
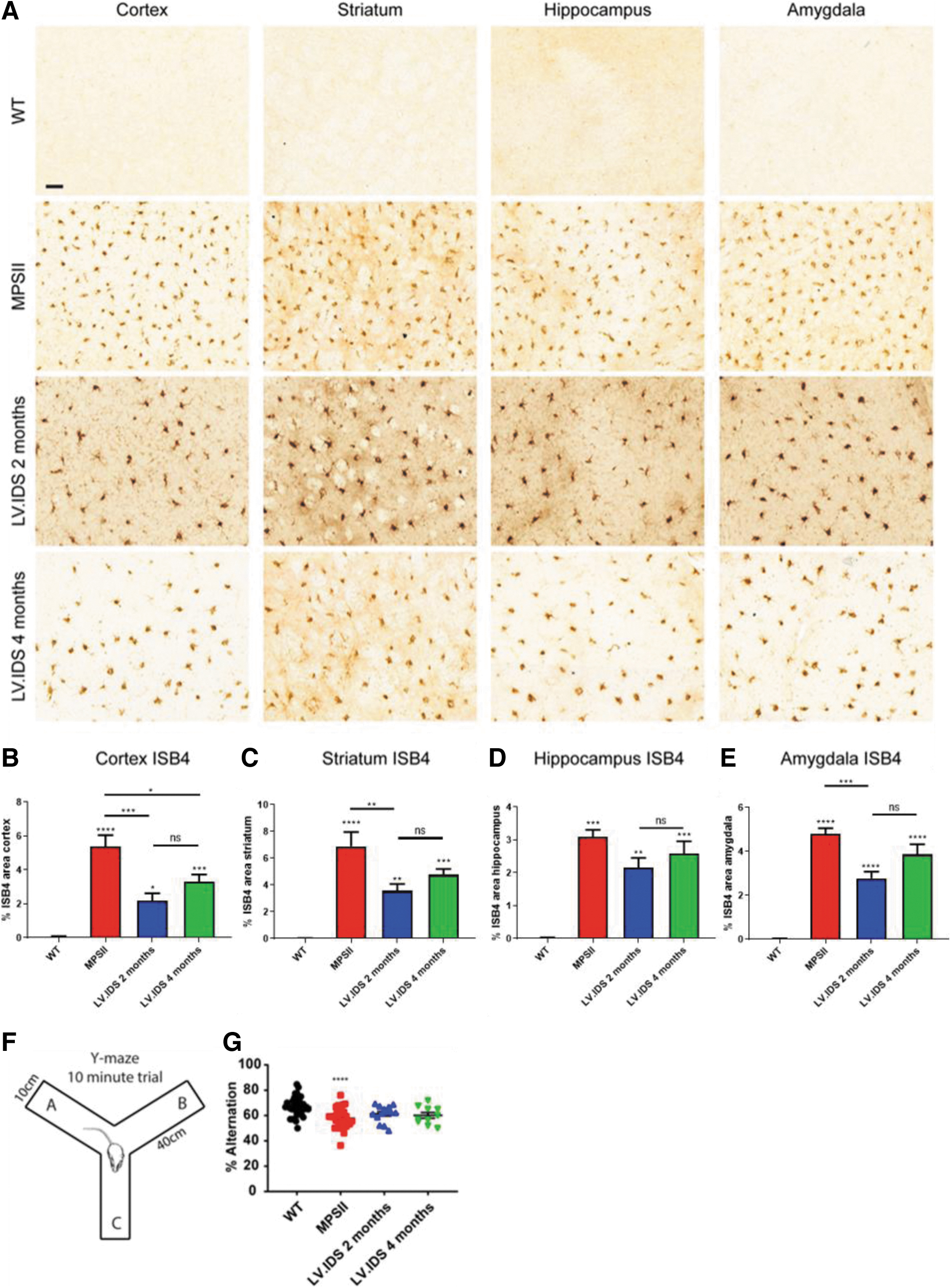
Microglia form an important immune function in the brain, with their activation a hallmark of disease pathology. To assess neuroinflammation, ISB4 staining was performed. In WT animals, a few activated microglial cells were visible in cortex regions at all ages studied (Fig. 1C), with little staining in other regions of the brain (Supplementary Fig. S1). Conversely significant microgliosis was present in MPSII animals in all regions of the brain studied (Fig. 1C and S1).
Quantification indicated that no significant difference was present between 2-, 4- and 9-month animals (Fig. 1E). Co-staining for lysosomal swelling (LAMP2) and astrogliosis (GFAP) and quantification revealed extensive LAMP2 and GFAP staining present at 2 months of age in MPSII animals (Fig. 1D). A trend toward increased astrogliosis was indicated between 2- and 9-month animals, although this was not significant (Fig. 1G). Conversely, lysosomal size was significantly increased by 1.6-fold between 4 and 9 months of age (Fig. 1F).
Successful HSCGT at 4 months in MPSII animals
Given that neuropathology was similar between 2- and 4-month-old mice, an identical
Late LV.IDS HSCGT still provides supraphysiological levels of active IDS in peripheral organs, but displays lower efficiency in the brain
VCN and IDS enzyme activity was measured in MPSII animals treated at 4 months of age throughout the body and compared with previous results achieved following transplant at 2 months of age. Measured VCN was equivalent between transplant groups in many organs apart from the bone marrow, where a significant increase in VCN was seen in the 2-month HSCGT group compared to the 4-month group, and similarly, there was a small increase in VCN in the lungs of 2-month-old treated mice. Conversely, higher VCN values were observed in the brain of 4-month-old treated mice (0.06), when compared to 2-month-old treated animals (0.03) (Fig. 2A).

Late
Supraphysiological enzyme was achieved in all peripheral organs of
Late transplant decreases HS storage both peripherally and centrally, yet sulfation status of the HS chain remains high
To determine the efficiency of IDS enzyme in transplanted animals at eliminating storage material, HS was measured peripherally in liver tissue and centrally in the brain (Fig. 3). In the liver, HSCGT at 4 months resulted in effective normalization of HS levels to WT (Fig. 3A), with coincident restoration of HS disaccharide patterning (Fig. 3B). In the brain, a significant 1.8-fold reduction in HS levels was achieved following late transplant, which was equivalent to the levels of reduction achieved by transplant at 2 months of age (Fig. 3C). However, when the patterning of the HS chain was studied, the effectiveness of the later transplant was reduced compared to early treatment, with significant increases in the most sulfated disaccharide types in 4-month treated animals (Fig. 3D).
Late
Late HSCGT appears to be less effective at reducing neuropathology in MPSII mice
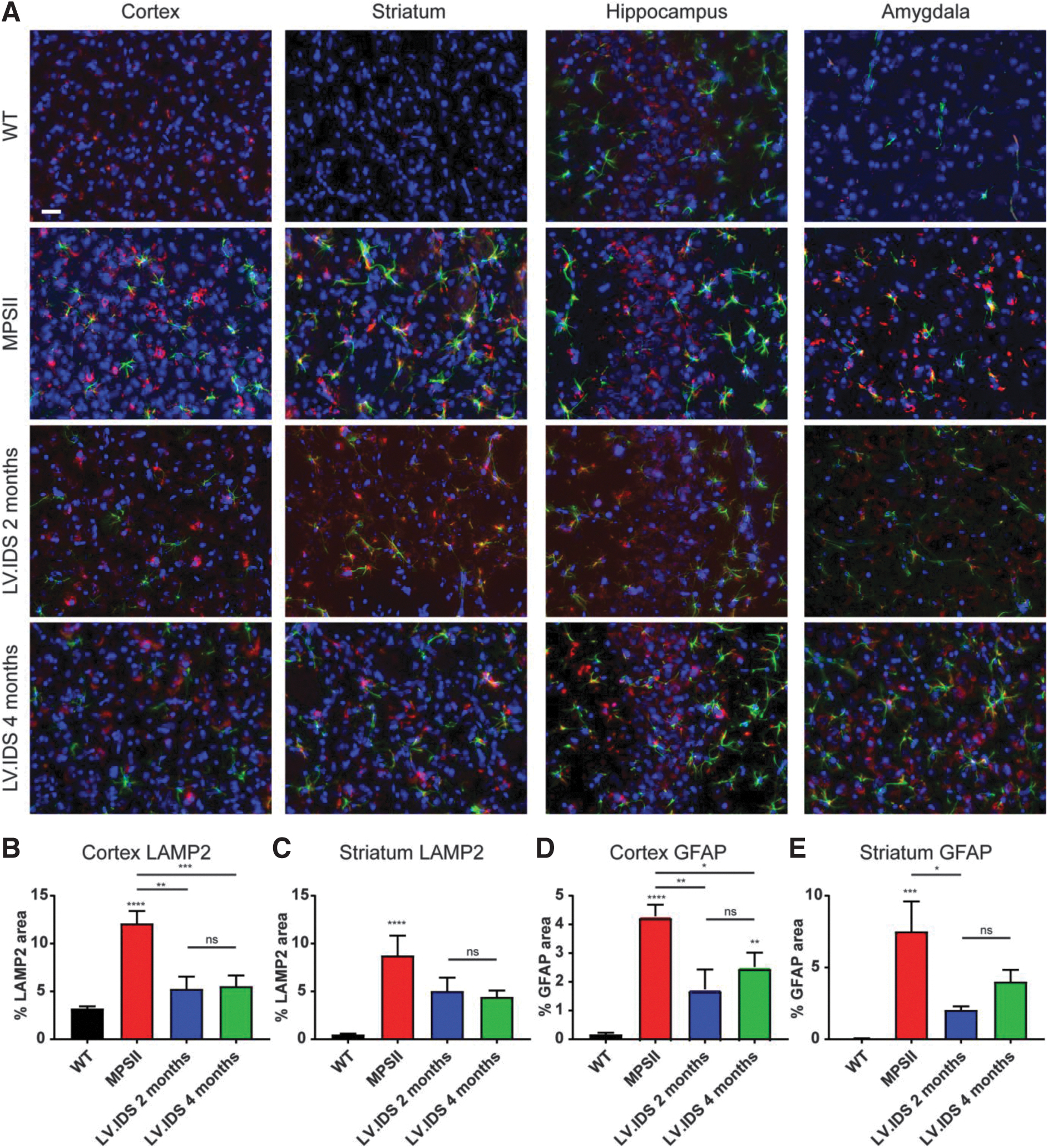
Given the extensive neuropathology in mice at 2 months of age and similarities between animals at 2 and 4 months of age (Fig. 1), staining for GFAP, LAMP2, and ISB4 was performed in the brains of control and 2- or 4-month HSCGT-treated animals at harvest (Figs. 4 and 5). Following HSCGT, staining for LAMP2 and GFAP suggested that HSCGT, either early or late, could effectively reduce lysosomal swelling to near WT levels in the cortex region of the brain (Fig. 4A, B). However, in other regions of the brain, including the striatum, reductions of ∼50% compared to untreated MPSII animals were apparent (Fig. 4C), with levels remaining elevated over WT.
Late
Late
Astrocytosis remained evident in HSCGT-treated animals, with clear astrocytic activation in all regions of the brain studied (Fig. 4A), yet they were noticeably reduced compared to untreated MPSII animals. Reductions in astrocytosis were more pronounced with earlier treatment (Fig. 4D, E); however, there was no significant difference between 2- and 4-month HSCGT-treated animals.
Activated microglial cells were also abundant in 2- and 4-month HSCGT-treated animals. Two-month HSCGT treatment improved outcomes in three out of four brain regions over untreated MPSII controls, while mice treated at 4 months were not significantly different to MPSII-untreated controls in any brain region (Fig. 5). A trend toward increased reversibility of neuropathology was apparent with early treatment, but no significant difference was measured between 2- and 4-month HSCGT-treated groups (Fig. 5B–E).
The extensive neuropathology observed in MPSII patients usually results in severe behavioral problems. When comparing 2- and 4-month HSCGT, neither treatment was able to correct the existing spatial working memory deficits present in MPSII animals (Fig. 5G), although this was expected based on the relatively low enzyme activity data in the brain (Fig. 2G).
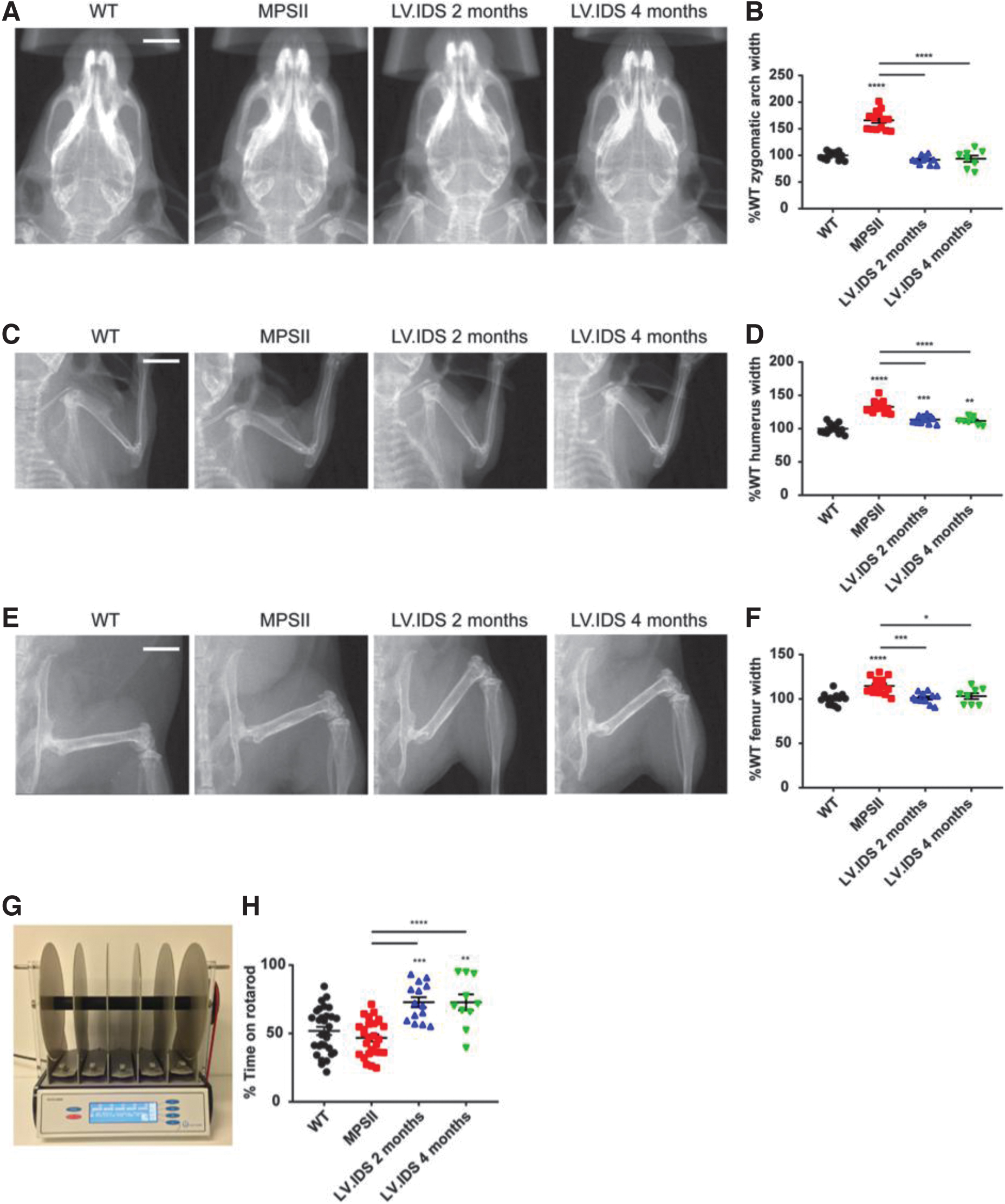
Reversal of bone abnormalities and sensorimotor coordination following LV.IDS-HSCGT at 4 months of age
All MPSII patients exhibit significant skeletal abnormalities and limited joint motility, which can be modeled in MPSII animals. To evaluate the somatic efficacy of HSGCT, skeletal abnormalities were assessed using total body X-rays at 8 months of age (6 or 4 months
Late
A significant decrease in width of the humerus compared to untreated MPSII was also seen in both HSCGT-treated groups (Fig. 6C, D). To investigate the functional outcomes of therapy, sensorimotor coordination was measured using an accelerating rotarod apparatus at 6 or 4 months post-transplant/8 months of age (Fig. 6G). Compared to MPSII animals, both
DISCUSSION
Currently, finding a suitable therapeutic option to address neuropathic MPS diseases is still a challenging field. While ERT has proved to be a suitable option to rescue the somatic symptoms associated with MPSII, 21 HSCGT provides a promising therapy to treat the brain, 16,17 although time of intervention might still pose a threat to its efficacy. In MPSI patients, HSCT is typically given to patients before 2 months of age, with the benefit from HSCT treatment decreasing proportional to transplant age. 22 After this age, reductions in efficacy in the brain, versus the risk of complications from HSCT treatment, preclude this treatment option. Thus, in this study, MPSII disease progression was investigated to elucidate optimal therapeutic windows.
Using the MPSII mouse model, we set out to determine the degree of brain HS storage and neuropathology in 2- (young), 4- (middle aged), and 9-month (old) animals. In this study, we have demonstrated that significant neurodegeneration due to accumulation of highly sulfated HS and presumably other secondary storage materials was evident in MPSII animals by 2 months of age, before delivery of HSCGT. Notably, a significant increase in HS accumulation was observed in the brain between 2 and 9 months of age, which was complemented by progressive enlargement of the lysosomal compartment.
However, changes in sulfation patterning, as well as glial activation, seemed to mostly occur before 2 months of age. This could be explained by the fact that the accumulation of highly sulfated GAGs potentially triggers neuroinflammation through Toll-like receptor 4 (TLR4) binding, a receptor that is present on the surface of microglia, ultimately determining its activation. Once activated, microglia are known to produce several proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, which can further drive astrogliosis and potentially neuronal degeneration. 23,24 While the high level of neuroinflammation observed in the MPSII mice matches the significant neuroinflammatory burden that may be manifesting as an early developmental delay in MPSII children, 25 mouse models of MPSI, IIIA, and IIIB displayed a slower progression of neuropathology, with astrogliosis and microgliosis reaching a peak at 9 months of age. 26
As would be expected based on the progressive nature of the disease, early HSCGT was predicted to be far more effective than later treatment. Our results indicate that, while this is true for CNS pathology, somatic disease can be equally reversed when delivering HSCGT at 4 months of age. In fact, somatic tissues were largely undistinguishable between early and late HSCGT-treated groups, despite differences in the length of treatment of animals at the time of comparison (early animals treated at 2 months are 6 months post-therapy, whereas animals treated later at 4 months are only 4 months post-therapy).
Notably, similar levels of lysosomal enzyme delivery to bone marrow and peripheral tissues were achieved when comparing the two treatments; these levels were also comparable to the ones reported following early HSCGT in other MPS disorders. 9,10 Bone abnormalities are considered one of the trickiest aspects of disease to treat, presumably due to limited enzyme penetration into musculoskeletal tissues due to limited blood flow. 27 Equally, bone damage may be too extensive for repair.
Significantly, late HSCGT provided full recovery of the bone defect apparent at the time of treatment 20 both at a structural (skeletal defects) and functional (sensorimotor coordination) level, with increased benefits when compared to late ERT. 28 ERT is not only costly but also restrictive to patients, requiring a lifelong commitment to weekly intravenous enzyme delivery, often in the clinic. Thus, a one-shot HSCGT therapy, resulting in significantly increased delivery of the missing enzyme to peripheral tissues compared to standard HSCT, may offer significant benefit for older MPSII patients. It is of course possible that we are not measuring subtle enough outcomes to determine if somatic disease is completely corrected by either treatment, and notably, we did not look at cardiac outcomes in this study, a notable manifestation of disease in patients.
With regard to CNS pathology, the percentage of WT brain IDS enzyme achieved trended lower in animals treated at 4 months of age compared to 2 months. This was complemented by poorer correction of HS sulfation patterning and gliosis levels. On the other hand, similar reductions in brain HS and lysosomal swelling, and cerebellar function normalization were observed following both treatments. This is in line with the neurologic recovery observed in both MPSI and MPSII mice when treated at advanced disease stages with AAV9 vector administered intracerebroventricularly and intravenously. 29,30 There are many possible reasons for the decrease in CNS enzyme expression.
First, lower recorded VCN in the bone marrow of the 4-month treated HSCGT group may account for the difference in brain enzyme. In this regard, defects in homing and migration pathways have been observed in a mouse model of MPSI-Hurler (MPSIH), 31 resulting in decreased engraftment following HSCT. This was proved to be caused by highly sulfated HS-mediated sequestration of multiple cytokines and chemokines, such as CXCL12. The latter is in fact a key mediator of HSC homing to the bone marrow following transplant, and it normally relies on HS as a coreceptor to bind its cognate ligand. Notably, in MPSIH, sequestration of CXCL12 seems to be caused by excessive accumulation of 2-O-sulfated HS, for which the chemokine already has a binding preference under normal conditions.
Although this has not been investigated yet, highly sulfated HS may play a similar role in other MPS subtypes, such as MPSII. In this respect, our analysis of disease progression indicates HS progressive accumulation between 2 and 9 months of age, inevitably resulting in an increase of its sulfated forms. As a result, delaying the time of therapeutic intervention might have repercussions on the homing of hematopoietic progenitors to bone marrow, following transplant. In a similar way, increased accumulation of highly sulfated HS might also alter key signaling pathways involved in the migration of engrafted monocytes to the brain, resulting in reduced brain enzyme, and eventually reduced disease correction.
In addition, it should be also noted that as stated above, there are differences in time post-transplant between groups (4 vs. 6 months in late vs. early treated animals, respectively); thus, enzyme levels may rise further with increased time post-transplant as further migration may occur. Finally, increased neuronal damage in the brain may alter the survival of engrafting cells, reducing the level of enzyme expression.
On the one hand, our results imply a clear benefit from HSCGT with both early and late transplant and provide a clear advantage of late HSCGT as a one-time therapy over ERT for somatic organs. On the other hand, our data suggest reduced correction of brain pathology following late HSCGT, hence supporting the view that the earlier the patients are treated, the better the overall therapeutic outcome.
Footnotes
ACKNOWLEDGMENTS
We thank The Bioimaging Core Facility at the University of Manchester for study support.
AUTHORs' CONTRIBUTIONS
O.M. performed the majority of in vitro experiments and wrote the article. A.L. performed the majority of the in vivo work. E.S. helped with in vitro experiments. C.O.L helped with in vivo work. R.J.H. supervised the project, helped with in vivo work, reviewed the data analysis, and made the figures. B.W.B. conceptualized the project, acquired funding, supervised the project, and contributed to writing and reviewed the article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
B.W.B. acknowledges support from Innovate UK (Innovate Manchester Advanced Therapy Centre Hub—iMATCH) and The Isaac Foundation.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.