
Abstract
Adoptive T cell therapy using natural T cell receptor (TCR) redirection is a promising approach to fight solid cancers and viral infections in liver and other organs. However, clinical efficacy of such TCR+-T cells has been limited so far. One reason is that syngeneic preclinical models to evaluate safety and efficacy of TCR+-T cells are missing. We, therefore, developed an efficient viral vector strategy mediating expression of human major histocompatibility complex (MHC)-I in hepatocytes, which allows evaluation of TCR-T cell therapies targeting diseased liver cells. We designed adeno-associated virus (AAV) and adenoviral vectors encoding either the human-mouse chimeric HLA-A*02-like molecule, or fully human HLA-A*02 and human β2 microglobulin (hβ2m). Upon transduction of murine hepatocytes, the HLA-A*02 construct proved superior in terms of expression levels, presentation of endogenously processed peptides and activation of murine TCR+-T cells grafted with HLA-A*02-restricted, hepatitis B virus (HBV)-specific TCRs. In vivo, these T cells elicited effector function, controlled HBV replication, and reduced HBV viral load and antigen expression in livers of those mice that had received AAV-HBV and AAV-HLA-A*02. We then demonstrated the broad utility of this approach by grafting macaque T cells with the HBV-specific TCRs and enabling them to recognize HBV-infected primary macaque hepatocytes expressing HLA-A*02 upon adenoviral transduction. In conclusion, AAV and adenovirus vectors are suitable for delivery of HLA-A*02 and hβ2m into mouse and macaque hepatocytes. When recognizing their cognate antigen in HLA-A*02-transduced mouse livers or on isolated macaque hepatocytes, HLA-A*02-restricted, HBV-specific TCR+-T cells become activated and exert antiviral effector functions. This approach is applicable to any MHC restriction and target disease, paving the way for safety and efficacy studies of human TCR-based therapies in physiologically relevant preclinical animal models.
INTRODUCTION
Despite the availability of an effective vaccine, hepatitis B virus (HBV) infection represents a major health concern with 296 million humans chronically infected worldwide. 1 Chronic HBV infection is associated with a high risk of developing liver cirrhosis and hepatocellular carcinoma (HCC) and more than 800,000 people die from HBV-associated diseases every year. 1 Liver cancer incidence has tripled since 1980, and HCC is the third leading cause of cancer deaths in the world with HBV infection being a main driver. 2 Current antiviral therapies control but rarely cure HBV infection, as the viral reservoir of covalently closed circular DNA (cccDNA) is not targeted. Life-saving therapeutic options for HCC are very limited, 3 highlighting the urgent need for new therapies with curative potential.
HBV-specific immunity, especially by T cells, appears to be essential to achieve HBV cure. 4 HBV-specific T cells, however, are very scarce and partially dysfunctional in the blood of chronic hepatitis B patients. 5 New immunotherapeutic approaches therefore aim to restore the patient's adaptive immune response to recognize and eliminate virus-infected cells and cells that have integrated the HBV genome and serve as cancer precursors or have transformed into malignant cells. 6 Clinical evidence highlights the potential of adoptive T cell therapy as patients with leukemia and persistent HBV infection were able to clear HBV after bone marrow transplantation from donors with an HBV-specific T cell response. 7,8
In adoptive T cell therapy the patient's own T cells are engineered to express an HBV-specific, natural T cell receptor (TCR) 9,10 or chimeric antigen receptor (CAR). 11,12 While CAR T cell therapy of hematologic cancers has already made its way into standard patient care, the treatment of solid cancer with CAR- or TCR-T cell therapy turned out to be much more difficult because of the tumor microenvironment, the anatomic separation of tumors from blood circulation and the low density of the major histocompatibility complex (MHC) on tumor cells. 13,14 In contrast to CARs, which recognize a target antigen that is expressed on the cell surface, TCRs recognize their target peptide in the context of MHC molecules. 14 Thus, transfer of T cells redirected with an MHC-restricted TCR is only possible in preclinical animal models or in patients expressing the respective MHC molecule. The lack of appropriate, clinically relevant animal models is the major drawback for systematic development of TCR-T cell therapies.
First clinical trials using TCR-redirected T cells demonstrated some success for different cancer types when treating patients with TCRs specific for tumor-associated antigens such as NY-ESO-1, MAGE-A3, or MART-1, or viral antigens such as HPV. 15 The limited success, however, certainly requires further systematic improvement.
With respect to a potential TCR-T cell treatment for chronic HBV infection and HBV-associated HCC, we have isolated and characterized a set of 11 HBV-specific, HLA-A*02-restricted TCRs with different avidities suitable for further clinical development. 10 In proof-of-principle experiments, human T cells expressing either of two selected high-avidity TCRs showed striking antiviral activity against HBV-infected cell cultures or in HBV-infected, humanized uPa/SCID mice. 16 However, such xenograft models do not allow large-scale comparisons of different TCRs or different transfer conditions. In addition, only a syngeneic model provides species compatibility of cytokines and cytotoxicity to completely address safety and efficacy. In the case of HBV, usage of syngeneic models has so far precluded a real HBV infection, including cccDNA formation as HBV only infects humans and humanoid primates. 17
In this study, we aimed at providing new syngeneic animal models for investigation of HBV-specific T cells restricted to human MHC-I molecules. We compared transgenic mice expressing an HLA-A*02-like, chimeric molecule composed of human β2m-HLA-A2.1-H-2Db (HHD 18 ) with mice transduced with liver-directed adeno-associated viral (AAV) vectors expressing the human HLA-A*02 or the HHD molecule after transduction with AAV-HBV. 19 Presentation of endogenously processed HBV peptides on hepatocytes of HHD transgenic mice was insufficient to activate HBV-specific T cells. This could be overcome by AAV-mediated expression of HLA-A*02 on mouse hepatocytes and allowed us to determine the in vivo potential of HLA-A*02-restricted HBV-specific TCRs. To show transferability of this system to other species, we expressed HLA-A*02 and the HBV entry receptor human Na+-taurocholate cotransporting peptide (NTCP) 17 on macaque hepatocytes through transduction with an adenovirus (Ad) vector, infected the macaque hepatocytes with HBV, and demonstrated that they were recognized by HBV-specific macaque T cells.
Taken together, we show that a transfer of HLA-A*02 permits studies of human TCRs in physiologically relevant preclinical animal models.
MATERIAL AND METHODS
Animal experiments
Mouse experiments were conducted in accordance with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA) and were approved by the local authorities (Regierung von Oberbayern). Mice were bred and kept in-house in specific pathogen-free animal facilities. Seven- to nine-week-old, male HHDII-HLA-DR1 mice 20 (HHDII = human β2 microglobulin—human α1 and α2 of HLA-A*02—murine α3 of H-2Db, founder line II) and Rag2−/−IL-2Rgc−/− mice were used as recipients. Heterozygous HHD mice were generated by backcrossing to CD45.2+ C57BL/6J mice. CD45.1+ C57BL/6J or homozygous HHD mice were used as T cell donors. Indicated numbers of viral genome equivalents of AAV diluted in phosphate-buffered saline (PBS) to a final volume of 200 μL were injected intravenously. T cells were adjusted for the indicated number of TCR+ T cells in 200 μL PBS and injected intraperitoneally.
For isolation of peripheral blood mononuclear cells (PBMCs) from rhesus macaques (Macaca mulatta), blood was obtained in line with the German Animal Welfare Act. Animals had been cared for by experienced staff at the German Primate Center (DPZ) complying with the European Union guidelines on the use of nonhuman primates for biomedical research and the Weatherall report. The DPZ has the permission to breed and house nonhuman primates under license number 392001/7 granted by the local veterinary office. For splenocyte and hepatocyte isolation, macaques were cared for at the Oregon National Primate Research Center (ONPRC) with the approval of the ONPRC Animal Care and Use Committee using the standards of the NIH Guide for the Care and Use of Laboratory Animals.
Vector generation
Adeno-associated virus
The expression cassettes described below were flanked by inverted terminal repeats derived from AAV serotype 2 and packed with an AAV serotype 8 capsid as described in details in the Supplementary Data. Genes encoding for HHD or beta-globin-intron-HHD originated from the HLA-A2/HHD Zac2.1 plasmid 21 and were cloned into pAAV in between a transthyretin (TTR) promoter and a bGH poly A (bovine growth hormone polyadenylation) sequence. Hβ2m was amplified from human cDNA and fused to HLA-A*02 by a P2A element. Primer sequences are given in the Supplementary Data. For all cloning steps, the Phusion Hot Start Flex 2 × Master Mix (NEB, Frankfurt, Germany) was used.
Final insert and vector sizes were: TTR-HHD bgH-PolyA: 2056 nt, total 4730 nt; TTR-betaglobin-intron-HHD bgH-PolyA: 2585 nt, total 5259 nt; TTR-HLA-A2-P2A-b2m-bgH-PolyA: 2107 nt, total 4811 nt. The AAV-HBV carried a 1.2-fold overlength genome of HBV genotype D (GenBank MN645906.1). As a control, an “AAV-empty” construct containing a GFP gene without a promoter was used. All final constructs were verified by sequencing. Production of AAV is described in the Supplementary Data.
Adenovirus
HBV1.3X- 22 consists of a 1.3-fold overlength genome of HBV (genotype D, GenBank MN645906.1) in which codon 7 of the X open reading frame was mutated to a stop codon by a CG exchange. HBV1.3X-, human NTCP, and HLA-A*02 were cloned into pEntry plasmids and inserted into the E1 region of adenovirus (Ad5ΔE1/E3) backbone plasmid pAd/PL-DEST through the Gateway recombination system following the manufacturer's instructions (Gateway system, Invitrogen, Karlsruhe, Germany). Linearized recombinant adenoviral genomes were transfected into HEK293T cells for initial vector generation. After four passages for amplification, vector stocks were prepared, purified, and quantified (pfu/mL) as described previously. 23
Isolation of primary hepatocytes
Primary mouse hepatocytes (PMH) and primary rhesus macaque hepatocytes (PRH) were isolated as detailed in the Supplementary Data and as described previously. 17
Isolation of leukocytes from blood, spleen and liver
For isolation of mouse PBMC, peripheral blood was collected into Microvette 500 LH-Gel (Sarstedt, Nümbrecht, Germany) and 15 μL of full blood were incubated with 250 μL ACK lysis buffer (8 g NH4Cl, 1 g KHCO3, 37 mg Na2EDTA, add to 1 l H2O, pH 7.2–7.4) for 2′ at room temperature (RT). After centrifugation at 450 g for 5′ at 8°C, cells were resuspended in FACS buffer (PBS with 0.1% bovine serum albumin, Carl Roth, Karlsruhe, Germany). Human and macaque PBMC (Fig. 3) were isolated from citrate blood as detailed in the Supplementary Data.
Spleens of mice or pieces of spleens of macaques were mashed through a 100-μm cell strainer (BD Biosciences, Heidelberg, Germany), washed and erythrocytes lysed using 2 mL ACK lysis buffer for 2′ at RT, and washed with 28 mL of RPMI wash (RPMI, 1% Pen/Strep; Life Technologies, Darmstadt, Germany). All washing steps were carried out at 450 g for 5′ at 8°C using RPMI wash. Livers of mice were first perfused with PBS through the portal vein to eliminate circulating lymphocytes in blood, then mashed through a 100-μm cell strainer and washed. The liver tissue pellet was digested with 4500 U collagenase type 4 (Worthington, Lakewood) in 12 mL RPMI wash for 20′ at 37°C. Leukocytes were purified in an 80%/40% Percoll (GE Healthcare, Solingen, Germany) gradient (1400 g, 20′, RT, without brake) and washed. Cells were maintained in murine T cell medium (RPMI dutch modified, 10% fetal calf serum (FCS), 1% glutamine, 1% Pen/Strep, 1% sodium pyruvate, and 50 μM β-mercaptoethanol; Life Technologies).
Stimulation and retroviral transduction of T cells
Briefly, murine splenocytes were stimulated at the presence of IL-2 and transduced using Platinum-E (Plat-E) cells carrying gag, pol, and env genes of Molony murine leukema virus (MoMuLV) as described previously. 24 Supernatant from untransfected Plat-E cells was used for mock-transduced T cells. Stimulation and transduction of human and also macaque PBMC (Fig. 3) were performed with supernatant from RD114-based packaging cells, 293Vec-RD114™ (BioVec Pharma), as described before. 16 The transduction procedures for murine and human T cells are detailed in the Supplementary Data.
For transduction of macaque splenocytes (Fig. 4), up to 5 × 106 splenocytes were stimulated in 1 mL R15 medium (RPMI 1640, 12.5% FCS, 1% Pen/Strep, 1% L-glutamine; HyClone, Omaha) supplemented with 100 U/mL IL-2, 2 μg/mL Staphylococcal enterotoxin b (Sigma-Aldrich, St. Louis), 0.3 μg/mL anti-monkey CD3 (Mabtech, Cincinnati), 1.5 μg/mL anti-monkey CD28 (Mabtech), and 1.5 μg/mL anti-hCD49d (eBioscience, San Diego) at 37°C overnight. The next day, cells were collected and washed three times with 5 mL RPMI full medium (RPMI 1640, 10% FCS, 1% Pen/Strep, 1% L-glutamine, 1% nonessential amino acids, 1% sodium pyruvate; HyClone). Finally, cells were resuspended in 1 mL R15 medium supplemented with 100 U/mL IL-2 and incubated at 37°C overnight. On the following day cells were counted and transduced a single time in the presence of 180 U/mL IL-2 as described for human PBMC. 16
Flow cytometry
Staining of cells was performed for 30 min in the dark on ice in FACS buffer. Antibodies were purchased from different suppliers: mCD4, mCD8, mCD45.1, mIFN-γ, hIFN-γ (all BD Biosciences), hCD4, Granzyme-B (GrzB) (both Thermo Fisher Scientific, München, Germany), hCD8 (Dako, Waldbronn, Germany), TCR-Vβ 5.1, TCR-Vβ 13.1, TCR-Vβ 14 (Beckman Coulter, München, Germany), and HLA-A2 clone BB7.2 (Santa Cruz Biotechnology, Dallas, TX). Dead cells were excluded from analysis by staining 20′, at 4°C in the dark, with the fixable viability dye eF780 diluted 1:5000 in FACS buffer (eBioscience). For intracellular cytokine staining, T cells were stimulated as indicated below. Cells were permeabilized using Cytofix/Cytoperm (BD Biosciences) before incubation with antibodies following the manufacturer's instruction. Total cell numbers were determined by the addition of CountBright™ Absolute Counting Beads (Thermo Fisher Scientific) before acquisition on a CytoFLEX S (Beckman Coulter) and analysis using FlowJo software (Tree Star, Ashland, OR).
Specific stimulation of murine T cells
For ex vivo peptide stimulation up to 4 × 106 freshly isolated splenocytes or LALs were plated into a U-bottom 96-well plate and stimulated with indicated peptides (C18–27: FLPSDFFPSV, S20–28: FLLTRILTI, S172–180: WLSLLVPFV, GPC3367–375: TIHDSIQYV; JPT, Berlin, Germany) in a final concentration of 1 μg/mL.
For stimulation with peptide-loaded cells, HLA-A*02+ transporter associated with antigen processing (TAP)-deficient T2 cells were adjusted to 1 × 106 cells/200 μL human T cell medium (RPMI, 10% FCS, 1% glutamine, 1% Pen/Strep, 1% sodium pyruvate, 1% nonessential amino acids, 10 mM HEPES, 16.6 μg/mL gentamicin; Life Technologies) and respective peptides were added (1 μM). After incubation at 37°C for 2 h, cells were washed three times and 1 × 105 peptide-loaded T2 cells were used for coincubation with respective effector cells.
For both types of stimulation, after 1 h at 37°C, Brefeldin A was added (1 μg/mL; Sigma-Aldrich, Taufkirchen, Germany). Stimulation was continued for 16 h at 37°C until an ICS was performed on the following day.
Coculture of redirected T cells with HepG2-hNTCP cells or primary hepatocytes
For coculture experiments, 5 × 104 HepG2-hNTCP cells were differentiated for 15 days and infected at MOI 200. HBV stocks were produced as described previously. 25 In short, the cell line HepAD38 was cultivated in hyperflasks, and the HBV-rich supernatant was collected every 3–4 days and pooled. The supernatant was purified using heparin affinity chromatography followed by sucrose gradient ultracentrifugation. HBV-rich fractions were supplemented with 50% FCS, aliquoted and HBV-DNA was extracted and quantified by quantitative PCR (qPCR) on a NeuMoDx 288 system with the NeuMoDx HBV Quant Test Strip. 16
For coculture experiments with primary hepatocytes, 2.5 × 105 or 5 × 105 cells per well were seeded in collagen-coated 12-well or 6-well plates, respectively. The next day medium was supplemented with 1.8% DMSO and adenoviral transductions and HBV infection were performed at the MOI and time points indicated in the figure legend. For all coculture experiments, medium without hydrocortisone was used. The number of effector T cells/well was adjusted according to the transduction efficiency of each receptor to identical numbers of TCR-expressing cells. Mock T cell numbers equaled the total T cell number of the TCR with the lowest transduction rate.
Enzyme-linked immunosorbent assay
Interferon-gamma enzyme-linked immunosorbent assay
To determine the concentration of interferon-gamma (IFN-γ) in supernatant of coculture experiments, commercially available ELISA kits were used (Mouse IFN-γ uncoated enzyme-linked immunosorbent assay [ELISA], Thermo Fisher Scientific; or the Monkey IFN-γ ELISA Development Kit [HRP]; Mabtech). Experiments were performed on Nunc MaxiSorb ELISA 96-well plates (Thermo Fisher Scientific) following the manufacturer's instructions. TMB substrate conversion was determined by measurement of OD450 subtracted by OD560 on an ELISA-Reader infinite F200 (Tecan, Crailsheim, Germany).
HBV antigens
For in vitro experiments, hepatitis B e antigen (HBeAg) in the supernatant of transduced PMH was measured using the Enzygnost HBe Monoclonal Kit on the BEP III platform (Siemens Healthcare Diagnostics, Eschborn, Germany). For supernatants of infected PRH, the HBeAg BioAssay ELISA Kit and the hepatitis B surface antigen (HBsAg) BioAssay ELISA Kit (US Biological LifeSci, Salem) were used according to the manufacturer's instructions.
Serological analyses
Peripheral blood was collected into Microvette 500 LH-Gel and centrifuged to separate serum (10′, 5000 g, RT). Serum HBsAg and HBeAg were quantified on an Architect™ platform (Abbott, Wiesbaden, Germany) using the quantitative HBsAg test (Ref.: 6C36-44; Cutoff: 0.05 infectious units [IU]/mL), and the HBeAg Reagent Kit (Ref.: 6C32-27) with HBeAg Quantitative Calibrators (Ref.: 7P24-01; Cutoff: 0.20 PEIU/mL). Serum alanine aminotransferase (ALT) activity was measured in a 1:4 dilution in PBS using the Reflotron® GPT/ALT test (Roche Diagnostics, Mannheim, Germany).
Intrahepatic AAV- and HBV-DNA copies
DNA was extracted from ∼20 mg of liver tissue using the NucleoSpin Tissue Kit (Macherey-Nagel, Berlin, Germany) following the manufacturer's instructions. qPCR was performed with SyBrGreen on a LightCycler® 480 II (Roche Diagnostics) using the following primers: AAVfw: AACCCGCCATGCTACTTATCTACGT; AAVrev: CACACAGTCTTTGAAGTAGGCC; HBVfw: GCCTCATCTTCTTGTTGGTTC; and HBVrev: GAAAGCCCTACGAACCACTGAAC. Results were normalized to the single copy gene PrP: PrPfw: TGCTGGGAAGTGCCATGAG; PrPrev: CGGTGCATGTTTTCACGATAGTA. The amount of AAV-HLA-A*A2 in livers could not be determined as the AAV primer bound in the AAV-HBV overlap. 19 Quantification of HBV-DNA in macaque hepatocytes was performed as described previously 17 and outlined in the Supplementary Data.
Immunohistochemistry
Liver pieces were fixed in 4% buffered formalin (Santa Cruz Biotechnology) for 24 h and then paraffin embedded. After antigen retrieval at 100°C for 30 min with EDTA, liver sections (2 μm) were stained with rabbit anti-HBcAg (#RB-1413-A; 1:50 dilution; Thermo Scientific) as primary antibody and appropriate horseradish peroxide-coupled secondary antibodies. Immunohistochemistry was performed using a Leica Bond MAX system (Leica Biosystems, Nussloch, Germany). For analysis, tissue slides were scanned using an Aperio AT2 slide scanner (Leica Biosystems).
Statistical analysis
Data are reported as mean values ± standard deviation or standard error of the mean. Experiments performed in technical replicates per mouse (Fig. 2) were analyzed with the nonparametric Mann–Whitney test. Data of macaque and mouse experiments were tested for normal distribution by Shapiro–Wilk test and analyzed by unpaired t-test. For calculation Prism 8.0 (GraphPad Software, Inc., La Jolla, CA) was used. A p-value <0.05 was considered statistically significant.
RESULTS
HHD-transgenic mice do not support activation of murine T cells in the liver
To identify an animal model that allows studying functionality of human TCR-redirected T cells in the liver, we first employed HHD-transgenic (HHD-tg) mice. HHD is an artificial chimeric molecule composed of hβ2m, human α1, and α2-domains of HLA-A*02, and the murine α3-domain of H-2Db. 18 HHD-tg mice were transduced with AAV-HBV and after one month, when persistent HBV replication had been established, received one or two consecutive adoptive T cell transfers (Fig. 1A).
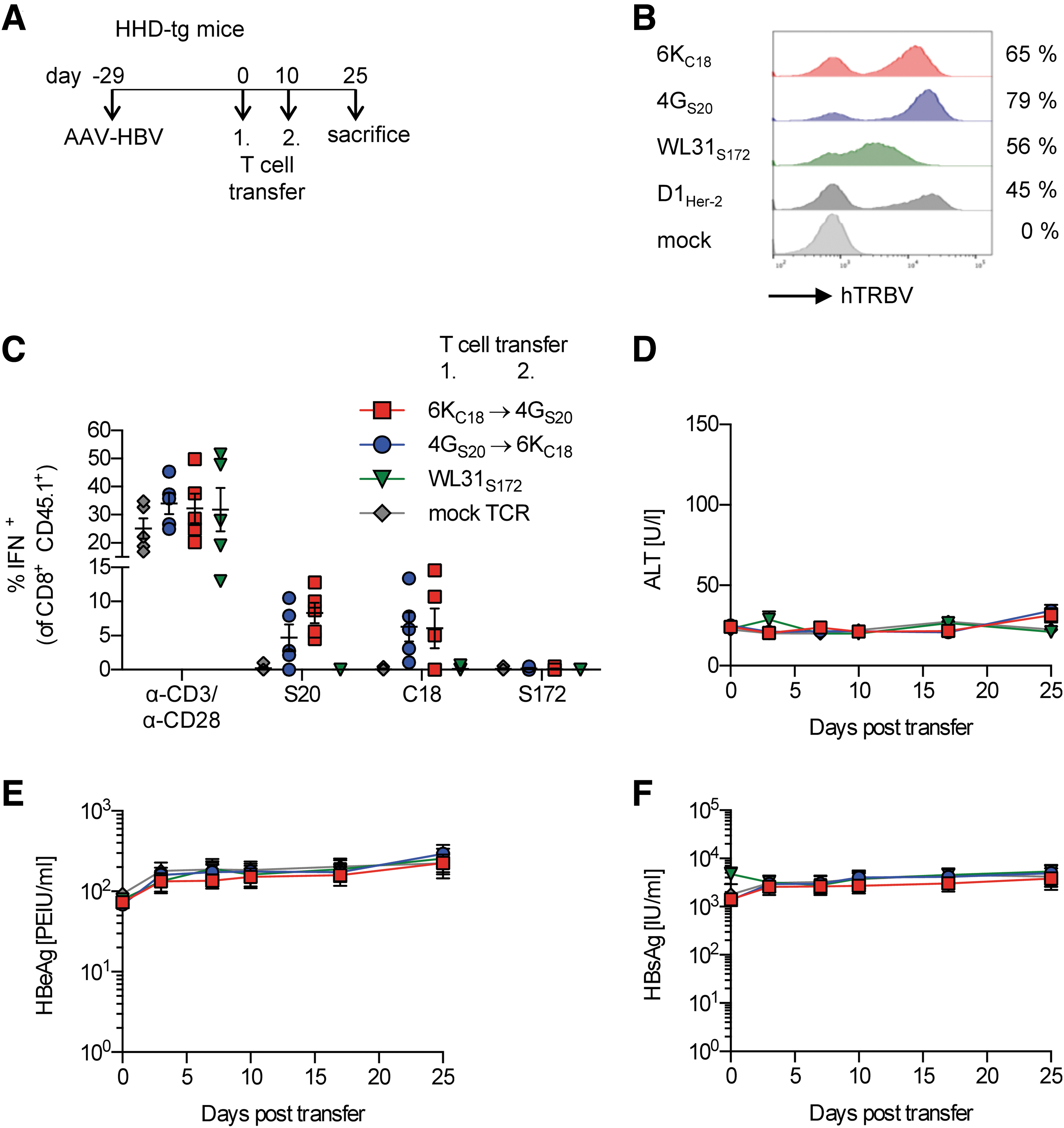
Transfer of HLA-A*02-restricted, HBV-specific TCR+-T cells into AAV-HBV-transduced HHD-tg mice.
HLA-A2-restricted, HBV-specific TCRs had been isolated previously by targeted peptide stimulation of CD8+ T cells from donors with resolved HBV infection and had been cloned into the myeloproliferative sarcoma virus-based retroviral vector MP71 (Supplementary Fig. S1). 10 Murine HBV-specific T cells or unspecific control T cells were generated by retroviral transduction with MoMuLV pseudotyped particles, resulting in expression of the core18–27 (C18)-specific TCR 6KC18, the S20–28 (S20)-specific TCR 4GS20, the S172–180 (S172)-specific TCR WL31S172, 10,16 or the Her-2-specific control TCR D1Her-2 26 (Fig. 1B). The congenic marker CD45.1 expressed in donor mice allowed differentiation of transferred cells from endogenous CD45.2+ cells of the recipient mice.
Transferred C18- and S20-specific T cells were present and functional in the liver 25 days post-transfer, as evidenced by their ex vivo activation by cognate peptides (Fig. 1C). However, we did not observe any effect in vivo (Fig. 1D–F). Serum alanine transaminase (ALT) levels, as a marker of hepatocyte cytotoxicity, (Fig. 1D and Supplementary Fig. S2A) as well as serum HBeAg and HBsAg (Fig. 1E, F; and Supplementary Fig. S2B, C) as markers for viral persistence remained unchanged despite the T cell transfer. These data suggested that insufficient antigen presentation by hepatocytes of HHD mice rather than a lack of T cell functionality was responsible for the missing antiviral activity of the HBV-specific T cells in vivo.
AAV-delivery mediates expression of human MHC-I molecules on murine hepatocytes
To address the hypothesis of insufficient antigen presentation by HBV-replicating hepatocytes, we aimed at expressing sufficient human MHC-I on hepatocytes. Therefore, we generated three AAV vectors encoding for different versions of HHD or HLA-A*02 under control of the liver-specific TTR promoter: (1) HHD (pAAV-HHD), (2) HHD downstream of a β-globin intron to enhance expression (pAAV-βGI-HHD), and (3) HLA-A*02 coexpressed with hβ2m (pAAV-HLA-A*02) (Fig. 2A). HHD mice were transduced with the respective AAVs and primary mouse hepatocytes (PMH) were isolated 17 days later (Fig. 2B).

Activation of HLA-A*02-restricted T cells by different HLA-A*02/HHD variants expressed in primary murine hepatocytes.
Expression of HHD or HLA-A*02 was analyzed by flow cytometry using an antibody that we verified to detect both HLA-A*02 and the HHD molecule on transfected cells and splenocytes of HHD-tg mice (Supplementary Fig. S3A, B). PMHs from HHD-tg mice as well as mice additionally transduced with AAV-HHD or AAV-βGI-HHD expressed the HHD molecule at a low level (Fig. 2C). In contrast, strong expression of HLA-A*02 could be detected on up to 99% of PMH of mice transduced with AAV-HLA-A*02 (Fig. 2C).
Next, freshly isolated PMH were cocultured with HBV-specific TCR+ or mock T cells and activation of T cells was determined by analyzing IFN-γ secretion into the supernatant at day 2 after start of coculture. When loaded with excess amounts of cognate peptide, all PMH cultures expressing HHD or HLA-A*02 strongly and specifically activated TCR+ 4GS20 or 6KC18 T cells, respectively (Supplementary Fig. S3C).
Next, PMH were transduced with Ad-HBV as murine cells cannot be infected with HBV (Fig. 2B, D). Presentation of intracellularly processed peptides clearly activated T cells when PMH were derived from AAV-transduced mice. However, PMH derived from both AAV-HHD- and AAV-HLA-A*02-transduced mice released threefold or over tenfold higher levels of IFN-γ compared to PMH derived from AAV-bGI-HHD mice (Fig. 2D).
To determine the antiviral effect of TCR 4GS20- and 6KC18-redirected T cells, HBeAg secreted by Ad-HBV-transduced hepatocytes was analyzed (Fig. 2E). In cocultures with PMH derived from untransduced HHD mice or animals transduced with AAV-βGI-HHD, reduction of HBeAg levels were about 20% and 40% for TCR 4GS20- and 6KC18-engrafted T cells, respectively. In accordance with the more pronounced IFN-γ secretion, antiviral T cell activity was most pronounced when PMH stemmed from mice that had been transduced with AAV-HLA-A*02 resulting in a 70–75% decline of HBeAg (Fig. 2E). In summary, transduction with AAV-HHD and in particular AAV-HLA-A*02, and subsequent Ad-HBV transduction generated hepatocyte target cells that activated antiviral effector functions of murine HBV-specific, HLA-A*02-restricted TCR+-T cells.
Adenovirus-mediated delivery of HLA-A*02 into HBV-infected macaque hepatocytes allows recognition by HBV-specific, TCR-redirected T cells
Vector-mediated delivery of human MHC molecules for preclinical assessment of TCR-based T cell therapies could also be applied to animals that support establishment of a physiological HBV infection with cccDNA formation, such as rhesus macaques and pigs. 17,27 Using our TCRs 4GS20 and 6KC18, we generated HLA-A*02-restricted, HBV-specific T cells from macaque primary cells. The retroviral MP71 vectors encoding the TCRs and pseudotyped with the RD114 envelope were able to transduce both CD8+ and CD4+ macaque T cells at an efficiency of up to 90%, even exceeding the transduction efficiency for human T cells (Fig. 3A). When cocultured with HBV-infected HepG2-NTCP cells (HLA-A*02+), human and macaque TCR+-T cells were specifically activated (Fig. 3B, C). Around 4% of CD4+ and 10–12% of CD8+ human TCR+-T cells (Fig. 3B) in comparison to 2% and 20–28% of TCR+ macaque T cells (Fig. 3C), respectively, produced both IFN-γ and granzyme B indicating full effector function.
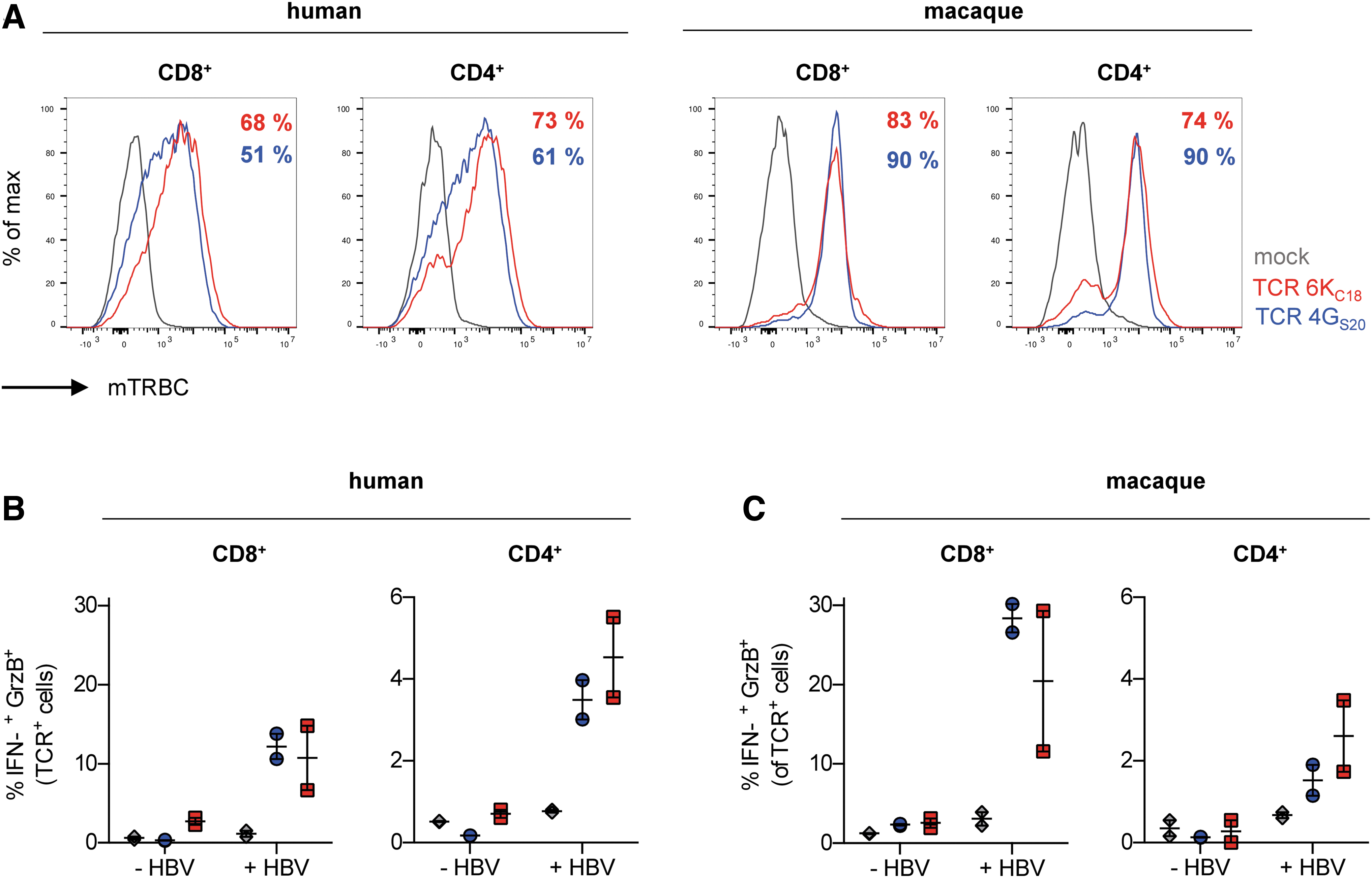
Activation of TCR+ primary macaque T cells by HBV-infected human hepatoma cells.
Having demonstrated HLA-A*02-restricted activation of macaque T cells transduced with a human TCR, we next investigated the antiviral efficacy of HBV-specific macaque T cells toward HBV-infected hepatocytes. To this end, primary macaque hepatocytes were isolated and cotransduced with Ad-NTCP and Ad-HLA-A*02, infected with HBV, and finally cocultured with macaque T cells transduced to express TCR 4GS20 or 6KC18, or with mock T cells (Fig. 4A). Using a relatively high MOI of 200 IU/cell of Ad-HLA-A*02, finally allowed reliable expression of HLA-A*02 on PRH (Fig. 4B). After 2 days of coculture, core-specific 6KC18- but not S-specific 4GS20-redirected T cells had secreted ∼400 pg/mL of IFN-γ when cocultured with HLA-A*02+, HBV-infected macaque hepatocytes (Fig. 4C).

Activation and antiviral efficacy of TCR+ macaque T cells on HBV-infected primary macaque hepatocytes.
Nevertheless, both, 6KC18- and 4GS20-grafted T cells were able to reduce HBeAg and HBsAg levels by day 6 of coincubation by 25% compared with the start of coculture, or by 40–50% compared with mock T cells, as viral replication still increased in controls until day 6 (Fig. 4D, E). The HBV-specific TCR+-T cells also reduced intracellular HBV-DNA by twofold (Fig. 4F) and cccDNA by more than fourfold compared with mock treatment (Fig. 4G). Again, the antiviral effect was more pronounced when TCR 6KC18 was expressed on T cells compared with TCR 4GS20 (Figs. 2C, D and 4C–G). The fact that 4Gs20 (damit es einheitlich ist) T cells reduced viral markers in the absence of detectable IFN-γ secretion hinted at T cell cytotoxicity being more relevant in this coculture set up. In conclusion, HBV-infected PRH-expressing HLA-A*02 were generated in vitro and allowed studying the antiviral activity of HBV-specific TCR+ macaque T cells restricted by HLA-A*02.
AAV-HBV and AAV-HLA-A*02 cotransduced mice can be used to study the antiviral activity of TCR-redirected murine T cells
As AAV- or adenovirus-vector-mediated expression of HLA-A*02 led to sufficient presentation of HBV-derived peptides on murine and macaque hepatocytes to activate HBV-specific, HLA-A*02-restricted TCR+-T cells in vitro, we asked whether this concept could also be applied for preclinical testing of TCR-T cell therapies in vivo. Therefore, AAV-HBV and either AAV-HLA-A*02 or an “AAV-empty” vector were injected in fivefold excess into immunocompetent HHD mice. Four weeks later either a 1:1:1 mixture of CD8+ TCR+-T cells expressing TCR 4GS20, 6KC18, and WL31S172 T cells or mock T cells were adoptively transferred (Supplementary Fig. S4A). ALT levels transiently increased in two of four mice on day 7 after T cell transfer when mice were cotransduced with AAV-HLA-A*02 and AAV-HBV (Supplementary Fig. S4B). Except for the animal with the highest ALT elevation, in which serum HBeAg and HBsAg levels decreased by ∼40%, viral antigens stayed stable in all groups for 1 month after adoptive transfer (Supplementary Fig. S4C, D).
Since we could detect neither significant expansion nor antiviral activity of transferred T cells, we hypothesized that the human TCR variable domains could be recognized as foreign antigen by the recipient's immune system. Indeed, we found that mice developed an antibody response against all TCRs investigated (Supplementary Fig. S4E–G) as well as a cytotoxic CD8 T cell response against TCR 6KC18 or TCR WL31S172 (Supplementary Fig. S4H).
Therefore, we next transduced immunodeficient Rag2−/−IL2rg−/− mice with AAV-HLA-A*02 and/or AAV-HBV. Eighteen days later, when a persistent HBV replication had been established, we treated the mice with either a mixture of TCR+-T cells containing TCR 4GS20, 6KC18, and WL31S172 in a 1:1:1 ratio according to multimer staining (Supplementary Fig. S5A, B), or with mock T cells (Fig. 5A). Interestingly, numbers of transferred TCR+-T cells in blood dropped 2 weeks post-transfer solely in the group that was cotransduced with AAV-HLA-A*02 and AAV-HBV (Fig. 5B and Supplementary Fig. S5C), which could indicate homing of HBV-specific T cells to the liver.

Transfer of TCR+-T cells into mice cotransduced with AAV-HBV and AAV-HLA-A*02.
Very late (60 days) after transfer, liver-associated lymphocytes were isolated and stimulated with HBV-peptides ex vivo. One to five percent of CD8+-T cells isolated from livers of mice cotransduced with AAV-HBV and AAV-HLA-A*02 produced IFN-γ upon peptide stimulation (Fig. 5C). The amount of HBV-reactive, CD45.1+ T cells retrieved from the liver was around four times higher, when only one AAV had been injected and hence HLA-A*02-HBV-peptide complexes could not form (Fig. 5C). Conversely, the expression intensity of PD-1 on TCR+-T cells was around four times lower in these mice (Fig. 5D).
Coinciding with a transient but profound rise in ALT levels up to 1300 U/L on day 7 post TCR-T cell transfer into AAV-HBV mice (Fig. 5E and Supplementary Fig. S5D) that were cotransduced with AAV-HLA-A*02, HBeAg, and HBsAg levels significantly declined by nearly 3 logs and remained low until the end of the experiment (Fig. 5F, G; and Supplementary Fig. S5E, F). In addition, TCR+-T cells substantially reduced AAV- and HBV-DNA in cotransduced animals about 10-fold compared with the control groups suggesting elimination of HBV-positive hepatocytes (Fig. 5H). The persistence of low-level HBV DNA and antigens indicated that a low percentage of hepatocytes might not have been transduced with HLA-A*02 and thus escaped T cell recognition (Fig. 5F–H). However, hepatocytes in the liver of AAV-HBV-transduced mice that were cotransduced with AAV-HLA-A*02 could no longer be stained for HBcAg, whereas more than 50% of hepatocytes of the mice in control groups stained positive (Fig. 5I).
Taken together, hepatocytes could be cotransduced in vivo with HLA-A*02 or HBV when two vectors were coinjected i.v. This allowed proper presentation of cognate antigen and activated the effector function of adoptively transferred, HBV-specific T cells restricted by HLA-A*02 that elicited a profound antiviral effect. This renders the HLA-A*02 and hβ2m coexpressing AAV- and adenoviral vectors as interesting tools for further preclinical assessment of other TCR-T cell therapies.
DISCUSSION
Preclinical animal models that can be used to predict safety and efficacy of TCR-T cell therapy are urgently needed. In this study, we show that vector-mediated expression of the human HLA-A*02 molecule on HBV-replicating murine and macaque hepatocytes allows recognition by syngeneic HLA-A*02-restricted, HBV-specific T cells and testing of their effector function. When native HLA-A*02 and hβ2m were coexpressed on murine hepatocytes, presentation of endogenously processed peptides was superior compared with the chimeric HHD molecule. 18 HLA-A*02-restricted, HBV-specific T cells secreted cytokines and led to a decrease of viral markers upon activation by HBV-infected PRH transduced with an adenoviral vector encoding HLA-A*02, hβ2m, and human NTCP. Finally, the strong antiviral activity of adoptively transferred HBV-specific T cells in mice expressing HLA-A*02 and HBV exemplified the in vivo applicability of this approach.
HHD mice, which express a chimeric MHC-I molecule consisting of a monochain of hβ2m, human α1 and α2 domains of HLA-A*02, and a murine α3 domain, 18 have been widely used for HBV vaccination studies. 28 They are considered to elicit superior HLA-A*02-restricted cytotoxic T cell responses compared with other HLA-A*02 transgenic mice, because they do not express murine MHC-I genes that would compete for peptide presentation, and because of murine T cells being able to bind to the α3 of the HHD molecule with their CD8 coreceptor (reviewed in Pascolo 28 ). However, in our study HBV-replicating hepatocytes of such HHD-tg mice barely activated redirected HLA-A*02-restricted, HBV-specific T cells in vitro (E:T 1:2) and not at all in vivo (E:T 1:25). The overall low antiviral activity of T cells in the HHDtg mouse model might be attributed to insufficient engraftment and in large part to a relatively low expression level of HHD on hepatocytes, which has also been observed for other HLA-A2.1 transgenic mice. 29
HHD expression has been demonstrated to be more stable when expressed from an AAV9-based vector than from a transgene. 21 Therefore, using an AAV8-based vector with liver tropism for delivery of the chimeric HHD molecule could increase antiviral T cell activity.
The observation that excessive amounts of exogenously added HBV peptides could activate HBV-specific T cells isolated from HBV-replicating HHD-tg mice, argued either for insufficient antigen processing or for lack of transport to or presentation on MHC on the hepatocyte surface. Strong T cell responses were detected when we used AAV-HLA-A*02 to transduce mouse livers allowing for physiological presentation of endogenously processed peptides after transduction with AAV-HBV. Hence, the overall amount of peptide-presenting HHD molecules on hepatocytes seemed to be too low to create a profound T cell activation in vitro and in vivo. In line with our data, adoptive transfer of high amounts of HLA-A*02-restricted CMV-specific T cells into mCMV-infected immune incompetent HHD mice hardly reduced viral titers in the liver. 30
It has been proposed that the murine TAP system has different peptide binding preferences compared with the human TAP system and would thereby generate different immunogenic peptides. 31 This, however, seems unlikely in our case because the murine TAP system provided HBV peptides to the HLA-A*02 molecules. The key structural difference between HHD and HLA-A*02 is that in HHD the MHC molecule is fused to hβ2M in a single protein chain, which may affect proper association of chaperones like calnexin and calreticulin involved in preparing MHC-I molecules for peptide loading or alter the structure of the MHC-peptide complex preventing sensitive detection by TCRs raised against the native molecule. Nevertheless, antigen processing and HHD expression on antigen-presenting cells was sufficient to prime CD8 T cell responses after immunization. 20,32 –34 Interestingly, in these and our own therapeutic vaccination studies (unpublished data), the induced cytotoxic T cells also did not exhibit direct antiviral activity toward HBV-replicating hepatocytes. This argues that the low overall amount of transgenic HHD molecules presenting endogenously processed, viral peptides particularly on hepatocytes is insufficient for efficient recognition by HLA-A*02-restricted T cells.
AAV-mediated expression of HLA-A*02 in AAV-HBV cotransduced mice was able to overcome the lack of functional MHC-I HBV-peptide complexes being presented in vivo as indicated by the profound antiviral activity of HLA-A*02-restricted, HBV-specific T cells observed in our study. Although HBcAg could no longer be stained in liver tissue, low levels of intracellular HBV-DNA and circulating viral antigen persisted. As observed previously for transferred CAR+ T cells, 24,35 this might be explained by a high expression of PD-1 on T cells and functional exhaustion after a strong activation. As we used two separate AAVs to deliver the HBV or HLA-A*02 transgene, there is the option that some hepatocytes had been transduced with only one of the AAVs preventing their elimination. We tried to exclude this by using an extremely high titer of AAV-HLA-A*02 aiming to reach almost all hepatocytes. However, to completely avoid this, a one-vector system such as a gutless adenovirus with a high transgene capacity would have to be used.
We have previously shown in a proof-of-concept study that human TCR-redirected T cells have the potential to mediate clearance of HBV infection in liver-humanized mice. 16 Humanized mice and especially those reconstituted with human immune cells, 36 are not available at a larger scale due to the intense effort to generate them. Therefore, we are convinced that the model established in our study will be broadly applicable for in vivo studies using human TCRs to compare different receptors, T cells, and transfer conditions as well as transgenic or knockout mouse strains. It would allow for studies in immunocompetent mice when combined with tolerization for human-derived sequences of HBV-specific receptors. 35 By adapting the setting systemically 21 or to a certain target organ by using AAV vectors with different cell tropism, 37 it can be more broadly applied.
Although mouse models mimicking persistent HBV infection proved to be very useful in determining functionality of adoptive T cell therapy, evaluating the safety and efficacy of immunotherapeutic approaches in preclinical animal models with natural HBV infection, cccDNA formation, still being immune competent would be of high importance. Successful infection with HBV virions after transducing hepatocytes with human NTCP has been reported for different macaque species in vitro 27 and also in vivo 17 and paved the way enabling in-depth investigation of HBV infection, associated pathogenesis, as well as therapeutic approaches in nonhuman primate models. T cell therapy approaches using a ROR1-CAR 38 or an anti-CD27-CAR 39 took advantage of the macaque model to address safety issues before first clinical trials. Thus, macaque models are promising models to evaluate immune therapy of HBV infection. Due to pre-existing immunity, macaques can hardly be transduced using AAV vectors 40 and hence we used an adenoviral vector to deliver human HLA-A*02 into primary macaque hepatocytes.
HLA-A*02 expression allowed presentation of viral peptides on HBV-infected macaque hepatocytes and to activate antiviral activity of TCR-redirected T cells. Also, in this model, HBV was not eliminated completely, which again, was most likely attributed to cells being infected with HBV but not transduced with the Ad-HLA-A*02. In future experiments, an adenovirus encoding for both NTCP and HLA-A*02 should be used, rendering only HLA-A*02+ cells susceptible for HBV infection. This vector could also be used to test antiviral potential, efficacy, and safety of immunotherapies in HBV-infected rhesus macaque in vivo as a last step before clinical use. This concept could also be explored in porcine hepatocytes, which are also susceptible to natural HBV infection when expressing human NTCP. 27 Pigs already serve as important animal models for viral infections 41 and would also be an ideal system to evaluate HEV-specific TCRs. 42
Taken together, in this study, we show that vector-mediated delivery leads to the expression of the human MHC-I molecule HLA-A*02 on murine and macaque hepatocytes and allows evaluation of activation and antiviral activity of HLA-A*02-restricted HBV-specific T cells in different species. This approach will also be applicable for other MHC restrictions or other viral diseases paving the way for safety and efficacy studies of human TCR-based therapies and vaccination studies in physiologically relevant preclinical animal models.
Footnotes
ACKNOWLEDGMENTS
The authors thank Thomas Michler for providing the AAV-empty control vector, Angela Krackhardt for the D1Her-2 control TCR plasmid, and Moriya Tsuji for the Zac2.1 plasmid. The authors are grateful to Antje Malo for practical support and advice and to Natalie Röder for helping with mouse breeding and mouse experiments.
AUTHORs' CONTRIBUTIONS
J.F., M.M.F., and T.A. performed experiments. J.M.W., M.A.M.-H., C.S.-H., B.J.B., J.B.S., and S.A. provided critical reagents, protocols, or infrastructure. J.F., M.M.F., B.J.B., U.P., and K.W. designed experiments. J.F., M.M.F., and K.W. analyzed the data. J.F., J.B.S., U.P., and K.W. drafted and finalized the article.
AUTHOR DISCLOSURE
U.P. is cofounder, shareholder, and board member of SCG Cell Therapy Pte. Ltd. K.W. is shareholder of SCG Cell Therapy Pte. Ltd. that brings HBV-specific TCRs into the clinics. K.W. is employed by SCG Cell Therapy GmbH. U.P. received personal fees from Abbott, Abbvie, Arbutus, Gilead, GSK, J&J, MSD, Roche, Sanofi, Sobi, and Vaccitech. The laboratory of U.P. receives research support by SCG Cell Therapy. The other authors have declared that no conflicts of interest exist.
FUNDING INFORMATION
This work was supported by the German Research Foundation (DFG) through TRR179 SFB-TRR 179/2, project No. 2983813 (to U.P.) and SFB-TRR 338/1, project No. 452881907 (to U.P.), by the German Center for Infection Research (DZIF), TTU Hepatitis project 05.806 to U.P. and young investigator grant 05.812 to K.W. M.F.'s research stay in Portland was supported by the TUM Graduate School. This work was also supported by the National Institutes of Health grant P51 OD011092 awarded to the Oregon National Primate Research Center (ONPRC).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.