
Abstract
Base editing technology, developed from the CRISPR/Cas9 system, is able to efficiently implement single-base substitutions at specific DNA or RNA sites without generating double-strand breaks with precision and efficiency. Point mutations account for 58% of disease-causing genetic mutations in humans, and single nucleotide variants are an important cause of tumorigenesis, and the advent of base editors offers new hope for the study or treatment of such diseases. Although it has some limitations, base editors have been continuously improved in terms of editing efficiency, specificity, and product purity since their development. In this review, we examine the main base editing technologies and discuss their applications and prospects in tumor research and therapy, as well as elaborate on their mode of delivery.
INTRODUCTION
With the continuous development of DNA sequencing technology, the application of DNA sequence data in various fields has become more and more extensive. Nonsynonymous single nucleotide variations (nsSNVs) account for the majority of genetic variations in humans, 1,2 and about one-third of nsSNVs have been found to be life threatening, 3 nsSNVs can cause at least 6,000 diseases, such as cancer, epilepsy, and myofibrillar myopathy, etc. 4,5 Cancer is a major threat to human health. 6 Twenty-two solid metastatic cancers were subjected to genome-wide sequencing, which identified 60 million single nucleotide variants (SNVs) and 840,000 multiple nucleotide variants (MNVs). 7 The emergence of gene editing therapies will bring therapeutic approaches to diseases caused by SNVs.
In 2007, Barrangou et al. demonstrated that clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) evolved in bacteria and archaea to resist invasion by phages and exogenous DNA. 8 In 2012, researchers successfully constructed the CRISPR-Cas9 system in vitro using artificially modified Cas9, in vitro transcriptionally matured CRISPR RNA (crRNA), and trans-activating crRNA (tracrRNA). 9 Later, the CRISPR/Cas9 system was successfully applied in mammalian cells and is widely used in various fields due to its simplicity and practicality. The CRISPR gene contains numerous repetitive and nonrepetitive sequences, and the CRISPR/Cas9 system can use homology-dependent repair for single-base substitution. However, because of the HNH enzyme and RuvC active site on the Cas9 protein cutting the complementary and noncomplementary strands when the CRISPR/Cas9 system acts, resulting in double-strand breaks (DSBs), base substitution is highly inefficient. 10
In 2016, Liu's team creatively developed a base editor that can replace single bases without relying on DSBs based on CRISPR/Cas9 technology, making precise gene editing a reality. 11 The RuvC and HNH functional domains of the Cas9 nuclease were inactivated or altered by Qi et al. to create dead Cas9 (dCas9) or nickase Cas9 (nCas9), which can accurately target the DNA without resulting in DSB. 12 Then the mutant Cas9, cytosine deaminase or adenine deaminase are fused to form a fusion protein, which is constructed as a base editor. 11,13 Base editing technology is able to efficiently and precisely replace bases in individual DNAs without creating DSBs. Many human diseases occur due to single-base mutations, and base editing technology has emerged to correct pathogenic mutations in genetic diseases, as well as to introduce point mutations in cells for plant and animal breeding.
BASE EDITOR
Cytosine base editor
The cytosine base editor (CBE) generally consists of nCas9 or dCas9 with cytosine deaminase. The cytosine editor works by targeting the fusion protein to specific genomic DNA under the guidance of sgRNA, and using cytosine deaminase and uracil glycosylase inhibitor to remove the amino group of cytosine (C), thus making cytosine (C) into uracil (U) after deamination, and then transforming uracil (U) into thymine (T) through DNA repair or replication, and finally cytosine (C) to thymine (T) base substitution 11 (Fig. 1A). Since CBEs were developed, several laboratories have constructed CBEs with different functions and different characteristics from improving editing efficiency, changing window width, and improving editing specificity and precision, respectively.
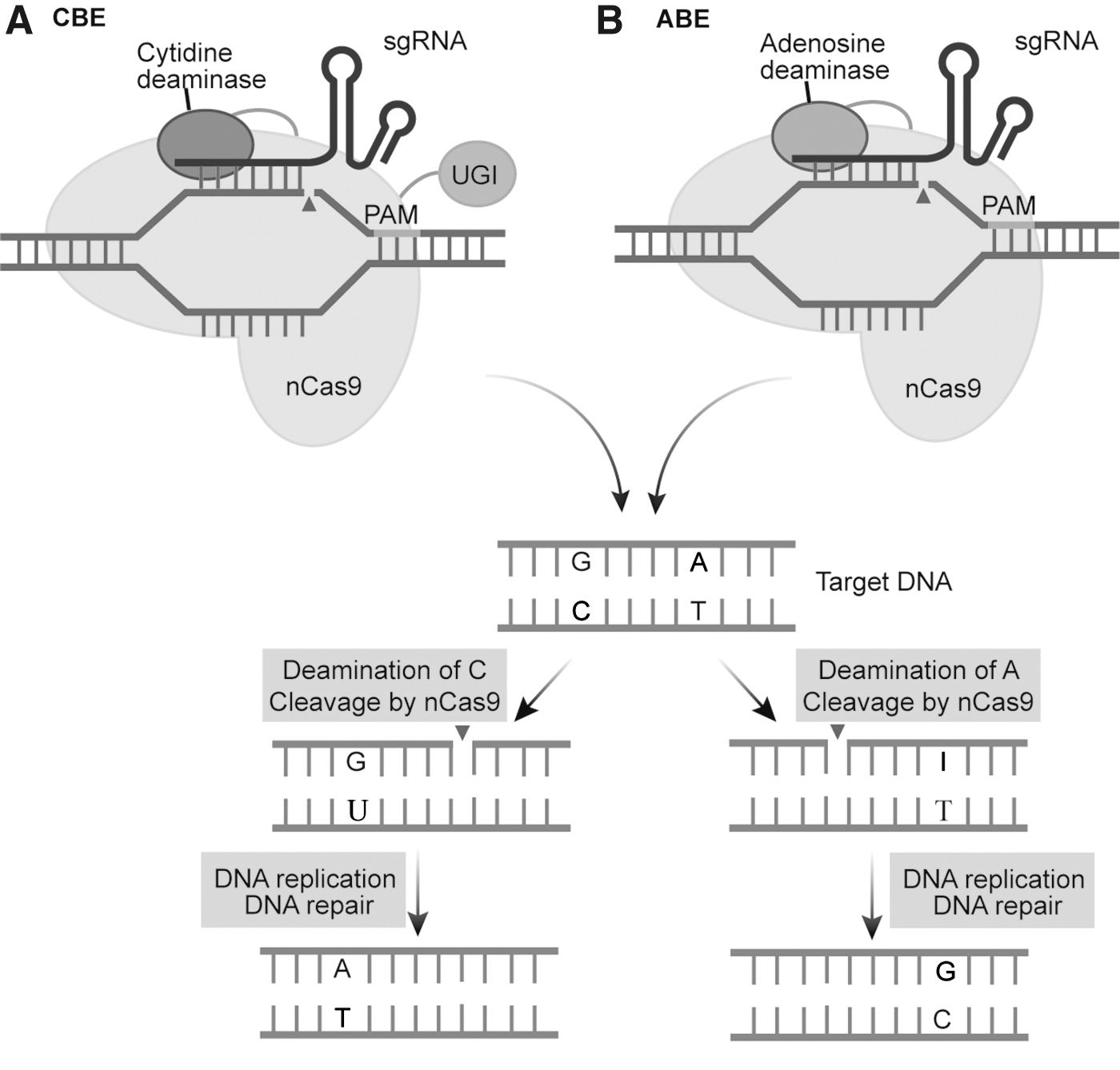
Mechanism of CBE and ABE.
Adenine base editor
Adenine base editor (ABE), a fusion of nCas9 or dCas9 and adenine deaminase to form a dimeric protein, is guided by gRNA to the target site to play the role of base substitution. The process involves removing the amino group on cytosine (C) or adenine (A) by deaminase, converting them to uracil (U) or hypoxanthine (I), respectively, and converting them to thymine (T) or guanine (G) during DNA replication or repair (Fig. 1B). The average editing efficiency of ABE1.2 in nurse animal cells was 3%, and after several modifications, the editing efficiency of ABE7.10 was increased to 58%. 13 Koblan et al. replaced the SV40 nuclear localization signal (NLS) with bipartite NLS (bpNLS) and added bpNLS to the N side of TadA7.10, optimizing it to obtain ABEmax, which has higher editing efficiency than ABE7.10. 14 Richter et al. screened the mutant TadA8e with eight new mutation sites added to ABE7.10 by the phage-assisted continuous evolution (PACE) selection system, and the editing efficiency of the constructed ABE8e was increased by 590-fold compared with ABE7.10. 15
Recently, Chen et al. created ABE9 that precisely catalyzes A > G transformation within a 1–2 nt nucleotide editing window without inducing C > T transformation in cells and embryos, and this study demonstrated that it precisely corrects nearly 50% of pathogenic SNVs and induces little off-target effects. 16 Table 1 summarizes the structure and characteristics of some major BEs.
Base editor and characteristics
AID, activation-induced cytidine deaminase; dCas9, dead Cas9; dCDI, deoxycytidine deaminase inhibitor; nCas9, nickase Cas9; NLS, nuclear localization signal; UGI, uracil DNA glycosylase inhibitor.
Dual-base editor
The single-base editor has some limitations on the modification of the target sequence, and to improve the base editing ability, and researchers constructed a dual-base editor. By optimizing the codon, adding NLS, and optimizing the uracil DNA glycosylase inhibitor tandem form and linker sequence, Zhang et al. successfully constructed the dual-base editor, A&C-BEmax, which has slightly lower activity for adenine and higher activity for cytosine, higher the editing window was expanded, and the RNA off-target activity was significantly reduced. 17 Grünewald et al. fused adenosine and cytidine deaminases (Tad A and Pm CDA1) to the N and C termini of Cas9n, respectively, and developed SPACE, a two-base editing system that allows simultaneous editing of adenine and cytosine. Compared with single-base editors, this base editor is comparable to CBE for C > T and slightly less efficient than miniABEmax-V82G for A>G, enhancing the scope and total editing efficiency of DNA editing, and showing very low off-target effects during editing. 18
Liang et al. fused ABE with CGBE to develop the multifunctional two-base editor AGBE, which can induce four base transitions of A-G and C-G/T/A at the same target under the guidance of a single sgRNA, improving gene editing diversity. 19
RNA base editor
Cox et al. fused the ADAR2 deaminase structural domain (ADAR2DD) to catalytically inactivated CRISPR/Cas13 to construct the RNA editor REPAIR, which enables base editing of A to I(G) in RNA 20 (Fig. 2A). Subsequently, the team further optimized the REPAIR system by evolving the adenine deaminase structural domain (ADAR2dd) of ADAR2 in the system into a cytidine deaminase, and constructed the RESCUE system with cytidine deaminase function of ADARDD fused to Cas13b, which retains A to I editing activity and enables multiple editing from C to U and from A to I. It can be repaired and corrected in mammalian cells and is a potential method for genetic disease treatment 21 (Fig. 2B).
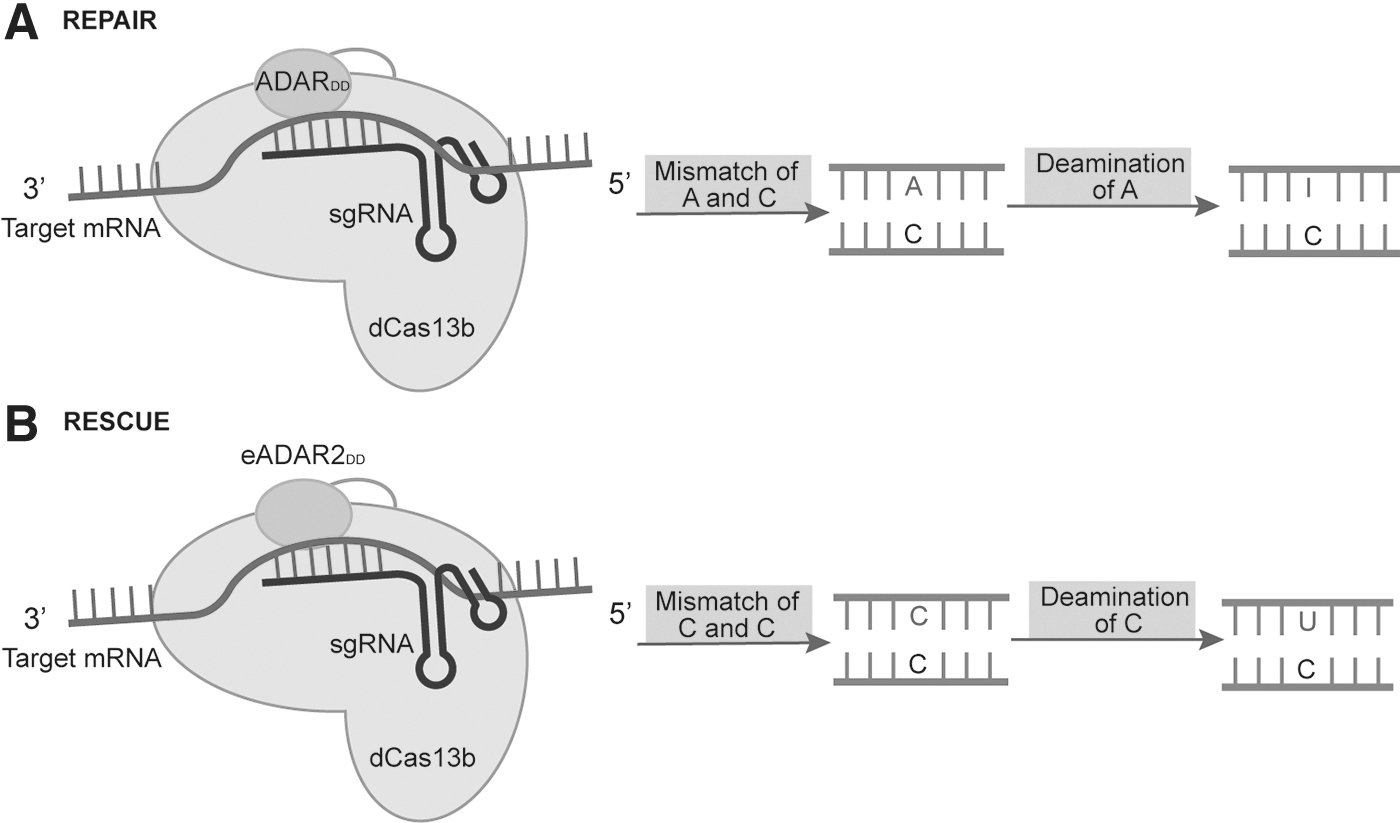
Mechanisms of RNA base editors.
Mitochondrial DNA editor
Single-base mutations in mitochondrial DNA (mtDNA) may cause systemic inflammatory response syndrome, cardiovascular disease, cancer, and many other human diseases. 22 –24 Mitochondria are susceptible to oxidative damage to mtDNA caused by ROS attack, resulting in mtDNA mutations and copy number changes, and consequently, abnormal proliferation and expression. 25,26 First- and second-generation gene editing technologies, zinc finger nucleases, and transcription activator-like effector nucleases, can target the target mtDNA and thus cleave the target loci. 27,28 CRISPR technology lacks gRNAs that can efficiently introduce exogenous DNA into mitochondria.
Mok et al. successfully constructed DdCBE using a bacterial toxin—DddA, which can catalyze C > U in dsDNA, fused with activator-like effector array proteins (TALE) and a uracil glycosylase inhibitor, making it possible to precisely edit mtDNA. 29,30 The team developed DddA6 and DddA11 with higher editing efficiency and broader editing ranges by PACE and related phage-assisted non-continuous evolution (PANCE). 31 Recently, a team constructed mitochondrial ABEs (TALEDs) with a fusion of TALE, TadA8e, and DddAtox, allowing TALEDs to perform CRISPR-free editing of mtDNA. 32 MtDNA is expected to be a target for tumor therapy, and the emergence of a mitochondrial base editor will bring new hope for the treatment of such tumors.
APPLICATION OF BASE EDITOR FOR ONCOTHERAPY
In recent years, tumor therapy has made significant breakthroughs and advances. However, the morbidity and mortality rates of many tumors remain persistently high. Humans cannot yet stop short of tumor therapy and research, and base editors are becoming more widely used for tumors. Various studies have shown that base editors are able to halt tumor development in multiple ways, providing new directions for tumor therapy. This section mainly summarizes applications of base editors to tumor research.
Reversal of gene mutations
KRAS and TP53
Activation of proto-oncogenes and loss of function (LOF) of tumor-suppressive genes are the key factors leading to tumorigenesis. 33 For example, Kirsten rat sarcoma viral oncogene (KRAS) mutations are one of the most common oncogenic factors in a variety of tumors. 34 Once mutated, KRAS loses its GTP hydrolase activity, which in turn continues to activate signaling pathways such as RAF-MEK-ERK, PI3K-AKT-mTOR, resulting in uncontrolled cell proliferation and carcinogenesis. KRAS has a mutation rate of 97% in pancreatic cancer, 52% in colorectal cancer, 42% in multiple myeloma, and 32% in lung cancer. 35 The most common mode of KRAS gene mutation is a point mutation, and the common forms of mutation are KRASG12X and KRASG12C. 36,37 TP53 is an oncogene, and TP53 mutations are present in more than 50% of cancer genomes, 80% of which are point mutations. 38 Base editing technology, which can reverse single nucleotide mutations, is the most promising approach to cure such diseases in the future.
Sayed et al. constructed NG-ABE, a base editor combining nCas9 and ABE8e, to reverse some mutations commonly found in cancer cells. After lentiviral (LV) transfection of cells, NG-ABE corrected the G12S mutation in the non-small cell lung cancer cell line A549, the G13D mutation in the colorectal cancer cell line HCT116, and the pancreatic cancer cell line PANC-1, which induces G0 to G1 cell cycle arrest and reduced cell proliferation. Also, a base editor was designed that corrects TP53 mutations with 79% A > G substitution, enabling efficient editing. 39
Mutations in KRAS and P53 genes cause resistance to gemcitabine in patients with pancreatic ductal adenocarcinoma. Won et al. transfected human pancreatic cancer cell line PANC1 by nano-lipid particles loaded with ABE7.10-nCas9, resulting in reduced expression levels of the mutant proteins, P53 gene at 53.6% and KRAS gene at 51.2%; the cell survival was significantly reduced, and the tumor suppression was also obtained during in vivo treatment. 40
Pyruvate kinase muscle
Pyruvate kinase muscle (PKM)1 and PKM2, produced by selective exon mutually exclusive variable splicing of the PKM gene, are rate-limiting enzymes in the glycolytic pathway, mainly found in early embryonic and tumor formation, and are an important target for cancer therapy. 41 PKM1 was shown to promote oxidative phosphorylation and inhibit tumor cell proliferation to suppress tumorigenesis, while it was found that PKM2 was overexpressed in malignant tumor tissues such as colorectal cancer and liver cancer. 42,43 Lin et al. were able to precisely and efficiently mutate gRNAs targeting the splice sites (flanking exons 9 and 10) of the PKM gene in a human colorectal cancer cell line (HCT116 cells) using the ABE (ABEmax-NG) and CBE (BE4max) with a mutation efficiency of 37–93% and no byproducts were produced. This study demonstrated the opposite roles of PKM1 and PKM2 in tumorigenesis. 44
Telomerase reverse transcriptase
The telomerase reverse transcriptase (TERT) gene is one of the important genes encoding the telomerase complex, and TERT promoter mutations (hTERTp-mut) are involved in the development and progression of a variety of tumors, including urologic tumors, lung cancer, liver cancer, and breast cancer. 45,46 Point mutations in the TERT promoter have been detected in 51% of gliomas and 44% of hepatocellular carcinomas. 47 To treat primary glioblastoma (GBM), Li et al. used sgRNA-guided and catalytically impaired Campylobacter jejuni CRISPR-associated protein 9-fused ABE (CjABE), which effectively corrected the 123/124C>T mutation in the TERT promoter of GBM cells, and intracranially injected expression of CjABE significantly inhibited GBM growth in mice. 48
Combined chimeric antigen receptor T cell for treatment
Allogeneic chimeric antigen receptor T cell (CAR-T) treatment is a genetic engineering method that transforms immunological T cells obtained from autologous or allogeneic blood into T cells with chimeric receptors that identify tumor antigens. It produces CARs that selectively target tumor antigens, killing and eliminating cancer cells. 49,50 Therefore, it is one of the most promising treatment modalities for hematological malignancies at present.
Stadtmauer et al. first clinical trial of CAR-T therapy for cancer treatment using CRISPR/Cas9-based technology to knock down T cell receptor (TCR) and programmed death-1 (PD-1) on T cells, confirming the feasibility of CAR-T therapy in cancer treatment. 51 However, recent studies point to the loss or damage of some genetic material when cells are treated, and the study used single-cell RNA sequencing to verify that chromosome aneuploidy and chromosome truncation occurred in the chromosome where the target gene was located after CRISPR/Cas9 technology edited human primary T cells, and that such changes might lead to instability of the cellular genome, leading to the development of disease. 52
Base editing enables gene editing without causing DSBs, which theoretically offers higher safety and feasibility.
Base editing has been used to create allogeneic CAR-T cells, and Diorio et al. used CBE to construct a CD7-directed allogeneic CAR-T, 7CAR8, that allows for four simultaneous and efficient base edits to block the expression of the proteins CD52, CD7, PD1, and TCRa. This approach does not lead to adverse events such as DNA damage or chromosomal translocations. The key immunotherapeutic target for T cell acute lymphoblastic leukemia (T-ALL) is the surface receptor CD7, and in an animal model of T-ALL, mice treated with 7CAR8 have longer survival significantly, indicating that this therapy has great potential for clinical application. 53
Webber et al. used CBE3 and CBE4 to disrupt the splice donor and splice acceptor sites of programmed cell death 1 (PDCD1), β-2 microglobulin (B2M), and T cell receptor ɑ constant (TRAC) locus in allogeneic CAR-T cells targeting CD19. Transcript levels and protein expression levels were significantly reduced, translocation frequency and DSB induction were substantially lower compared with CRISPR/Cas9 editing, and these knocked-out cells' cytokine functions were also preserved. 54 Gaudelli et al. constructed to obtain ABE8s with a significantly higher editing efficiency and a 3.5-fold increase in γ-globin expression in human CD34+ cells by editing the promoters of γ-globin genes HBG1 and HBG2, while multiple edits were targeting B2M, class II major histocompatibility complex transactivator, and TRAC in human primary T cells to reduce MHC class I and II, respectively, already TCR expression, both with editing efficiencies of 98–99%. 55
Georgiadis et al. demonstrated specific cytotoxicity against leukemia cell lines and primary T cell acute lymphoblastic leukemia (T-ALL) cell lines using a codon-optimized cytidine base deaminase (coBE3)-edited 3CAR or 7CAR primary T cells, with a dramatic reduction in the frequency of chromosomal translocations, and were able to successfully inhibit disease progression in a mouse model of human-derived leukemia. 56 Preece et al. introduced premature stop codons by delivering a hepatitis B-virus (HBV)-specific recombinant TCR and a codon-optimized BE3 to disrupt endogenous TCRs, which produced increased cytokines and intact antigen-specific function in a hepatocellular carcinoma model of HBV antigen expression. 57
Tumor modeling
Single-base editing technology can also generate tumor models, facilitating the study of related diseases. Liu et al. demonstrated in vitro that ABE can generate A > G mutations in mouse-derived Neuro-2a (N2a) cells and that these mutations do not affect embryonic development. Select sgRNAs targeting Ar and Hoxd13 were microinjected into mice to generate mouse models with relevant mutations at the target site, exhibiting androgen insensitivity syndrome (sex reversal) and Syndactyly (fused digits) symptoms. This experiment also combined ABE and saBE3 with simultaneous base editing so that saBE3 targets the tyrosine receptor (Tyr) gene, and the Tyr mutation can turn black hair into white hair, successfully constructing a mouse model with complex clinical symptoms of Syndactyly and white hair, 58 creating an important condition for the study of related diseases.
Annunziato et al. successfully constructed BE3 cytidine base editor Cre conditionally expressed knock-in mice by LV loading of sgRNA targeting Akt1 exons to establish missense mutations, allowing in situ base editing of the mammary gland. We confirmed that activation of PIK3CA and AKT1 and loss of phosphatase and tensin homolog function are important causes of BRCA1-related tumorigenesis, such as breast cancer. 59
Rosello et al. first constructed a model of albinism in zebrafish using CBE4max-SpRY, followed by the injection of CBE4max SpRY mRNA, tp53 sgRNAs, and Nras NAN protospacer adjacent motif (PAM) into an embryonic cell targeting the G12 amino acid of Nras, which is mutated in human melanoma, showing that the MAP kinase pathway downstream of Nras signaling was activated and tp53 was successfully targeted, resulting in a zebrafish melanoma susceptibility model that more closely resembles human genetic events. 60
Screening for oncogenes
Point mutations in the sequence encoding the structural domain of ABL kinase cause imatinib resistance during chronic granulocytic leukemia (CML) treatment, Ma et al. designed the targeted activation-induced cytidine deaminase (AID)-mediated mutagenesis (TAM) system incorporating the human AID variant hAIDx, expressed in the CML cell line K562 cells, and identified a number of known and unknown mutant loci, 61 providing a new strategy to resist drug resistance. sgRNAs targeting BRCA1 and BRCA2 were designed by Huang et al. After transfection of eHAP cells using the LV packaging CBE AncBE4max and ABE ABEmax, deep sequencing and screening of the sgRNAs yielded 910 BRCA1/2 LOF variants. 62
Hanna cloned the library into a LV vector expressing sgRNA and loaded with BE3.9max, and screened the sgRNA library in MELJUSO (melanoma), OVCAR8 (ovarian cancer), HA1E (immortalized kidney), and HAP1 (near-haploid CML-derived) cell lines, and the BE screen efficiently identified sgRNAs that introduced LOF mutations in both positive and negative selection analyses, and efficiently screened and validated BRCA1 and LOF mutations in BRCA2, and also identified LOF variants in many DNA damage repair genes. 63
Cuella-Martin et al. generated variants in the DNA damage response (DDR) gene and altered cellular fitness by LV packaging of the BE3 CBE and sgRNA targeting the DDR gene, analyzed the response of these mutations to DNA damaging agents, determined the gain, loss, and segregation of function of 53BP1, clarified the LOF mutation of Checkpoint kinase 2 (Chk2) and accelerated the study of DDR gene function. 64
Sánchez-Rivera et al. identified multiple TP53 missense mutations as drivers of cancer cell proliferation and tumorigenesis development through base editor screening and detection of cancer-related SNVs and identified Trp53T208I and TP53T211I as valid mutation loci for the construction of a mouse model of pancreatic cancer. 65
Regulation of tumor microenvironment
The primary immune cells of the glioma microenvironment are tumor-associated microglia/macrophages (TAMs), and various tumor microenvironments can cause macrophage development into various subtypes, 66,67 mainly including the classically activated type (M1-TAM) and alternatively activated type (M2-TAM). TAM is involved in the immune presentation, secretes proinflammatory cytokines, and exerts antitumor effects, whereas M2-TAM has the function of inhibiting T cell responses, promoting angiogenesis, and promoting tumor cell growth, invasion, and metastasis. 68,69
Ma et al. constructed an isocitrate dehydrogenase one mutation (IDH1R132H/WT) human glioma cell model by CBE, which resulted in the downregulation of 80% of immune response-related genes and signaling pathways, and was able to promote macrophage polarization toward M1 type and enhanced phagocytosis and migration of macrophages, and in an in situ xenograft mouse model, IDH1R132H/WT was able to increase TAM recruitment, induce TAM antitumor, reduce TAM M2 polarization, and significantly inhibit tumor growth. This study also confirmed that silencing of ICAM-1 in glioma is the key to inhibiting tumor growth. 70
DELIVERY METHOD
Whether in vitro or in vivo, gene therapy must be efficient in targeting the cells or tissues to be treated and achieve adequate safety and efficacy, and common delivery methods include physical methods, nonviral vectors, and viral vectors (Fig. 3).
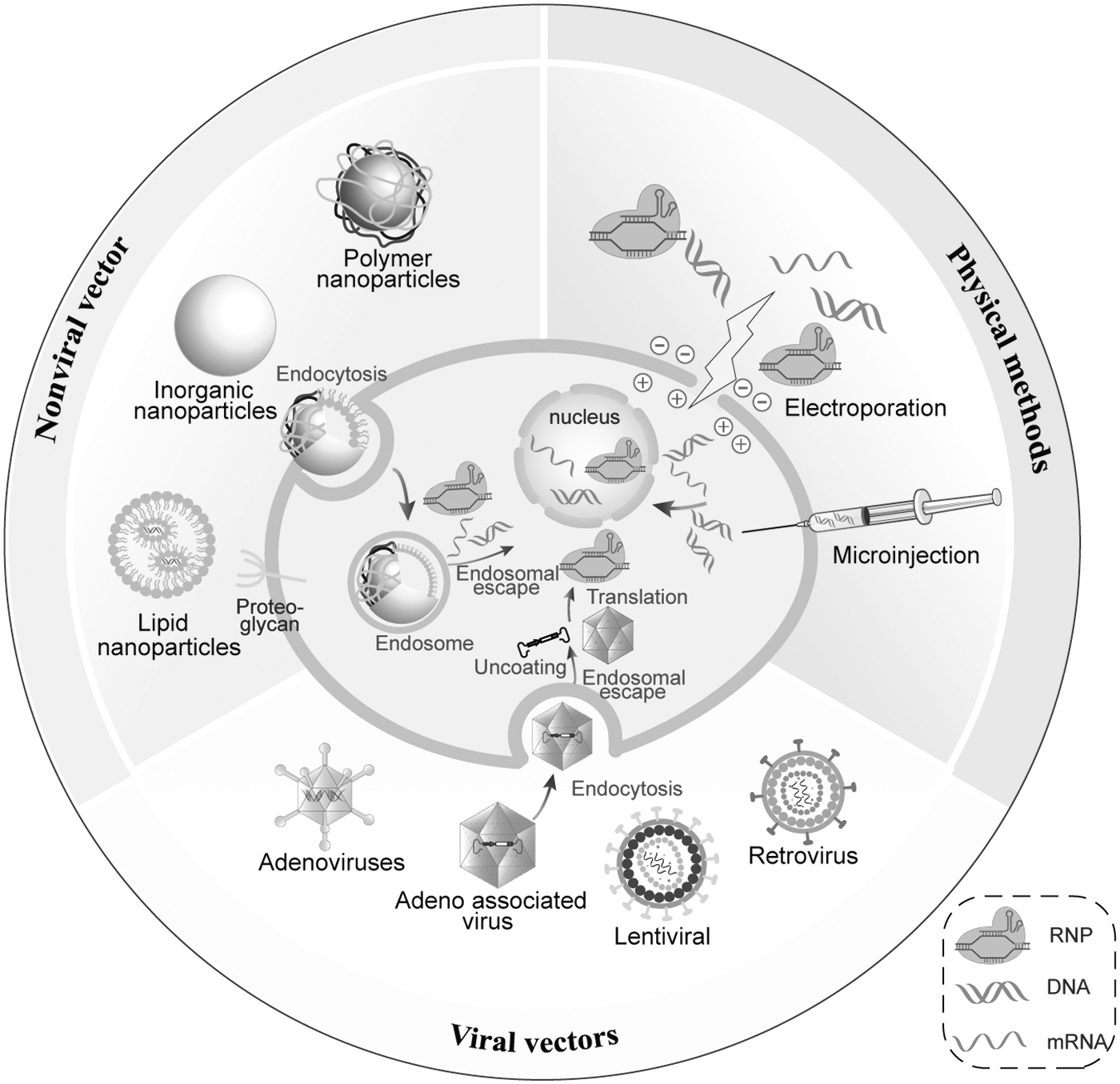
The main delivery methods for gene therapy include physical methods, nonviral vectors, and viral vectors, and this diagram also shows the main categories and delivery methods for each method.
Physical methods
The most direct method of delivering gene editing systems into cells is by physical methods, such as microinjection and electroporation. Microinjection is able to deliver mRNA, plasmids, and ribonucleoprotein (RNP) directly into the cell. This method is not limited by the molecular weight and genome size of the delivered system, but the number of cells treated at a time is limited, making it difficult to apply on a large scale. 71 Electroporation achieves efficient delivery of nucleic acids or proteins by instantaneous disruption of the phospholipid bilayer on the cell membrane, but the delivery process may be hazardous such as loss of cellular activity or even death, so the voltage and current parameters need to be continuously optimized, 72 and this method is often used in cell therapy and in the generation of targeted mutations in fertilized eggs. 73,74
Nonviral vectors
Nonviral vector-based drugs have been widely used for the delivery of microRNA, siRNA, shRNA, and CRISPR/Cas9, which have the advantages of high vector capacity and low immunogenicity, and have shown great potential for application. Nonviral vectors mainly include polymeric nanoparticles, liposomes, and inorganic nanomaterials. In addition, some nonviral vectors based on zeolitic imidazolate framework (ZIF), 75 functionalized peptides, and proteins have been shown to be used for the delivery of nucleic acids. 76
Cationic polymers deliver drugs across membranes by endocytosis and are effective in protecting drugs from degradation by nucleases. Commonly used cationic polymers include polyethyleneimine and chitosan (CS). 77,78 In addition, many novel and powerful nanomaterials have been designed for nucleic acid delivery, and Chen et al. synthesized poly(beta-aminoesters) (PBAEs) nanoparticles with higher transfection efficiency to deliver ABEmax-NG and sgRNA to human brain astrocytoma (U87-MG) cells and xenograft gliomas. EGFP was used to determine the efficiency of base editing in mouse tumors, and the conversion of EGFP signals demonstrated successful base editing in vivo and in vitro using PBAEs. 79
Lipid nanoparticles (LNPs) are one of the most commonly used nucleic acid delivery systems and are widely used in areas such as gene editing and vaccine development, and the recently developed New Crown Pneumonia vaccine also belongs to the LNP formulation (BNT162b1 and BNT162b2). 80 –82 LNPs are prone to aggregation in the liver and have been approved by the Food and Drug Administration (FDA) for intravenous administration of liver-targeted siRNAs, as well as by intramuscular injection during delivery of vaccines. 81,83 Negatively charged nucleic acids are compounded with positively charged lipids through electrostatic interaction to form LNPs, which can protect nucleic acids from destruction by nucleases by joining cells through giant cell drinking and endocytosis, and LNPs have the advantages of high encapsulation rate, strong tissue penetration, and safety and simplicity. 84 LNPs mostly accumulate in the liver, and the rational use of this property would be of great significance for the treatment of liver diseases.
Zhang et al. designed a series of lipid-like nanomaterials using functionalized N1, N3, N5-tris(2-aminoethyl) benzene-1,3,5-tricarboxamide derivatives (FTT5) for the delivery of long mRNAs in the liver. Like-nanomaterials (LLN) for delivery of long mRNAs showed that in vivo delivery of ABEs and sgRNAs targeting the PCSK9 gene achieved ∼60% of A > G mutations in the liver. 85 Furthermore, due to the natural targeting of LNPs to the liver, making LNPs target other tissues is extremely challenging, and researchers are continuously developing LNPs for different tissues, 86 which may be of great help in expanding the application of base editors.
Inorganic nanomaterials are also widely used for nucleic acid delivery with the advantages of noninfectious, nonvolume limiting, wide source of materials, and easy to prepare in large quantities. In particular, gold nanoparticles (AuNPs) have been regarded as promising carriers for nucleic acid delivery due to their multifunctionality, high stability, and ease of synthesis. 87 The interaction between AuNPs and contents can produce nanoparticles that can rapidly bind cell membranes and release Cas9 protein and RNP into the cytoplasm. For example, AuNPs can easily crosslink with substances with sulfhydryl (-SH) groups through Au-S bonds, which changes the surface charge of nanoparticles, enhances the binding ability to contents, and changes the hydrophilicity and targeting of nanoparticles. The powerful loading ability and unique optical properties make AuNPs one of the most promising carriers for drug delivery. Wang et al. compressed Cas9 on TAT peptide-modified AuNPs through electrostatic interactions and encapsulated lipids on AuNPs.
After intravenous administration, the thermal effect of AuNPs was triggered by laser, which allowed Cas9 and sgRNA to enter the cytoplasm and nucleus of tumor cells, and successfully targeted and knocked down the Polo-like Kinase 1 (Plk-1) gene of melanoma in vitro and in vivo, which induced apoptosis and inhibited the growth of tumor cells. 88 Lee et al. developed CRISPR-Gold23 to target the mGluR5 gene and deliver Cas9 and sgRNA to the brain of a fragile X syndrome mouse model. mGluR5 expression was successfully reduced by 40–50%, rescuing the abnormal behavior of the mice. 89
Viral vectors
Viral vectors are the most commonly used vectors for delivering base editors and have the advantage of being highly efficient and targetable. Most of the drug vectors used in clinical trials are viral vectors, mainly lentiviruses, adenoviruses, adeno-associated viruses, and retroviruses (Table 2).
Characteristics of viral vectors
LV vectors are derived from the HIV-1 virus and consist of single-stranded RNA that replicates through a reverse transcription process with a large loading capacity and the ability to be stably inherited. Suh et al. corrected pathogenic mutations in vivo and in vitro by subretinal injection of ABEmax packaged by lentivirus and sgRNA targeting the Rpe65 gene, restoring Rpe65 protein expression as well as rd12 vision in mice. However, the authors also suggest that more in-depth applications require replacement with safer vectors. 90
Adenovirus (AdV) vectors have been successfully used to deliver nucleic acids, and Rossidis et al. used AdV to deliver BE3 in utero, resulting in reduced levels of PCSK9 and improved liver function. 91 However, due to the high immunogenicity of AdV, there are significant limitations in its clinical use.
Adeno-associated virus (AAV) has been commonly used in various animal models and clinical trials because of its stable expression and high safety profile, lower immunogenicity compared with other viruses, and different capsid serotypes that can target different tissues. 92 However, its loading capacity is a great challenge when using AAV, which is less than 4.7 kb, 93 which limits the application of AAV, and for this reason, researchers have created a series of methods to enable the smooth loading of gene editing tools into AAV.
Cas9 protein is split in half, packaged in separate AAVs, and reassociated into full-length Cas9 in cells through intrinsic peptide, rapamycin-controlled FKBP/FRB system, light-induced system, and sgRNA mediated. 94 –97 Lim et al. constructed an intrinsic peptide-based trans-splicing system and introduced nonsense mutations in the superoxide dismutase 1 (SOD1) gene by intrathecal injection of CBE into mice, which prolonged the survival time of Amyotrophic Lateral Sclerosis mice. 98 The investigators also chose Staphylococcus aureus Cas9 (saCas9), with a genome size of only 3.2 kb, to replace spCas9, which allowed the editor to be packaged all in a single AAV, avoiding the dosage problem created by double AAV. 99,100
An RNA delivery vector, selective endogenous encapsidation for cellular delivery, was recently developed by Segel et al. to exploit the potential of the mouse and human retrovirus-like protein PEG10 untranslated region to bind to specific RNA molecules and to modify it for loading specific RNAs and delivering them into cells, providing a new idea for the development of gene therapy. 101
CHALLENGES AND PROSPECTS
CRISPR/Cas9 gene editing technology is advancing rapidly, and laboratories in many parts of the world are competing to conduct clinical trials. Lu et al. conducted the first human clinical trial based on CRISPR/Cas9 gene editing technology to knockout the PD-1 gene from immune cells extracted from patients' blood and transfuse it back into patients, demonstrating the feasibility of CRISPR/Cas9 in the treatment of non-small cell lung cancer. 102 Maeder et al. developed EDIT101 therapy by loading the gene for SaCas9 and two gRNAs into an AAV5 vector and delivering the gene editing system to photoreceptor cells by subretinal injection to restore the expression of the CEP290 gene. 103 How to address the potential risks of gene therapy will be a major challenge for future research.
Base editors based on the CRISPR system take gene editing to a new level and will help in understanding the mechanisms of disease occurrence and developing potential diagnostic and therapeutic approaches. Precise editing can help correct SNV. However, base editing systems remain challenging. The deamination window of the base editor is generally within 4–5 nucleotides (nt), which can easily lead to bystander editing, and base editing also needs to be done within a specific range of the PAM sequence (generally 15 ± 2 nt). At the same time, the base editing system also lacks functions such as deletion and insertion of bases, 104 and how to improve editing efficiency, product purity, and reduce off-target effects are issues that need to be continuously addressed. The advent of C > G base editors (CGBE1) enabled base editing from pyrimidines to pyrimidines, 105 and tBEs systems constructed using a cleavable deoxycytidine deaminase inhibitor can effectively reduce both dependent off-target effects and nondependent single-stranded DNA off-target effects during base editing. 106
By optimizing base editors, base editing technologies have demonstrated lower off-target effects and more accurate editing, showing great potential for application in gene therapy. The base editing system can also be selectively delivered to tumors through vectors, reducing accumulation in other organs or tissues, and greatly reducing the systemic toxicity associated with the therapeutic system.
Gene-level editing involves many potential issues. Genetic alterations cause permanent changes in genetic material, and the application of gene editing to embryos and reproductive systems may cause permanent changes in heritability that are passed on to the next generation, raising intense ethical concerns. 107 A study has detected antibodies against SaCas9 and SpCas9 in human serum, suggesting that the body can develop an immune response to the Cas9 protein. 108 Different therapeutic regimens need to be designed for different diseases. AAV is expensive to produce and its efficacy will gradually weaken when AAV is injected for several years. The first high dose of AAV treatment will stimulate the body's immune system and the level of antibodies will increase significantly, and the efficacy of the second AAV injection will inevitably decrease. LNP can only achieve efficient targeted delivery to organs such as liver and lung. Therefore, the safety and efficacy of the therapy need to be assured before gene editing can be expanded for production.
The CRISPR-Cas9 system may cause large fragments of gene deletion at the target site, as well as genomic rearrangement, but studies have demonstrated that the base editing system does not induce large fragments of deletion and is a more precise and safer gene therapy. 109,110 We believe that base editing technology has important applications and will become a new tool for tumor therapy after continuous optimization and updating.
Footnotes
ACKNOWLEDGMENTS
Authors thank all the participants enrolled in the study, and the staff.
AUTHORs' CONTRIBUTIONS
L.H. conceived and wrote the article; H.D. drew the figures; Y.C. contributed database tools; C.Y. revised and edited the article; Z.L. guided the review; H.X. aided in conceptualization and the supporting funding. All authors contributed to the article and approved the submitted version.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was financially supported by the Science and Technology Program of Sichuan Province (no. 2022NSFSC0792, 2020YFS0417); Beijing Xisike Clinical Oncology Research (no. Y-QL202101-0125); Technology Innovation Research and Development Project of Chengdu Science and Technology Bureau (no. 2022-YF05-01591-SN); Foundation of Sichuan Province Key Laboratory of Individualized Drug Therapy (no. 2021YB03); and Research Project established by the Chinese Pharmaceutical Association Hospital Pharmacy department (no. CPA-Z05-ZC-2022-002).