
Abstract
Duchenne muscular dystrophy (DMD) is a debilitating genetic disorder that results in progressive muscle degeneration and premature death. DMD is caused by mutations in the gene encoding dystrophin protein, a membrane-associated protein required for maintenance of muscle structure and function. Although the genetic mutations causing the disease are well known, no curative therapies have been developed to date. The advent of genome-editing technologies provides new opportunities to correct the underlying mutations responsible for DMD. These mutations have been successfully corrected in human cells, mice, and large animal models through different strategies based on CRISPR-Cas9 gene editing. Ideally, CRISPR-editing could offer a one-time treatment for DMD by correcting the genetic mutations and enabling normal expression of the repaired gene. However, numerous challenges remain to be addressed, including optimization of gene editing, delivery of gene-editing components to all the muscles of the body, and the suppression of possible immune responses to the CRISPR-editing therapy. This review provides an overview of the recent advances toward CRISPR-editing therapy for DMD and discusses the opportunities and the remaining challenges in the path to clinical translation.
INTRODUCTION
Duchenne muscular dystrophy (DMD)
It has been known for over 35 years that mutations in the DMD gene cause the disease. 3 The DMD gene is located on the X chromosome, and it is one of the largest genes of the genome. It consists of 79 exons that encode the dystrophin protein that is located beneath the sarcolemma and connects the cytoskeleton of muscle fibers with the extracellular matrix. 4 Dystrophin functions as a shock absorber, reducing the mechanical stress induced by muscle contraction, thereby maintaining sarcolemmal integrity and supporting muscle function. 5
Due to the extensive length of the DMD gene, more than 7,000 mutations causing DMD have been identified. 6 Approximately 70% of DMD patients harbor a large deletion ( = or >1 exon) in the DMD gene, and the other 30% of mutations include duplications, point mutations, or small insertions or deletions (INDELs). Most of the exon deletion or duplication mutations cluster into two hotspot regions spanning between exons 6 and 7, and exons 43 and 53, whereas other small mutations occur randomly through the DMD gene. 6 Mutations in the DMD gene generally result in a shift in the open reading frame (ORF), rending the DMD gene out-of-frame, and ultimately introducing a premature stop codon that leads to the absence of functional dystrophin protein in skeletal muscle myofibers and in cardiomyocytes.
While the amino- (N) and carboxyl- (C) termini of the protein are essential regions, deletions in the rod-like domains of the central region that preserve the correct dystrophin ORF are tolerated and result in a Becker muscular dystrophy (BMD) phenotype that can be a relatively benign muscular dystrophy compared to DMD. 7
Unfortunately, at present, there is no curative therapy for this lethal disease. Corticosteroid treatments are available for the mitigation of the secondary symptoms of DMD, such as inflammation, fibrosis, impaired angiogenesis, altered calcium homeostasis, and mitochondrial dysfunction. 8,9 However, long-term use of corticosteroids causes many adverse effects, despite minimal amelioration of the DMD phenotype. An efficient strategy to treat DMD could be to express a semifunctional dystrophin to mimic the BMD-like phenotype. Truncated dystrophin lacking exons encoding the central rod domains can fit the limited packaging capacity of adeno-associated virus (AAV) for systemic delivery, and this led to the development of a wide range of mini- and microdystrophin variations for therapeutic use. 10
Currently, the utilization of these truncated dystrophins is under evaluation in several clinical trials. 11 Other pharmacological approaches focus on the restoration of the disrupted DMD ORF using antisense technology. Eteplirsen is the first drug specific for DMD approved by the U.S. Food and Drug Administration (FDA). It is an antisense oligonucleotide that induces the skipping of exon 51 of the DMD gene resulting in the expression of a semifunctional dystrophin and BMD-like mild symptoms in a small cohort of DMD patients. 12 Two other oligonucleotides (golodirsen and viltolarsen) are under evaluation for treating DMD patients that could be therapeutically beneficial by promoting skipping of exon 53. 13,14 However, all these approaches have shown only minimal clinical benefit, and none removes the underlying genetic cause of the disease to enable lifelong expression of dystrophin.
GENE-EDITING THERAPEUTIC STRATEGIES
Gene editing with CRISPR-Cas9 has the potential of permanently correcting mutations causing DMD and ameliorating the pathology of the disease. The CRISPR-Cas9 system was originally discovered in bacteria as an adaptive defense system against viruses and has been engineered for the editing of the genome of eukaryotic cells. 15 –17 The Cas9 endonuclease can be directed to a specific DNA sequence of the genome, using a short guide RNA (sgRNA). The target sequence is specified by the complementary of a region of 20 nucleotides of the sgRNA called the protospacer. The most studied and used Cas9 is derived from the CRISPR system of Streptococcus pyogenes (SpCas9). The SpCas9 endonuclease recognizes a protospacer adjacent motif (PAM) in DNA consisting of the sequence 5′-NGG-3′ and cuts the target DNA 3 base pairs (bp) upstream of the PAM, generating a DNA double-stranded break (DSB).
The actual gene editing can occur in two ways: (1) in the presence of a DNA donor template, homology-direct repair (HDR) can introduce the desired modification in the target locus. However, the HDR mechanism is only active in proliferative cells, with limited efficiency in postmitotic cells, such as myofibers and cardiomyocytes; (2) alternatively, nonhomologous end joining (NHEJ) works in both quiescent and proliferating cells and can imprecisely repair the DNA generating short INDELs at the site of the DNA DSB. In recent years, CRISPR-Cas9 has shown great potential for gene editing of mutations causing DMD. We use the term “myoediting” to refer to CRISPR-mediated gene editing in muscle to permanently correct genetic mutations causing DMD and restore muscle function, and here, we describe different myoediting strategies developed for treatment of DMD (Fig. 1, “Gene-editing strategies”). 18 –20
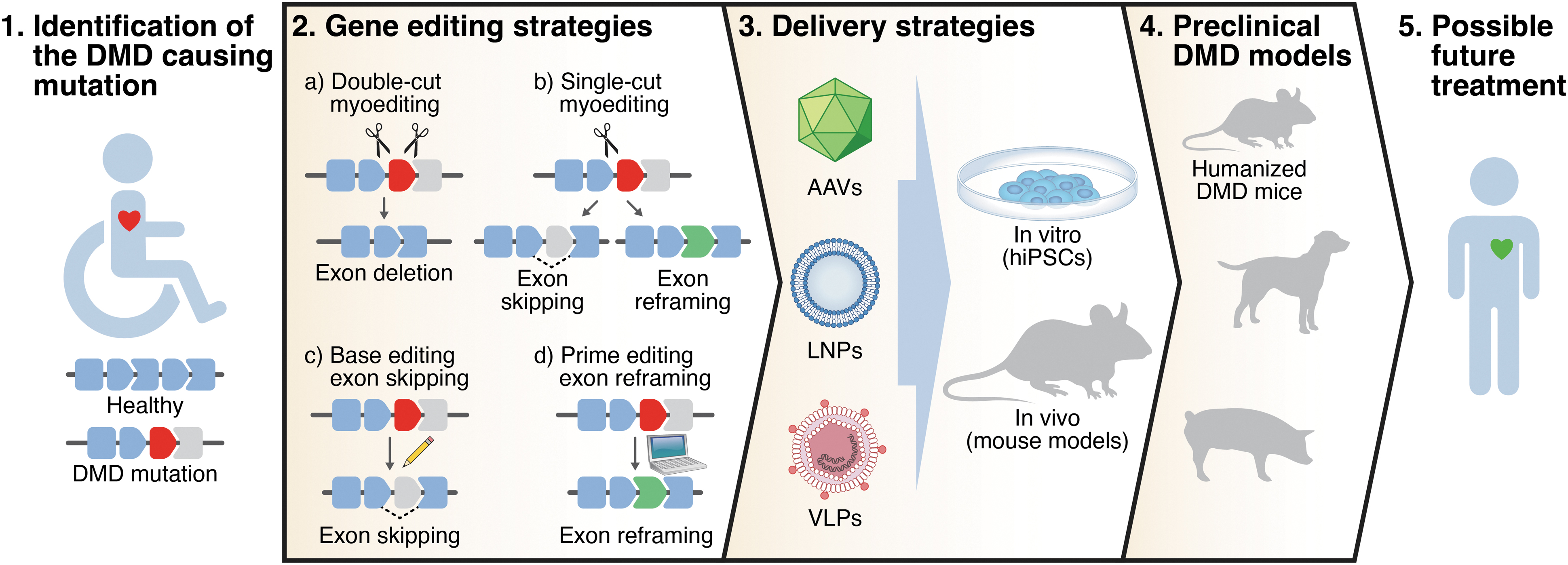
Overview of strategy to correct DMD by myoediting. DMD mutations are identified in DMD patients. Different gene-editing strategies and delivery systems are tested in vitro (in human hiPSCs from patients) and in vivo (in DMD mouse models) to assess myoediting by restoration of dystrophin. Efficacy and safety of myoediting is tested in the appropriate preclinical models with the goal of proposing a therapeutic approach for DMD patients. DMD, Duchenne muscular dystrophy; hiPSCs, human induced pluripotent stem cells.
Double-cut myoediting
Deletion of one or more exons can be used to restore the ORF of DMD. In the double-cut strategy, removal of a target exon(s) is accomplished using two sgRNAs to simultaneously target the introns flanking the exon(s) to be deleted. This approach was extensively applied to restore the DMD ORF in human induced pluripotent stem cells (hiPSCs) from patients and human myoblasts harboring different mutations. 21 –28 Exon deletion is especially suitable to correct exon duplication mutations, as it can restore full-length dystrophin. 25,29 –31 Moreover, as a general approach, double-cut myoediting can excise a multiexonic genomic region, deleting the mutational hotspot of the central rod domain (exons 45–55) and producing a truncated but functional form of dystrophin. 21,32
However, the deletion of numerous exons can impact dystrophin function, and when two sgRNAs are used, exon deletions can generate diverse and unpredictable genome modifications, including exogenous DNA integration or aberrant splicing at the DSB cut sites. 33 Another weakness of the double-cut strategy is the low editing efficiency, due to the necessity of simultaneous cutting using two sgRNAs and the subsequent joining of distant free DNA ends. Two sgRNAs also increases the probability of additional off-target effects. 33
Single-cut myoediting
Limitations of the double-cut myoediting approach can be partially overcome by single-cut gene editing. In this strategy, only one sgRNA generates a single DSB in the proximity of the splice site of the target exon. Repair of the DSB through the NHEJ pathway generates INDELs in the target locus and reconstitution of the correct ORF can occur: (1) by exon skipping if the INDELs destroy the splice consensus site of the exon to be skipped, or (2) by exon reframing if the appropriate number of INDELs occurs in the exonic region. Based on the Leiden DMD mutation database, almost 80% of the DMD patients could benefit from exon skipping, so designing a relatively small number of optimized sgRNA allows efficient correction of most DMD mutations. 34
Indeed, several studies demonstrated high editing efficiency using the single-cut myoediting strategy to restore dystrophin expression in muscle cells derived from hiPSC of DMD patients. 25,35 –39 Another advantage of the single-cut approach is that it decreases the likelihood of off-target mutations since only one sgRNA is used. However, this strategy relies on the endonuclease activity of the Cas9, and generation of DNA DSBs has been shown to be deleterious to cells and can lead to integration events in the genome, if AAV is used as a delivery vector. 33,40 –43
Base editing myoediting
Base editing allows the permanent and precise modification of the genome, without generating a DNA DSB. 44 At the moment, two major classes of base editors are available: cytosine base editors that convert DNA C•G base pairs to T•A base pairs, and adenine base editors (ABEs) that convert DNA A•T base pairs to G•C base pairs. 45,46 Base editors can be used to correct not only point mutations in the DMD gene, but also to induce beneficial exon skipping via an approach termed “single-swap” editing of splice sites. 47 –49 Importantly, while moderate bystander editing is an inherent downside of base editing, this is negated in the single-swap approach as the bystander edits occur in the intron or to-be-skipped exon and not in the mature transcript.
However, as discussed further below, a current major weakness of base editors is their large size that impinges on the limited cargo capacity of the AAV vectors commonly used as delivery systems for myoediting strategies.
Prime editing reframing myoediting
Another nucleotide editing technology, prime editing, can permanently modify the genome without cutting DNA. 50 It consists of a Cas9 nickase fused with an engineered reverse transcriptase. This, in combination with a prime editing guide RNA, can perform targeted and precise small insertions, deletions, or base changing. This technology has been used to correct point mutations in myoblasts of DMD patients. 51,52 In a more general approach, prime editing was recently used to precisely insert the correct number of nucleotides to reframe the DMD ORF in human cardiomyocytes harboring an exon deletion mutation in the DMD gene and restoring dystrophin expression. 48 However, despite the great potential of prime editing to correct DMD causing mutations, up to now, there is no report of its applicability in vivo.
IN VIVO DYSTROPHIN RESTORATION BY GENE EDITING
To test the therapeutic potential of DMD gene editing in vivo, several DMD mouse models representing the most commonly deleted exons in DMD patients (including deletion of exon 43, 44, 45, 50, 51, or 52) were generated by CRISPR technology. 36,37,48,53 These animal models permitted validation of the myoediting strategies initially described in quiescent cells. Plus, they provided an estimate of the in vivo recovery of dystrophin, as well as, evaluation of the safety of gene editing, and optimization of delivery of the gene-editing components.
Inarguably, one of the major challenges of postnatal myoediting is the efficient delivery of the CRISPR editor and the sgRNA to the target tissues: the skeletal and cardiac muscles. Both viral and nonviral delivery systems have been developed for this aim (Fig. 1, “Delivery strategies”). Among the viral systems, AAV is the most commonly used vector for myoediting as it has the following features: (1) low pathogenicity, (2) low immunogenicity, and (3) several serotypes that show tropism for skeletal and cardiac muscles (serotypes1, 6, 8, 9, rh10, rh74). 54 However, AAVs have a packaging cargo size limitation (<4.7 kb) close to the SpCas9 cDNA size (∼4.2 kb). Several groups used a dual-AAV system (one encoding the Cas, the other the sgRNAs), or a single-AAV system with a smaller Cas9 (i.e., Staphylococcus aureus Cas9, SaCas9, or Campylobacter jejuni Cas9, CjCas9) to restore dystrophin expression in different DMD mouse models by double- or single-cut myoediting strategies. 36 –39,53,55 –62
Importantly, AAV delivery permits the utilization of specific promoters (e.g., muscle creatine kinase promoter) to direct the expression of Cas9 only in muscle cells, adding an additional level of cell-specificity editing in addition to viral tropism. 53 One major disadvantage of AAV delivery is the high dosage of the AAV vector(s) necessary for efficient gene myoediting. As an optimization strategy to reduce viral dose, utilization of a self-complementary AAV has been used for expression of the sgRNAs, providing a 20-fold reduction of dose, compared to single-stranded AAV, to achieve efficient gene editing. 63,64
Despite their large size, base editors have been used effectively to restore dystrophin expression in different DMD mouse models. 48,65 –67 To correct nonsense point mutations, ABEs were split into two trans-splicing AAV vectors to overcome the packaging limitation of AAV and then delivered into muscle, or systemically, demonstrating the therapeutic potential of base editing in adult DMD animal models. 65,67 The same packaging strategy was used to demonstrate the efficient recovery of dystrophin in vivo by single-swap base editing of splice sites. 48 In another study, the utilization of the small SaCas9 permitted packaging of a functional base editor in a single AAV and sgRNAs in another AAV (similarly described for the dual-AAV system for single- and double-cut myoediting strategies). 66 However, the development and utilization of an efficient single AAV delivery solution for expression of both base editor and sgRNA (“all-in-one”) is desirable for meaningful and consequential preclinical studies.
As a nonviral delivery system, lipid nanoparticles (LNPs) were used to deliver in vivo gene-editing components to muscle cells to correct DMD causing mutations. Ribonucleoprotein complexes of CRISPR editing components for single-cut editing were encapsulated into LNPs and delivered to skeletal muscle, showing effective, although less efficient, gene editing to restore dystrophin expression. 68 Importantly, it was demonstrated that LNP delivery shows low immunogenicity, allowing repeated local administration into skeletal muscles to induce stable genomic exon skipping and restore dystrophin protein in DMD mice. 69 However, systemic gene-editing correction in skeletal muscles and heart of DMD mice using LNPs has not yet been demonstrated.
FUTURE CHALLENGES
CRISPR gene-editing technologies provide powerful tools for treating genomic mutations of DMD patients that cause the disease, theoretically enabling permanent restoration of dystrophin expression and function of muscle cells. Unlike replacement gene therapy approaches, the gene myoediting strategies enable endogenous regulation of dystrophin, thereby expressing appropriate amounts of protein in the correct tissues. Although myoediting strategies have been successfully applied in human iPSCs and animal models, there are several key obstacles that need to be overcome before considering clinical application. These issues include the following: (1) developing new editing strategies to improve efficiency and reduce dose; (2) affirming safety features of the gene-editing components; and (3) providing testing of myoediting in meaningful preclinical models.
New gene-editing strategies
With the exception of correcting point or duplication DMD mutations, all gene-editing strategies attempt to restore a functional, although truncated, form of dystrophin. These myoediting approaches result in converting a DMD lethal phenotype to a milder Becker phenotype of muscle cells. Fortuitously, in vitro studies report similar protein stability between full-length dystrophin and all in-frame exon-skipped isoforms in the mutational hotspot regions encoding the central rod domain. 70 However, when designing new exon skipping strategies for DMD, the functionality of the resulting Becker-type isoform must be taken into consideration.
Other gene-editing strategies for DMD target overexpression of utrophin, a paralog of dystrophin that can compensate for its functional deficiency. 71,72 In a recent study, a double-cut gene-editing strategy was applied to delete several microRNA binding sites of utrophin in DMD stem cells, thereby inducing overexpression of utrophin to compensate dystrophin deficiency. 73 The main advantage of this approach is that it is applicable to all DMD mutations. However, the level of utrophin overexpression has not been measured in vivo, and due to sequence differences between dystrophin and utrophin, this strategy may lead to a mild Becker phenotype. 74
In the future, gene-editing strategies that can efficiently and effectively restore full-length dystrophin are desirable. Homology-independent targeted integration (HITI) and single homology arm donor mediated intron-targeting integration (SATI) strategies were developed to perform robust DNA knock-in, in proliferating and quiescent cells both for in vitro and in vivo applications. 75,76 HITI technology was shown to edit the genome and restore full-length dystrophin protein in a DMD mouse model with an exon deletion mutation. 77 However, the efficiency was low, and minimal total dystrophin recovery was reported. 77
In addition, several groups have developed new tools and approaches for integration of large DNA fragments into the genome without introducing DNA DSBs, such as CRISPR-associated transposase, insert transposable elements by guide RNA-assisted targeting, twin prime editor, and programmable addition via site-specific targeting elements. 78 –81 Future developments of these technologies to improve their gene-editing efficiency and efficacy in vivo could lead to new potential therapies that restore the full-length dystrophin protein.
Safety
Up to now, the biggest obstacle of translating gene-editing therapies for DMD to the clinic is efficient and safe delivery of the gene-editing components to muscles. AAV has provided the most effective means of systemic delivery of gene-editing components; however, it is crucial to further examine the safety features of this viral vector for application of CRISPR for clinical application for DMD patients. In fact, a high dose of AAV is required for editing all the muscles of the human body. However, high dose of AAV can trigger adverse events in multiple organs, such as liver toxicity. 82 To lower viral dose, new AAV capsid variants with enhanced muscle tropism, such as AAVMYO and MyoAAV, are being developed. These AAV variants have the potential to lower the viral dose by at least 10-fold, offering a safer therapeutic dose. 83 –85
Moreover, as previously described, the dual-AAV system, which is the most popular method for in vivo delivery of the gene-editing components, necessitates using a high dose of AAV for efficient gene editing. 36,37,39,53,55 –60,62 The development of new “all-in-one” AAV vectors that combine small Cas9 variants and sgRNAs in the same vector would not only reduce the amount of viral dose but would also address the problem related to manufacturing different clinical-grade AAV vectors in high quantity. 38,61 However, several studies with the dual-AAV system have demonstrated that efficient systemic editing requires higher amounts of the AAV encoding the sgRNAs, and this ratio difference can only be achieved with the dual-AAV approach. 36,86,87
Besides AAV vectors, virus-like particles (VLPs) have recently been created to deliver mRNA of gene-editing components to correct mutations causing DMD in vivo. 88 The possibility of modifying the viral envelope to target specific cell types will allow further development of new delivery strategies based on VLPs. 89,90 However, due to the limited publications using this new delivery strategy, more studies are necessary to assess its safety.
Concerns over the immune response of humans to CRISPR gene-editing therapy components remain a safety issue. An immune response can be evoked not only by the AAV vectors but also by the Cas proteins. Cas9 is a bacterial protein and the presence of Cas9-specific antibodies has been detected in human plasma. 91 It was also reported that sgRNA may trigger an innate immune response within human cells in vitro, but it has not been demonstrated whether sgRNAs induce an immune response in vivo. 92,93 Furthermore, more studies are needed to assess specific post-transcriptional modifications of sgRNAs that were administered systemically to determine if these modifications could induce an immune response in vivo. 93 Importantly, an immune response against AAV and Cas9 has not been detected in neonatal mice, suggesting that an immune response may be avoided by treating humans at early ages. 33
Another safety concern of gene-editing therapy is the potential of off-target activity. Different bioinformatic investigative methods have been developed to detect off-target effects, such as Digenome-seq, GUIDE-seq, and CIRCLE-seq. 94 –96 Although off-target activity has been observed with gene editing in proliferating cells in tissue culture, it is documented to be minimal in animal models by in vivo studies, especially in postmitotic muscle cells. 97 Nonetheless, off-target activity by CRISPR gene editing can be further reduced using high fidelity Cas9 or by optimization of the sgRNA. 98 –105
Preclinical animal models
Generating and using appropriate preclinical DMD animal models will allow optimization of gene-editing strategies by assessing delivery of the gene-editing components, and evaluating the outcome of DMD myoediting in vivo (Fig. 1, “Preclinical DMD models”).
Mouse and human dystrophin proteins are highly conserved in exon composition and amino acid sequence. However, the mouse and human dystrophin genes vary at the genomic level and the differences in nucleotide sequence impede testing of human-specific sgRNAs in DMD mouse models. Creation of humanized DMD mouse models allows for preclinical trials to evaluate the efficiency of genome editing of human-specific sgRNAs in an animal model for eventual use of the sgRNA in clinical trials. A humanized mouse was generated, in which the entire human DMD sequence was integrated into mouse chromosome 5 and subsequently mutations were introduced to recapitulate the dystrophic phenotype. 60,106,107 However, a recent study revealed that this humanized DMD mouse model carries two copies of the human DMD transgene at the integration locus. 108
A new generation of humanized DMD mouse models were generated in which mouse exon(s) were replaced by human ortholog(s) within the endogenous genomic location on the X chromosome. 39,69,109 These chimeric mouse models containing human DMD genome sequence allow testing of gene-editing efficiency of human-specific sgRNAs for single-cut or base editing correction strategies. 39,69,109 In the future, it would be useful for preclinical studies to establish additional humanized DMD mouse models by substituting all the therapeutically relevant exons with human sequence.
DMD mouse models are helpful tools to test gene-editing strategies as they mimic the pathological features of the diseases. However, due to the compensatory effects of utrophin and muscle regeneration, they do not fully recapitulate the phenotype of DMD patients. 110 For this reason, therapeutic (and also delivery) strategies need to be optimized in larger animals and more pathologically severe DMD mammalian models, before clinical application. 111 Although one study suggested that Cas9 immunity was induced by AAV delivery vectors in large mammals, encouragingly two other large animal applications of gene editing, one in a DMD dog model and another in an induced DMD pig model, efficiently restored dystrophin protein after systemic administration of AAV9, validating therapeutic gene editing in large animals. 27,112,113
Another important variable to consider before moving DMD gene myoediting into the clinic is the timing of gene-editing correction. All in vivo DMD editing studies that show efficient dystrophin restoration have been performed by administering the editing components to young animals, suggesting that early intervention is effective and most likely better than delivery of AAV CRISPR to older animals. In fact, administration of AAV CRISPR to younger patients is preferable because (1) a lower amount of AAV CRISPR is needed for younger animals due to the smaller size; (2) young patients are less likely to have pre-existing immunity against AAV9; and (3) younger muscles are better preserved, showing minimal pathological features of dystrophic muscles. Following administration, the durability and longevity of the gene-editing correction has to be monitored.
Long-term studies after double-cut gene editing showed skeletal muscle editing at 12 or 18 months after editing. 33,86 Recently, a study using single-cut myoediting reported lifelong expression of dystrophin in myoedited mice and corrected skeletal muscle was highly resistant to necrosis and fibrosis, plus showed sustained dystrophin expression in response to chronic injury. 114
Due to the high turnover rate of DMD muscles, the ability to deliver the gene-editing components to satellite cells, the muscle stem cells, would likely improve and sustain dystrophin expression in myoedited muscle. In this regard, AAV infectivity of satellite cells has been controversial and needs further investigation and development. 56,115 –118 Encouragingly, MyoAAV transduces satellite cells with about three times higher efficiency compared to AAV9, and in the future, the utilization of a satellite cell-specific promoter to drive the gene-editing components could improve and advance gene editing of DMD. 84
CONCLUDING REMARKS
Different CRISPR myoediting strategies have been successfully applied in DMD human iPSCs and DMD animal models, providing new, promising, effective treatments for DMD. The efficacy of gene-editing technologies is no longer an obstacle for the treatment of DMD in clinical applications. Considering all the recent preclinical successes obtained with CRISPR systems, it seems reasonable that the remaining safety and delivery challenges will be overcome in the next few years.
Footnotes
ACKNOWLEDGMENTS
We thank J. Cabrera for his creative assistance with the graphics.
AUTHORs' CONTRIBUTIONS
F.C. wrote the initial draft, and E.N.O. and R.B.D. edited the article. All authors revised the article and approved it for publication.
AUTHOR DISCLOSURE
E.N.O. is a consultant for Vertex Pharmaceuticals and Tenaya Therapeutics. The other authors declare no competing interests.
FUNDING INFORMATION
This work was supported by grants from the National Institutes of Health (HL-130253, HD-087351, HL-157281) and the Robert A. Welch Foundation (grant 1-0025 to E.N.O.).