
Abstract
The ability to specifically, safely, and efficiently transfer therapeutic payloads to the striated musculature via a minimally invasive delivery route remains one of the most important but also most ambitious aims in human gene therapy. Over the past two decades, a flurry of groups have harnessed recombinant adeno-associated viruses (AAVs) for this purpose, carrying cargoes that were packaged either in one of the various wild-type capsids or in a synthetic protein shell derived by molecular bioengineering. In this study, we provide an overview over the most commonly used techniques for the enrichment of muscle-specific (myotropic) AAV capsids, typically starting off with the genetic diversification of one or more extant wild-type sequences, followed by the stratification of the ensuing capsid libraries in different muscle types in small or large animals. These techniques include the shuffling of multiple parental capsid genes, peptide display in exposed capsid loops, mutagenesis of individual capsid residues, creation of chimeras between two viral parents, or combinations thereof. Moreover, we highlight alternative experimental or bioinformatic strategies such as ancestral reconstruction or rational design, all of which have already been employed successfully to derive synthetic AAV capsids or vectors with unprecedented in vivo efficiency and/or specificity in the musculature. Most recently, these efforts have culminated in the isolation of unique clades of myotropic vectors called AAVMYO or MyoAAV that have in common the display of the amino acid motif RGD (arginine–glycine–aspartate) on the capsid surface and that exhibit the highest transduction rate in striated muscles of mice or nonhuman primates reported to date. Finally, we note essential looming improvements that will facilitate and accelerate clinical translation of these latest generations of myotropic AAVs, including the identification and utilization of capsid selection or validation schemes that promise optimal translation in humans, and continued efforts to enhance patient safety by minimizing hepatic off-targeting.
INTRODUCTION
Among the many possible combinations of clinical targets and indications with available gene delivery vehicles, the juxtaposition of striated musculature and adeno-associated virus (AAV) may be one of the most exciting and most promising, for a variety of reasons. First and foremost, these include the many debilitating and often life-threatening human disorders that originate in the musculature, including the skeletal muscle, heart, and diaphragm, such as Duchenne muscular dystrophy (DMD) or X-linked myotubular myopathy, and for which no cure exists, thus raising a large and urgent, unmet clinical need. The latter is reflected by the flurry of recent clinical trials targeting these diseases, especially DMD, such as CIFFREO (AAV9, NCT04281485; Pfizer, Inc.), ENDEAVOR (AAVrh.74, NCT04626674; Sarepta Therapeutics), or IGNITE (AAV9, NCT03368742; SOLID Biosciences), some of which have indeed already demonstrated functional improvements in treated individuals.
Second, skeletal muscle is not only a prime target for transfer of therapeutically relevant genes or gene editing machinery but also an excellent platform and reservoir for the expression and subsequent secretion of clinically relevant proteins such as blood clotting factors or neutralizing antiviral antibodies. Third, probably no other currently available, nonviral or viral gene delivery system provides such an enormous amenability to bioengineering and repurposing as does AAV, combined with roughly six decades of experience with the wild-type virus and over four with recombinant AAV gene transfer vectors.
This, in turn, raises significant hopes that optimized AAV vectors can achieve widespread, specific, and efficient delivery to a large volume of muscle types, ideally from limited doses, minimally invasive peripheral delivery, despite the presence of neutralizing anti-AAV antibodies as they circulate in the human population, and while detargeting non-muscle tissues, especially the liver that normally acts as a sink for circulating AAVs. The importance of the latter is drastically and tragically illustrated by a string of recent serious adverse events, including multiple fatalities in children, in different clinical studies that employed high doses of vectors based on wild-type capsids that were not yet optimized for muscle-specific gene delivery. 1
To date, an ideal AAV vector that concomitantly circumvents all these challenges does not exist; instead, the present variants typically merge biological characteristics that can be both beneficial and detrimental to clinical applications and affected patients. Still, there is every reason to believe that this goal will ultimately be reached in view of the ever expanding and improving portfolio of techniques for bioengineering and in vivo biopanning of next-generation myotropic AAV vectors, which is the topic of the present article.
LOW- OR HIGH-THROUGHPUT EVALUATION OF WILD-TYPE AAV VARIANTS EX VIVO OR IN VIVO
The simplest and, therefore, most widely used strategy to repurpose AAV vector particles including (re)targeting to the musculature is to explore the vast sequence space of naturally occurring AAV capsid variants (Fig. 1, top). To this end, a given AAV vector genome is cross-packaged into the desired wild-type capsid, by merely expressing the corresponding cap gene, for example, from an AAV helper plasmid during vector production. Owing to the ease of this approach, it was readily implemented and harnessed in the late 1990s and early 2000s, once a flurry of viral isolates had been identified beyond the AAV2 prototype and became available as molecular clones, and before the implementation of capsid engineering methodologies.
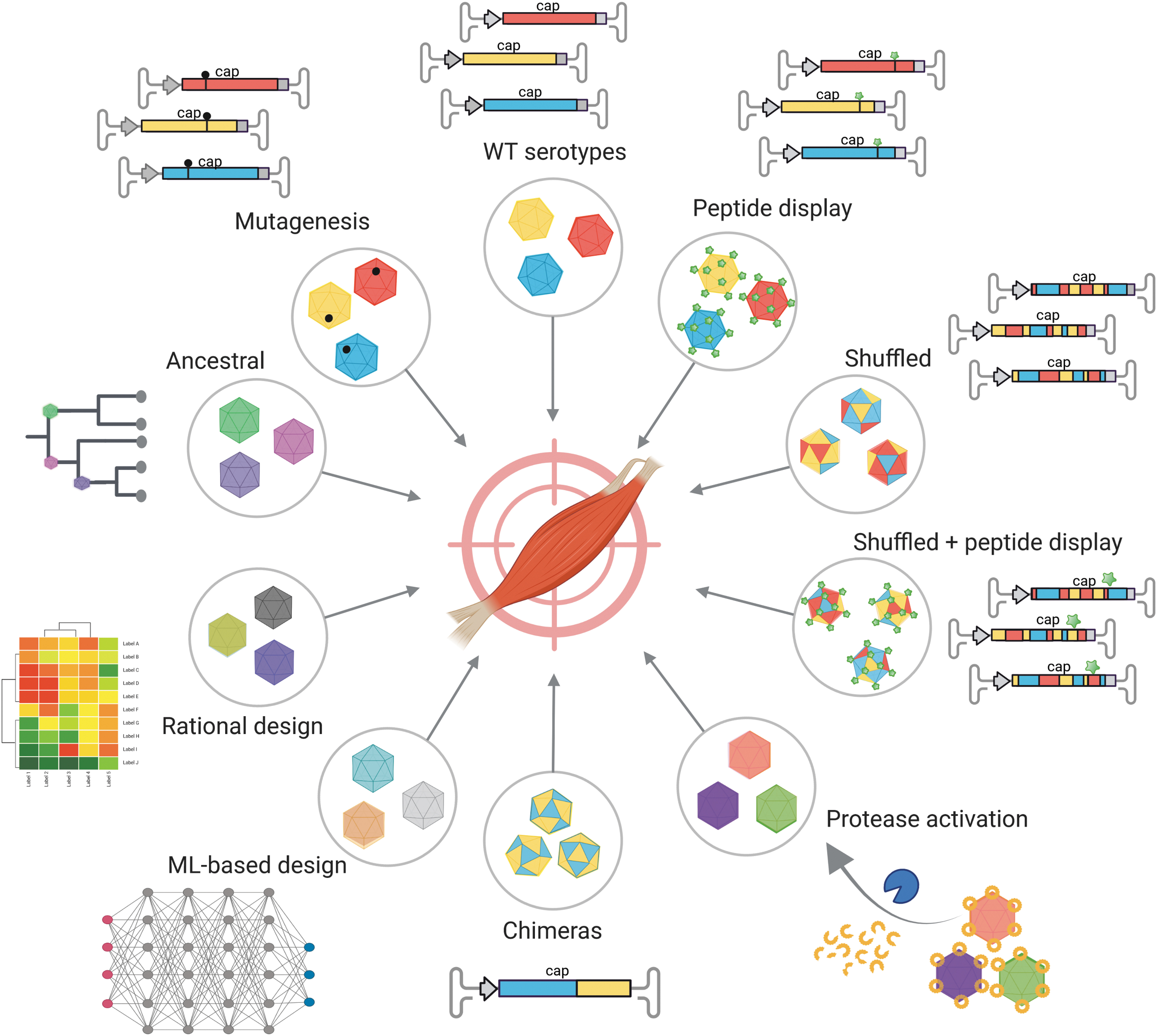
Technologies to derive AAV capsid variants for muscle gene transfer. Techniques to identify or derive myotropic AAV capsids include (clockwise, starting from top) the use of wild-type AAV isolates, peptide display in various serotypes, DNA family shuffling of AAV cap genes, combinations thereof, locking of capsids with protease-activatable peptides, creation of chimeras by domain-swapping between two parental viruses, rational capsid design including machine learning (ML)-based approaches, ancestral reconstruction of putatively extinct capsids, and mutagenesis of individual critical residues in AAV capsid proteins. AAV, adeno-associated virus. This figure was created with BioRender.com
Because of the wealth of studies that have capitalized on this technology over the past two decades, and for space reasons, we will only highlight a few selected examples below. A particular focus is put on studies that reported comparative evaluations of at least two different natural capsid variants under largely identical experimental conditions and that exemplify the parameters that typically govern the outcome of AAV muscle gene transfer, including delivery route, tests in cells or animals, healthy versus diseased study objects, vector dose, or expression strength and kinetics.
For instance, representative early studies in this category include the work by Xiao et al. 2 and Duan et al., 3 who demonstrated the superiority of AAV1 or AAV5, respectively, over AAV2 in the murine striated musculature following direct intramuscular vector administration. Interestingly, Duan et al. not only compared the two viral isolates in healthy mice but also in a mouse model of DMD. While AAV5 also outperformed AAV2 in these mice, overall vector efficiency was reduced compared with wild-type mice with a GFP (green fluorescent protein) but not a luciferase reporter, highlighting the pivotal role of the vector cargo in assessments of in vivo vector performance. A higher resistance of dystrophic mouse muscle to AAV transduction was also reported by, for instance, Liu et al., 4 illustrating the need to evaluate AAV vectors in the physiologically most relevant system.
Further notable in the study by Duan et al. is the analysis of these two AAV serotypes in undifferentiated versus differentiated murine myoblasts (C2C12 cells), which revealed a much higher efficiency of AAV5 in the differentiated cells, and a preference of AAV2 for the undifferentiated myoblasts. 3 These findings were independently confirmed by Mori et al. 5 and Hildinger et al., 6 who also reported a better performance of AAV5 over AAV2 ex vivo or in vivo, respectively.
Finally, a third interesting aspect of this work is that it is one of the first to provide insights into the mechanisms possibly underlying the differential performance of AAV capsid variants in the musculature. Specifically, data reported by Duan et al. imply that the higher efficiency of AAV5 does not necessarily correlate with better binding to muscle cells but instead appears to be related to improved intracellular processing.
Beyond AAV2 and AAV5, numerous groups have reported pairwise comparisons of other combinations of two natural AAV isolates in muscle cells and/or animals. Noteworthy examples include a study by Kawamoto et al., who demonstrated the superiority of AAV6 over AAV2 in the rat heart following intracoronary delivery of either vector. 7 This is consistent with even more comprehensive work by Blankinship et al., who found up to 500-fold better transduction of the murine musculature with AAV6 over AAV2 from multiple injection routes, including intramuscular, intraperitoneal (in neonatal mice), or intrathoracic; the latter resulted in particularly efficient gene transfer to the diaphragm. 8 Notable in this context is also a string of meticulous studies by Gregorevic et al., which consistently reported a very robust performance of AAV6 in different systems comprising healthy or dystrophic mice and dogs. 9 –11 Additionally, these authors showed an enhancing effect of vascular permeabilizing agents such as VEGF, and a further increase in muscle specificity through the use of muscle-specific promoters such as CK6. 10
Others such as Su et al. compared AAV1, a natural isolate that is very closely related to AAV6, with AAV2 in the mouse or pig myocardium and found AAV1 to be superior in both species. 12,13 An alternative pair are the cynomolgus macaque isolates AAV10 and AAV11, which were shown by Mori et al. to transduce the murine musculature following intravenous or intramuscular (only AAV10) delivery. 5 Ex vivo, both isolates preferred differentiated mouse C2C12 myoblasts over undifferentiated cells, again in contrast to AAV2 as also noted by others (see above). 3
As a third example, our own group compared the pig isolate po.1 versus AAV9 in peripherally injected mice. 14 An important finding was that po1, while being slightly inferior to AAV9 in striated muscles, exhibited a much lower degree of unwanted off-target transduction in the liver, implying a lower risk of adverse hepatotoxicity. This bias toward the musculature was further improved through combination with the muscle-specific promoter Spc5–12, again exemplifying the potential of merging transductional (capsid) and transcriptional (promoter) targeting. Of note, the Kobinger laboratory who had originally isolated po1 later also reported a good performance in the murine musculature for several other pig AAV isolates, including po2.1 and po4 through po6. 15
Two other important and promising AAV serotypes that were frequently compared with one or more alternative AAV isolates, and often found to be superior, are AAV8 and AAV9. For instance, Katwal et al. juxtaposed AAV9 with AAV1 in an intravenously injected mouse model of peripheral arterial disease and found AAV9 to work better in the ischemic muscle. 16 Intriguingly, it was inferior to AAV1 in the normal muscle, implying that ischemia-induced desialylation of galactosylated glycans (the AAV9 receptor) synergizes with higher vascular permeability and thereby fosters transduction with AAV9. Besides, also in this work, muscle specificity was improved further through incorporation of the myotropic CK6 promoter.
Multiple other groups, such as the Nakai or Duan laboratory, 17,18 directly compared AAV8 versus AAV9 in different animals and often found them to be similar, or one slightly better than the other. Intriguingly, however, Pan et al. not only detected a better performance of AAV8 over AAV9 in the neonatal dog heart but also a substantially lower activity of AAV9 in dogs versus mice, even when extremely high doses of almost 1 × 1015 vector genomes per kilogram were applied to dog puppies. 18 This surprising finding once again underscores the differences in preclinical animal models and the associated difficulties in identifying the physiologically most relevant species that is most predictive for translation in humans.
Finally, we note a plethora of studies in which three or more natural AAV isolates were compared side-by-side in different animal species, and whose power and potential we can again only highlight with selected examples due to limited space. These include several comparisons of AAV1 through AAV5 in healthy or ischemic mice, 12,19 –21 following intramuscular or intracoronary injection, or via ex vivo transduction of murine versus human cardiomyocytes. 20 Many of these studies consistently identified AAV1 as the best of these five viral serotypes, although there were notable exceptions such as the finding by Du et al. of a better AAV2 performance in neonatal murine cardiomyocytes. 20
The inferiority of AAV2 was further confirmed by us and others in expanded in vivo comparisons also including AAV6. 22,23 A particularly notable example is work by Müller et al. who improved cardiac targeting through the use of the myotropic myosin light chain promoter MLC1.5 (combined with a CMV enhancer) or by mutating two critical residues (R484E, R585E) in the AAV2 capsid. 23 The latter ablated binding to the AAV2 receptor heparan sulfate proteoglycan (HSPG) and thus resulted in liver detargeting, in turn increasing cardiac specificity.
Additional combinations of three or more AAV serotypes were dissected by numerous other colleagues, for example, Wang et al. (AAV1, 2, 5–8; delivered intraperitoneally, intravenously, or intramuscularly in neonatal or adult mice and hamsters), 24 Pacak et al. (AAV1, 8, 9; mice and nonhuman primates), 25 Louboutin et al. (AAV1, 2, 5, 7, 8; mice), 26 Wang et al. (AAV1, 2, 5 through 9; hemophilic mice), 27 Prasad et al. (AAV1, 2, 6, 8, 9; neonatal mice), 28 Rivière et al. (AAV1, 2, 5; mice), 29 Li et al. (bat AAV10HB, AAV2, 8; mice), 30 or Gao et al. (AAV1, 2, 5, 7, 8; mice; this is also the first report of AAV7 and 8). 31 Last but not least, we highlight studies that even compared eight or nine serotypes to each other, such as Zincarelli et al. who repeatedly assessed AAV1 through 8 (or 9) in mice, 32,33 or Palomeque et al. who compared AAV1 through 8 in the rat heart. 34
Notwithstanding the simplicity of the strategy to harness natural AAV serotypes, and the concurrent complexity enabled by the variation of seminal parameters such as injection route or test species, this technology also has a series of limitations that restrict its clinical potential. First, none of the extant endogenous AAV isolates appears to be perfect for use in humans, especially considering their frequently observed promiscuity in non-muscle tissues in the absence of additional measure such as transcriptional or post-transcriptional targeting. Second, the typical interanimal variability including the differential presence of neutralizing anti-AAV antibodies in higher species complicates comparison of data obtained with distinct AAV variants in different subjects, even when experiments are performed under standardized conditions and using high animal numbers per cohort.
Third, and related to this, these comparisons of individual AAV variants are inherently restricted due to their demand for large animal numbers and, in turn, due to their low throughput. Fortunately, over the past decade, a solution to most of these challenges has been implemented in the form of capsid barcoding technology, 35 –38 which nowadays enables a comparison of dozens or hundreds of capsid variants in the same animal and thus under identical conditions. Furthermore, despite the inherent limitations of the original screening approach, it must be pointed out that it has successfully identified natural AAV isolates that have yielded encouraging clinical data in patients with different muscle disorders. Still, the recent series of severe adverse events including multiple fatalities in children from high doses of wild-type AAV8 or AAV9 drastically illustrate the urgent need to bioengineer better—more efficient, more specific, ideally regulatable, and thus overall safer—synthetic AAV variants, using techniques that are outlined in the following paragraphs.
AAV CAPSID DIVERSIFICATION THROUGH CAP GENE SHUFFLING
One of the first techniques for high-throughput AAV capsid engineering that was implemented about two decades ago and has since been harnessed extensively for molecular evolution of AAV vectors in the musculature and elsewhere is DNA family shuffling. 39 –41 In essence, this technique utilizes enzyme-based fragmentation of at least two different parental AAV cap genes, followed by their reassembly into a library of chimeric sequences composed of various fragments of the parental viral isolates according to their partial DNA homology (typically more than 80% for most AAV isolates). This library is then iteratively selected in a target cell line or organ; in the latter case, typically through local or systemic administration in small or large animals, followed by rescue of all successful cap genes after, for example, 2 weeks, production of a secondary library and a new cycle of selection.
In one of the first representative studies that applied this technique, the Samulski laboratory shuffled AAV1 through 9 and then selected the library ex vivo on a Chinese hamster melanoma cell line. 41 Not surprisingly, following intramuscular delivery into mice, the lead candidate (a chimera of AAV1, 2, 8, and 9) was inferior to the AAV1 and AAV2 benchmarks, reflecting its selection in tumor cells. The same observations were made in our concurrent report in which we described a chimera composed of AAV2, 8, and 9 and selected ex vivo in liver cells, that likewise did not outperform the benchmarks in the mouse musculature (i.e., a heterologous target) following systemic delivery. 39 Curiously, another chimera called B1 and isolated from a library composed of 11 different capsids through selection in the mouse brain performed well in the musculature of mice and cats, and even surpassed AAV9 in mice at least on the level of vector genomes in the muscle. 42
Interestingly, when Yang et al. independently created a library composed of AAV1 through 9 and then purposely biopanned it in the skeletal muscle in mice, this yielded a predominant clone called M41 and assembled from AAV1, 6, 7, and 8. 43 Compared with the AAV6 and AAV9 benchmarks, M41 showed a better or comparable, respectively, transduction of the mouse heart from systemic administration, and in both cases, a substantially improved liver detargeting. Notably, the M41 chimera was also found to be more resistant to neutralization by pooled intravenous immunoglobulins (IVIg), raising hopes that it could also evade antibody-mediated neutralization in a wider patient population.
The latter was actively used as a selection pressure by Li et al., 44 who screened their original library of AAV1 through 9 chimeras directly in the mouse skeletal muscle in the presence of neutralizing anti-AAV antibodies. 41,44 This led to the successful isolation of six shuffled capsid variants that outperformed most of the wild-type benchmarks (AAV2, 6, 8, and 9) in the mouse skeletal or heart muscle following intramuscular or intravenous delivery, and of which five displayed the ability to largely escape neutralizing antibodies. Further notable about this work is the demonstration how phylogenetic analysis coupled with structural modeling of shuffled AAV chimeras can inform the identification of capsid motifs that govern tissue tropism or antibody resistance.
Finally, we highlight comprehensive work by Paulk et al. from the Kay laboratory, 45 who shuffled 10 different AAV wild types and then simultaneously selected the resulting library 6 times in pools of stem cells and myotubes derived from surgically resected human skeletal muscle cells from 5 patients. This yielded several chimeras that had maintained 6 of the initial 10 isolates, and of which the best producers were taken further. Notably, when benchmarked against several wild types (AAV1, 6, and 8) in four different cell types (human muscle stem cells or myotubes, normal or dystrophic mouse myoblasts), the chimeras mostly performed better, although results differed depending on species, cell type, and disease state.
Moreover, they were significantly more resistant than the parental wild types to neutralization by 50 serum samples from healthy adults. Importantly, the chimeras also performed well in primary muscle explants from five human individuals as well as in an explant from a rhesus macaque, underscoring their great clinical potential. Finally, akin to the earlier work by the Samulski laboratory, 44 the data reported by Paulk et al. enabled them to speculate on sequence or structure motifs that contributed to the observed seroreactivity and muscle tropism. 45
As a whole, these representative studies exemplify the enormous potential of DNA shuffling technology to concurrently optimize multiple capsid features with relevance for muscle gene therapy in humans, that is, tropism toward the musculature, detargeting from the liver and other non-muscle tissues, in turn ideally improving patient safety and relieving the burden on vector manufacturing, as well as improved escape from neutralizing anti-AAV antibodies. At the same time, these reports also showcase the inherent limitations of this technique that it shares with all other strategies for molecular AAV capsid evolution, such as the dependency on an artificial ex vivo or in vivo selection model whose translatability in humans cannot be guaranteed, and the need to go through multiple iterative selection rounds, which invokes labor, costs, time, and ethical concerns in cases where animals are used.
AAV CAPSID REPURPOSING THROUGH PEPTIDE DISPLAY
A second methodology for AAV capsid diversification that is technically simpler but still very powerful is peptide insertion, that is, the addition of short, usually around seven amino acids-long foreign sequences into exposed capsid loops, typically hypervariable regions (HVR-)IV or VIII. Originally established in AAV2 owing to the fact that its three-dimensional structure including the position of the most exposed residues in the two HVRs has been resolved first, we and others subsequently adapted this technique also for numerous other, wild-type, or synthetic AAV capsids. 39,46,47 Because of the simplicity of this technology that merely requires a single molecular cloning step granted the structure of the underlying capsid is known, and because it has been established already two decades ago, 48,49 the corresponding literature including its use for the isolation of myotropic vectors is enormous, and we can unfortunately only note a few selected studies in the context of this article that illustrate its principle and potential.
One is a study by Yu et al. from the Xiao laboratory in which the muscle-targeting peptide ASSLNIA, previously isolated from a phage peptide display library, was inserted in two adjacent positions within the AAV2 HSPG binding site, 587 and 588. 50 Indeed, in particular, the 587 insertion, which increases the spacing of the two HSPG-binding arginines R585 and R588, disrupted heparin binding, potentially explaining why it was slightly inferior to AAV2 upon direct intramuscular injection in mice. Notably, however, the 587 insertion mutant displayed a higher affinity for various muscles and heart after systemic delivery, combined with reduced transduction in the liver and other off-targets.
Similarly, Lee et al. inserted an acidic six-L aspartic acid motif (D6) into position 587 of AAV2 and showed that it likewise reduced heparin binding and liver targeting in mice, while increasing gene expression in the musculature. 51 Curiously, in vivo transduction occurred primarily in paraspinal and gluteus muscles, perhaps explained by the fact that the negative charges in D6 sequestered the particles to hydroxyapatite in the bone, in turn resulting in a temporal immobilization of the vectors and their subsequent uptake by adjacent muscle tissue, since AAV2–D6 does not infect the bone marrow itself. Irrespective of mechanism, this study is another example for the potential of this simple yet effective technology.
A third example that further corroborates this conclusion and concurrently documents its capability with high-throughput screening is recent work by Rode et al., in which iterative biopanning of an AAV2 library displaying randomized seven amino acid-long peptides in HVR-VIII (position 587) was performed in the mouse heart. 52 This yielded two lead candidates that outperformed the parental wild-type AAV2 in cardiomyocytes in vivo and that gave gene expression comparable to AAV9 in the heart but coupled with better liver detargeting, resulting in an improved target-to-noise ratio. Further beneficial with respect to clinical translation is the higher resistance of the peptide display variants to anti-AAV2 antibodies and their compatibility with AAV9 during in vivo vector readministration, as demonstrated in this work.
Finally, we point out a series of recent reports from our own laboratory in which we have engineered over a dozen AAV capsid variants for display of preselected or randomized peptides and have then harnessed the ensuing libraries for ex vivo or in vivo biopanning in different formats, including in combination with DNA/RNA barcoding technology. 35,36,38,46 The power of this technique, first introduced into the AAV field on the DNA level in 2014 by the Nakai laboratory, 35 is that the identical expression cassette (except for the barcode) is used in all capsids that are compared, ensuring that any differences observed primarily originate from the viral protein shell. In turn, this reduces interanimal variability and largely eliminates confounding effects from other hard-to-control parameters, such as variations in injection efficiency or accuracy for each individual variant.
With respect to this article's topic, most notable is the 2020 study by Weinmann et al. in which we had individually barcoded more than 180 capsid variants comprising numerous peptide display or shuffled capsids as well as wild-type benchmarks and then screened the ensuing libraries in all major tissues and numerous cell types in adult mice. 38 This yielded one particularly striking AAV9 mutant displaying the 7-mer RGDLGLS in HVR-VIII and dubbed AAVMYO, which mediated highly efficient gene expression from systemic delivery in all striated muscle types tested including forelimb and hind limb skeletal muscle, heart, and diaphragm.
This was combined with robust liver detargeting to an extent not seen with any of the natural or synthetic variants that were assessed in parallel. Importantly, AAVMYOs superior efficiency and specificity in the muscle are conserved across numerous mouse strains and injection routes [intravenous and intramuscular (manuscript submitted)], and it enabled phenotypic benefits in various mouse models of human muscle disorders, including X-linked myotubular myopathy and DMD. Mechanistically, the RGD (arginine–glycine–aspartate) motif displayed on AAVMYO likely interacts with integrin alpha and beta subunits on the surface of target cells, as experimentally supported by recent independent studies implying binding to βv and β3, β5, or β8. 53
Last but clearly not least, we note a comprehensive 2021 study from the Sabeti laboratory that further corroborated the importance of RGD motifs in an AAV9 context, as discussed together with our own preceding study in a recent commentary. 54 In their study, Tabebordbar et al. created and biopanned AAV9 libraries displaying either fully randomized 7-mers or peptides in which the RGD motif was already fixed (in the same position as in the AAVMYO peptide) 38 in mice or nonhuman primates. 37 This led to the enrichment of various RGDXXXX (X = any amino acid) motifs, of which selected candidates called MyoAAV mediated robust gene expression in the murine and/or primate musculature following peripheral delivery.
Notable about this work is not only the direct in vivo comparison of AAV variants of small and large animals but also the multiple mechanistic insights that were provided including potential binding to yet another integrin, αvβ6, which is expressed on the surface of mouse, human, and nonhuman primate muscle cells. Furthermore, in line with prior studies from us and others, 46 the authors provided compelling evidence for the importance of the amino acids flanking the inserted RGD motifs, which together informs future efforts at peptide-mediated AAV bioengineering. Finally, this work is additionally noteworthy for its use of a technology called DELIVER that enables the in vivo selection of functional AAV particles in a library on the (m)RNA level, as opposed to conventional polymerase chain reaction (PCR)-based rescue on the DNA level.
As recently shown by several groups 55,56 and confirmed by Tabebordbar and colleagues, 37 an RNA-based rescue substantially increases chances of enriching desired capsid variants that are capable of undergoing the full transduction cycle including uncoating and transgene expression in the nucleus. This is in stark contrast to DNA-based strategies that are prone to enriching false positives that merely attach to target cells or enter ineffective pathways without actually delivering the cargo to, and expressing it in, the nucleus.
SEMI-RATIONAL CAPSID BIOENGINEERING BY COMBINING DNA SHUFFLING AND PEPTIDE DISPLAY
A recent study by El Andari et al. from our own laboratory illustrates the synergism of combining two molecular evolution technologies, in this case, DNA family shuffling and peptide display, especially when merged in a semi-rational manner. 36 Briefly, we had originally shuffled four (AAV1, 6, 8, and 9) or five (the same four plus AAVpo1) wild types and then selected the ensuing two libraries in the skeletal muscle, heart, and diaphragm of mice over two to three rounds. This enriched a series of chimeric capsids primarily composed of AAV1, 6, 8, and 9, which, when simultaneously validated using barcoding technology, displayed robust activity in the musculature often combined with targeted transduction of the liver. Still, none of them surpassed the AAVMYO capsid that we had previously isolated in an independent study. 38
However, our subsequent notion that the most interesting chimeras all shared an exterior capsid portion derived from AAV9 with AAVMYO then motivated us to transplant the myotropic peptide RGDLGLS (also called P1) from the former lead into two selected chimeras from our latest study, resulting in capsids AAVMYO2 and AAVMYO3. Indeed, when delivered peripherally, these second-generation myotropic AAVs exhibit a desired and superior combination of (1) robust efficiency, (2) high specificity in all striated muscle groups, as well as (3) significant detargeting from the liver as the most frequent and most critical off-target in systemic gene therapies.
Accordingly, they enabled a substantial improvement of disease-associated phenotypes in two mouse models of X-linked myotubular myopathy and DMD, and one of them has recently also produced significant benefit in a novel rat model of Pompe disease (revised manuscript submitted). Last but not least, preliminary data imply that the ability of AAVMYO2 and especially of AAVMYO3 to detarget the liver is also maintained in nonhuman primates, to an extent not seen with an assortment of wild-type or synthetic myotropic AAVs that were tested in parallel (Zayas and Grimm, unpublished).
While this study and others that have merged multiple capsid evolution techniques illustrate the unique potential of such combinatorial approaches, it must also be noted that success is never guaranteed, in particular when heterologous targeting moieties such as peptides are transferred between different capsid scaffolds. In fact, while insertion of the RGDLGLS peptide retargeted most of the wild-type or chimeric capsids tested in our work to the musculature, it concurrently yielded unexpected effects in others, such as AAVrh10, which was broadly enhanced in many tissues. 36 This once again reflects our gaps in the understanding of AAV biology and the limitations of top-down screening approaches, whose results are mostly unpredictable and often surprising.
Ideally, these gaps will be closed in the future as our knowledge of the AAV infection or transduction mechanisms improves, and as our abilities to facilitate and accelerate AAV capsid evolution via artificial intelligence and machine learning will increase. 57 –59 Together, this will ultimately enable the implementation and broader use of rational strategies that will allow the bottom-up design of capsid with predictable properties, such as high efficiency and specificity in the musculature, combined with low seroreactivity.
ANCESTRAL RECONSTRUCTION OF EXTINCT AAV CAPSID VARIANTS
While all the aforementioned AAV capsid engineering techniques as well as those discussed below go forward in evolution, an intriguing and ingenious deviation from this concept was introduced in 2015 by the Vandenberghe and Schaffer groups. 60,61 Dubbed “ancestral sequence reconstruction,” this strategy uses maximum likelihood methods to infer putative extinct evolutionary intermediates of contemporary AAV capsids and thus goes back in evolution. For more details on this unique technology, which we have previously also referred to as “Back to the future,” we refer the reader to our former review article. 62
Here, we point out that the lead candidate from the original study by Zinn et al., Anc80L65, outperformed the AAV2 and AAV8 benchmarks when injected directly into the skeletal muscle of mice, and it also moderately surpassed AAV8 in the heart upon systemic delivery in mice. 61 A true assessment of its potential for muscle gene therapy was, however, hampered by the absence of the more relevant benchmarks AAV6 and AAV9.
This gap was filled in a more recent study by Katz et al., who compared Anc80L65 with AAV9 in neonatal rat cardiomyocytes ex vivo and in rat hearts in vivo, following intramyocardial or intracoronary injection. 63 Notably, in all scenarios, the ancestral capsids expressed faster, more robustly and more homogeneously than the extant wild-type control. A conclusion on vector specificity was prohibited in this work owing to lack of data on Anc80L65 activity outside the heart, but results reported in the original study and elsewhere imply that this capsid is not specific for cardiomyocytes. 61
Most recently, the same group has reported a multiparametric quantitative in vivo screen of their original library of 2,048 Anc80 variants, which they now barcoded using a new methodology called “CombiAAV.” 64 Strikingly, Zinn et al. identified a crucial role of residue 266 (position p3), which is 1 of the 11 variable positions (p1–p11) in the Anc80 library, in the efficiency and specificity of gene delivery (DNA) and vector transduction (messenger RNA [mRNA]) in the liver versus muscle. Specifically, a glycine at this position favored liver gene transfer, whereas an alanine led to a strong and significant liver detargeting while maintaining the ability to transduce skeletal and cardiac muscle, although at modestly reduced efficiency.
Notably, this liver toggle could also be grafted into two other, clinically relevant AAV wild types, namely AAV9 where mutation of an existing glycine in p3 to an alanine led to an over 100-fold detargeting from the mouse liver, and AAV3B where the reverse mutation of alanine to glycine enhanced liver transduction by 20-fold. It remains to be seen whether these effects can be recapitulated in nonhuman primates and ultimately in human patients, but together with the reasonable speculation on an involvement of the universal AAVR receptor in the function of this liver toggle, this latest study clearly illustrates the power of molecular AAV evolution to improve both our basic knowledge of AAV biology and our arsenal of gene therapy vectors.
Of note, this promise was independently confirmed by Santiago-Ortiz et al., who also created an ancestral capsid library and then isolated several extinct variants that exceeded the AAV1 benchmark upon intramuscular injection into the mouse gastrocnemius. 60 Also in this work, however, important additional benchmarks were missing, and the reproducibility of the mouse data in higher species including humans continues to remain open.
PROTEASE-ACTIVATABLE SYNTHETIC AAV CAPSIDS
An ingenious strategy that also relies on the insertion of foreign sequences into the AAV capsid but exploits them for deliberate particle inactivation rather than activation was reported by the Suh laboratory. 65 These authors created the so-called provector, by using sequence and structure information on AAV9 to semi-rationally insert a flexible peptide lock into the threefold capsid protrusion where it blocks AAV9 transduction. This lock can be cleaved off in vivo by extracellular metalloproteinases (MMP)-2 and -9, which are frequently found in the microenvironment of diseases tissues such as the infarcted heart, that is, the model used in this work. As hoped for, when delivered systemically into mice with myocardial infarction, this provector mediated targeted transduction of the damaged heart combined with reduced off-targeting in tissues lacking secreted MMPs. Moreover, limited evidence suggested a slightly increased resistance to neutralizing anti-AAV antibodies of the engineered AAV9 variant compared with the parental wild type.
Considering that this is a first proof-of-concept study that has only begun to explore the potential of peptide locks in AAV9, it is clear that numerous questions and room for improvement remain. This includes the exploration of other insertion sites and lock designs, as well as an expanded in vivo characterization in other species and disease models, which will then allow to better understand the promise and pitfalls of this system, including potential in vivo promiscuity caused by other MMPs that share common substrate sequences. Nonetheless, this is an original and promising approach that is compatible with other capsid engineering strategies as well as with (post-)transcriptional targeting measures, and which should thus hold great potential for the future of AAV gene therapies in diseased muscle tissues or elsewhere.
CHIMERAS OF TWO AAV WILD TYPES CREATED BY RESIDUE OR DOMAIN SWAPPING/MIXING
Multiple groups have deliberately swapped either one or more individual residues, or entire domains, between two parental wild-type AAVs with the aim to improve muscle targeting and efficiency of myotropic gene transfer in vivo. Most notable work includes several studies by Hauck et al. from the Xiao laboratory, who created multiple sets of chimeras between the AAV1 and AAV2 cap genes via domain swapping using conventional cloning techniques. 66 –68 Eventually, this allowed them to narrow down the critical region determining the high muscle specificity of AAV1 compared with AAV2 to amino acids 350–430. Evidence included that insertion of this portion from AAV1 boosted the muscle performance of AAV2, although not to the levels of wild-type AAV1, whereas the reverse insertion lowered the efficiency of AAV1 in the muscle of mice.
Notably, however, the AAV1-AAV2 chimera displayed an enhanced resistance to neutralizing anti-AAV antibodies, illustrating the potential of this approach. Additionally, the fact that the binding site for the AAV2 primary receptor HSPG is fully conserved in the AAV1-AAV2 chimeras permits its purification via large-scale heparin affinity chromatography columns while maintaining high efficiency in the musculature, and thus combining the best of both worlds (AAV1 and AAV2), as demonstrated with a therapeutic transgene in a follow-up study by the same authors. 68 Importantly, because of the similarity of AAV1 and AAV2, the chimeric vector differs only by nine amino acids from wild-type AAV2, which should further facilitate its clinical translation considering how well characterized the AAV2 prototype is.
This chimeric vector design was further validated by Murakami et al., who additionally substituted a surface-exposed tyrosine at AAV2 VP1 position 730 with phenylalanine, which is known to frequently enhance AAV transduction. 69 Also in the hands of these authors, AAV2 with an insert from AAV1 (residues 350–430, as before) plus the noted extra mutation could be purified by heparin affinity chromatography and mediated robust expression after intramuscular delivery.
In a permutation of this approach, Hauck et al. reported the strategy to combine the benefits of AAV1 and AAV2 not by capsid engineering, but by mixing the two corresponding helper plasmids during vector production. 66 In contrast to the aforementioned domain swapping strategy, this yielded vectors that performed well in mice in both primary target tissues, that is, muscle (AAV1) and liver (AAV2), and thus showed an expanded rather than a narrowed down tissue tropism. As expected, this chimeric vector was also susceptible to neutralization by both anti-AAV1 and anti-AAV2 antibodies. As seen before with the genetic AAV1-AAV2 chimera, the vector containing a mixture of both full-length capsid proteins performed better than wild-type AAV2 in the mouse muscle but did not reach the efficiency of wild-type AAV1.
As demonstrated in these exemplary proof-of-concept studies, this fairly simple approach especially of helper plasmid mixing holds significant potential for clinical muscle gene therapy, yet it also has a series of limitations that need to be overcome. These include that the in vivo efficiencies of the chimeric particles consistently remained subpar to AAV1, that not all genetic chimeras can be produced efficiently, that the mixed particles are in fact even more immunoreactive than the two separate viral parents (as they are detected by both antibody populations), and that the outcome of capsid mixing during particle production is hard to control.
RATIONAL DESIGN OF MYOTROPIC AAV CAPSIDS BY FUSION OR INSERTION
In this section, we will highlight three selected studies showcasing the potential of rational capsid engineering through fusion or insertion of known targeting moieties including some that are specific for muscle into different AAV capsids. We note an overlap with rational design studies by others who have mutated individual amino acids to improve AAV performance in the musculature and refer to a later paragraph for these reports.
In the first of the exemplary studies, Zhang et al. tethered FerA domains derived from the mouse Otoferlin, Dysferlin, or Myoferlin protein (together forming the Ferlin family of type II transmembrane proteins) onto different AAV serotypes (AAV6, 8, and 9; not all combinations of FerA domains and serotypes were tested). 70 In systemically injected mice, FerA tethering delayed blood clearance, while enhancing vascular permeability and globally improving gene transfer. Notable in the context of the present article is that AAV9-mediate muscle gene transfer was also enhanced upon Oto-FerA tethering and direct intramuscular delivery of the complex. Its simplicity and efficiency make this approach very promising, but questions remain about the immunogenicity in particular of the human Ferlins and the scalability of the approach, considering its dependency on FerA expression and purification. At the same time, optimism is provided by the fact that Myoferlin is highly expressed in the developing skeletal muscle and in the heart, implying that it may preferentially benefit gene transfer into these muscle tissues.
In a second and similar example, Finet et al. tested the hypothesis that cardiac tropism could be enhanced by fusion of cardiac ion channel-specific ligands onto the AAV capsid; specifically, of cardiac sodium channel (Nav1.5)-binding toxin Anthopleurin-B (ApB). 71 To this end, the authors fused ApB with AAV2 VP2 N-terminus (position T138) and additionally ablated AAV2s heparin binding capability through mutation of residues R585 and R588. In cultured immortalized or fresh cardiomyocytes, ApB fusion significantly improved cell attachment and transduction over the AAV2 mutant only carrying the ablated receptor binding site, showing the positive effect of ApB on Nav1.5-expressing cells.
Importantly, in peripherally injected rats, the receptor-blinded ApB-encoding AAV2 particle gave robust and highly specific expression in the heart in the nearly complete absence of off-targeting in the liver or elsewhere, while the ApB-lacking control showed a much broader transduction pattern. As encouraging as these results are, it should be noted that these effects were only observed upon ablation of the primary AAV2 receptor binding site; in its presence, binding to HSPGs dominated any influence of the ApB domain. Hence, further preclinical development needs to focus on a better dissection of these opposing effects in vivo and should also test this strategy in additional serotypes considering the general drawbacks of AAV2 for use in humans including its high seroprevalence.
Finally, we highlight a study by Jackson et al. in which a high-affinity binder of the insulin receptor (IR), that is, the insulin-mimetic, 36 amino acid-long peptide S519, was inserted into AAV9 and other serotypes and then found to improve transduction of IR-expressing primary human muscle cells ex vivo or mouse muscle in vivo. 72 For insertion, the authors originally tested the two most exposed capsid loops in HVR IV or VIII and eventually pursued the HVR-VIII variant (called eAAV9) as it outperformed the other insertion. Intriguingly, the best performance was obtained when particles were produced in the presence of a 10-fold excess of wild-type AAV9 capsid proteins, most likely because an overabundance of the bulky S519 peptide was incompatible with the assembly of functional capsids.
In primary human myotubes, eAAV9 surpassed the unmodified AAV9 control and could be blocked by an IR-Fc inhibitor, proving the dependency on the IR. Higher efficiency of gene transfer was also noted upon intramuscular injection in mice, but, surprisingly, not after systemic delivery, for reasons that remained unclear. Notably, the enhancing effect of the S519 insertion upon intramuscular delivery in mice was recapitulated in four other capsids (AAV1, 2, 8, and NP22; 45 the latter is an inherently myotropic shuffled capsid [see above]), but not in cultured cells as AAV2 and NP22 were not enhanced ex vivo. This is informative as it may suggest a competition of IR-binding with attachment of AAV2 and NP22 to the HSPG receptor, which is abundant in cultured cells but downregulated in differentiated skeletal muscle.
If true, this would not only explain the discrepant results obtained with these two specific capsid variants (that both bind HSPG), but it would also clearly underscore the necessity to screen AAV capsids or libraries thereof in the physiologically most relevant system rather than more artificial ex vivo cultures. Altogether, the strategy reported in this work is therefore very encouraging, as it improves vector efficiency in skeletal muscle, is portable to other AAV variants including synthetic capsids, and may be translatable to humans owing to the similar binding of S519 to murine and human IRs.
IMPROVEMENT OF MUSCLE TROPISM THROUGH MUTATION OF SINGLE AMINO ACIDS
A large body of work has already illustrated the power of targeted alteration of individual residues in the AAV capsid protein to increase the efficiency of muscle gene transfer and/or improve specificity in this tissue by reducing off-targeting in the liver and elsewhere. As in the other chapters, we can thus only highlight a few representative examples to highlight this concept, and we apologize to all colleagues whose equally pivotal we had to omit for space reasons.
Previously, we had already highlighted recent work by the Vandenberghe and Lisowski laboratories that had identified a crucial role of a single residue in Anc80 or AAV3B, which acts as a toggle between liver and muscle tropism. 64,73 Corroborating these data and hence the potential of mutagenesis as molecular AAV evolution technique is a prior study by Li et al., who comprehensively studied the possibility of using different wild-type or mutated AAV capsids for repeated gene transfer to the mouse muscle. 74 This study capitalized on earlier work also from the Samulski laboratory, in which Bowles et al. had reported encouraging clinical phase I data with a rationally designed AAV2 capsid called AAV2.5, carrying five mutations from AAV1 and exhibiting desirable properties from both parents. 75
Specifically, in this mutant, four residues including one at position 263 (previously also noted as important by Hauck et al. 68 ) were selected based on a series of criteria (e.g., location on the surface and in HVRs) and then substituted with AAV1 residues. Moreover, one additional AAV1 amino acid, a threonine, was inserted at position 265. The resulting retained the compatibility with purification by heparin affinity chromatography and, importantly, outperformed the AAV2 parent upon intramuscular injection in mice, although it did not reach the levels of AAV1. Notably, despite the fact that only five residues were exchanged in AAV2, this sufficed to provide the AAV2.5 capsid with a distinct immunological profile and to partially escape from neutralization with anti-AAV1 or anti-AAV2 antibodies, to better evade neutralization by human sera, as well as to enable vector readministration in mice that were pretreated with AAV2. This vector was later successfully evaluated in a clinical phase I trial, whose results will not be reviewed here as they are outside of the focus of this article.
In the aforementioned related study, Li et al. further dissected the contribution of the five residues in AAV2.5 to the increased muscle tropism and could show that most critical is the single insertion of the AAV1 threonine at position 265 of the AAV2 capsid. 74 Intriguingly, insertion of all 20 amino acids at this position revealed that three decreased, but eight including threonine enhanced muscle transduction in intramuscularly injected mice, with three amino acids even surpassing threonine. This suggests that the enhanced muscle transduction does not correlate with a specific amino acid but is rather related to common changes in capsid conformation, again illustrating the power of molecular evolution to not only breed better vectors but to also disentangle AAV biology. No correlation was noted between transduction efficiency and ability to escape antibody neutralization, further highlighting the complex life cycle and features of AAV.
Another set of studies has consistently exemplified the great power and potential of surface-exposed tyrosine-to-phenylalanine mutations to enhance in vivo muscle transduction with AAV. For instance, Qiao et al. showed that two such mutations (Y445F and Y731F) could substantially boost AAV6 efficacy at different doses after intramuscular administration in mice. 76 As has been hypothesized before, 77 this effect was likely caused by the ability of these mutants to evade phosphorylation and subsequent ubiquitination, thereby avoiding proteasome-mediated capsid degradation. This conclusion was confirmed and extended, for example, by van Lieshout et al., who introduced an additional F129L (the same as in AAV6.2) 78 mutation into AAV6 and showed a faster, up to 50- or 100-fold enhanced transduction of mouse lung or muscle, respectively, with the AAV6 triple-mutant. 79 Notably, however, there was no difference in the long-term expression levels between the mutant and the parental AAV6 wild type beyond 2 weeks post-administration, implying that the kinetics but not the strength of transduction was enhanced.
Complementing these data with AAV6 is, for instance, work by Hakim et al., in which the benefits of surface-exposed tyrosine-to-phenylalanine mutations were also confirmed for two other serotypes, AAV1 and AAV9. 80 Most notably, an AAV1 double-mutant provided excellent gene transduction in systemically injected skeletal and heart muscle of neonatal dogs, which makes this vector interesting for future preclinical work in canine models and ideally clinical application in humans, granted the results hold up in larger animal numbers. Besides, we note a potential additional benefit from tyrosine-to-phenylalanine mutations that was reported by Martino et al., namely a reduced targeting of transduced cells by capsid-specific CD8+ T cells. 81
One more example that illustrates the breadth and potential of mutagenesis as AAV evolution technique in the muscle comes from Pulicherla et al. from the Asokan laboratory, who used error-prone PCR to mutate the GH loop (residues 390–627) in AAV9 and then identified 10 point mutants based on sequence and structural analysis for further characterization. 82 Of these, two (9.45 and 9.61) are particularly interesting in the context of this article as they mediated robust expression in the mouse skeletal and heart muscle upon peripheral delivery, coupled with marked liver detargeting akin to that of other synthetic AAVs, for example, AAVM41. 43
Finally, we note an exciting earlier study by the Samulski laboratory in which Asokan et al. reengineered the receptor footprint of AAV2, by replacing a hexapeptide spanning two critical arginines (R585 and R588) known to interact the HSPG AAV2 receptor, with the corresponding sequence of other serotypes. 78 Most remarkably, one of the mutants, AAV2i8 (containing the hexapeptide 585–590 from AAV8 in AAV2), displayed a unique in vivo biodistribution in peripherally injected mice, where it targeted the skeletal muscle, heart, and diaphragm with high efficiency but was about 40-fold detargeted from the liver compared with the parental AAV2 and AAV8.
Moreover, it was more resistant to anti-AAV2 or human sera, corroborating its chimeric nature. In subsequent work, Shen et al. showed that additional engraftment of the galactose-binding footprint of AAV9 into AAV2i8 can further modulate in vivo transduction behavior, including faster kinetics of gene expression and a mild increase in skeletal muscle transduction. 83 However, expression in the heart was reduced compared with AAV2i8, and liver off-target transduction was fivefold higher, although still two orders of magnitude below AAV9.
Collectively, these and many other studies that could not be highlighted due to space reasons document the enormous potential of even the simplest of all AAV capsid evolution technologies, namely mutagenesis of as little as a single capsid residue. Particularly attractive is that this technology can be integrated synergistically with other evolution techniques and that it can also be easily upscaled by randomizing one or more positions and then screening the resulting libraries for desired phenotypes. In a most recent exciting development, the power of this methodology has been further potentiated by its combination with machine-learning approaches, such as those exemplified by the Church, Kelsic, and Zolotukhin laboratories. 57 –59 While the potential of this combination for rational design of efficient, specific, and safe myotropic AAVs remains to be demonstrated, the present data provide every reason to be optimistic that this goal can be achieved.
CONCLUSIONS AND OUTLOOK
In more than one way, AAV and muscle gene therapy are reminiscent of an old relationship that is now increasingly starting to show cracks, notwithstanding that, to our best knowledge, eight AAV capsid variants have already been or are currently being investigated in humans, including six wild types (AAV1, 2, 8, 9, rh10, rh74) and two synthetic capsids (AAV2i8, 2.5). The beginning estrangement is most impressively and most disturbingly illustrated by the recent fatalities among AAV-treated children in several clinical trials. Most likely, these resulted from the use of excessive vector amounts per kilogram, necessitated by the inability of the employed capsid variants to mediate specific and efficient muscle gene transfer from lower doses.
Together, this highlights the urgent need and responsibility of the AAV community to continue and further intensify efforts to bioengineer a new vector generation, capable of providing maximum clinical benefit in the absence of patient safety concerns. Fortunately, the technologies that we highlighted in this review and more that we had to omit for space reasons provide substantial optimism that our community can eventually provide these vectors, granted that a flurry of lingering questions on the levels of engineering methodology and virus biology are addressed and solved.
To this end, we are encouraged by the rapidly increasing number of new experimental strategies that now enable in vivo library biopanning and capsid validation on the (m)RNA level, filling in gaps that were left by prior, purely DNA-based methodologies and promising the looming discovery of truly functional capsid variants. 37,55,56 We are likewise thrilled by the accumulating evidence for the power of structure-guided and machine learning-based approaches at capsid design, which promise the eventual possibility to derive optimal AAV vectors bottom-up and in silico, thereby significantly reducing work, time, costs, and, most importantly, small and large animal numbers during preclinical screening. These exciting developments go hand in hand with concurrent improvements in massively parallel, high-throughput in vivo biopanning pipelines as well as in next-generation sequencing technologies such as PacBio/SMRT or Oxford Nanopore, whose accuracy and read depths are constantly increasing.
As the associated costs continue to go down, this will make these disruptive technologies available to a larger number of researchers especially in the academic environment. In turn, this will foster and accelerate innovation and thus ultimately benefit human patients with devastating disorders in the musculature and elsewhere. Further encouragement is provided by the increasing number of studies reporting combinatorial approaches at AAV bioengineering that merge two or even more technologies for capsid diversification or that combine transductional targeting with additional strategies on the transcriptional (promoters, enhancers) or post-transcriptional (microRNA [miRNA]-based detargeting) 84 levels. Based on promising data in other contexts, we also readily anticipate substantial benefit for muscle gene therapy from the insertion of alternative targeting moieties into AAV capsid variants, such as nanobodies or DARPins, 85,86 or from chemical modification.
Last but not least, as is the case with any clinical indication and target tissue, the biggest and most pivotal question will always remain the translatability of the preclinical data obtained with bioengineered AAVs in cultured cells or in small or large animal models, to human patients. This includes practical aspects, such as the scalability of the required up- and downstream manufacturing pipeline for a given capsid, which may be particularly challenging in the muscle gene therapy even with optimized capsids considering the sheer mass of the target tissue relative to the whole body. Moreover, findings that vector performance can differ between healthy versus diseased animals of the same species and strain raise concerns that AAV capsids evolved in a healthy preclinical animal model may not necessarily translate into a diseased human, besides worries about the species barrier.
Along these lines, more and more data also imply the possibility of differences in vector efficiency and/or specificity even within the same species, as first reported for the AAV-PHP.B capsid in various mouse strains and subsequently for others as well. Consequently, the debate in the field is currently intensifying which animal model may be the most predictive for clinical translation in diseased patients, and it is likely that this will dominate the field in the years to come. One possible solution may be to automatically incorporate a cross-species component into new molecular evolution stratagems, whose potential has recently been demonstrated by the Asokan laboratory through iterative library cycling in three species, that is, mice, pigs, and nonhuman primates. 87
Until then, we note preliminary, as-of-yet unpublished observations from our own laboratory implying that the marked liver detargeting and hence the beneficial target-to-noise ratio observed in various mouse strains with our latest AAVMYO2 and AAVMYO3 capsids is conserved in nonhuman primates, not guaranteeing but raising hopes that this pivotal characteristic may be exhibited across species and thus also translate well in humans. Notwithstanding this optimism about efficiency and specificity, patient safety always remains the top priority; hence, we hope that these latest and subsequent generations of myotropic AAV vectors will also prove to be safe in clinical trials and gene therapies of human muscle disorders.
Footnotes
ACKNOWLEDGMENTS
The authors thank their laboratory members for internal discussion of the topics of this article. We kindly ask all our colleagues working on AAV bioengineering and/or muscle gene therapy for understanding that space for text and references was limited in this article, forcing us to make a selection and unfortunately requiring the omission of numerous equally interesting reports.
AUTHORs' CONTRIBUTIONS
J.L.: Conceptualization (equal), writing—original draft (equal). T.W.K.: Conceptualization (equal), writing—original draft (equal). O.M.: Conceptualization, writing—original draft (equal). M.Z.: Conceptualization. D.G.: Conceptualization (lead), writing—original draft (lead).
AUTHOR DISCLOSURE
D.G. is an inventor on pending patent applications describing myotropic AAV capsids. All other authors declare no conflict of interest.
FUNDING INFORMATION
D.G. greatly appreciates support by the German Research Foundation (DFG) through the DFG Collaborative Research Centers SFB1129 (Project number 240245660) and TRR179 (Project number 272983813), as well as by the German Center for Infection Research (DZIF, Bundesministerium für Bildung und Forschung [BMBF]; TTU-HIV 04.819). D.G. is moreover grateful for funding through the program COMMUTE (BMBF). D.G. and M.Z. acknowledge a collaboration with Cure Rare Disease.