
Abstract
Muscular dystrophies (MDs) comprise a diverse group of inherited disorders characterized by progressive muscle loss and weakness. Given the genetic etiology underlying MDs, researchers have explored the potential of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) genome editing as a therapeutic intervention, resulting in significant advances. Here, we review recent progress on the use of CRISPR/Cas9 genome editing as a potential therapy for MDs. Significant strides have been made in this realm, made possible through innovative techniques such as precision genetic editing by modified forms of CRISPR/Cas9. These approaches have shown varying degrees of success in animal models of MD, including Duchenne MD, congenital muscular dystrophy type 1A, and myotonic dystrophy type 1. Even so, there are several challenges facing the development of CRISPR/Cas9-based MD therapies, including the targeting of satellite cells, improved editing efficiency in skeletal and cardiac muscle tissue, delivery vehicle enhancements, and the host immunogenic response. Although more work is needed to advance CRISPR/Cas9 genome editing past the preclinical stages, its therapeutic potential for MD is extremely promising and justifies concentrated efforts to move into clinical trials.
INTRODUCTION
Muscular dystrophy
Muscular dystrophies (MDs) are a collection of clinically and genetically heterogeneous disorders characterized by progressive muscle weakness and loss. 1,2 The disorders vary according to the affected muscles, age of onset, severity, and rate of progression. Currently, most medical interventions are limited to symptom management and delaying disease progression. 1,3
Here, we provide a comprehensive overview of the current state of genome editing strategies using clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) in models of MD. This work focuses on the utilization of CRISPR/Cas9 as an in vivo therapeutic strategy to treat Duchenne muscular dystrophy (DMD), congenital muscular dystrophy type 1A (MDC1A), and myotonic dystrophy type 1 (DM1). In addition, we discuss current in vivo delivery options, focusing on adeno-associated viral (AAV) vectors, recent advances made, challenges that lie ahead for the field, and what issues must be resolved for CRISPR/Cas9 genome editing to become a viable therapy.
DMD is the most prevalent pediatric neuromuscular disorder due to its X-linked recessive inheritance. 4 It is a life-limiting condition caused by mutations in the DMD gene, which encodes for dystrophin. 5,6 Dystrophin is a subsarcolemmal protein integral to the dystrophin-associated protein complex, which prevents contraction-induced muscle damage. 7 Most commonly, mutations that cause DMD generate out-of-frame transcripts, eliminating dystrophin expression. 8,9
MDC1A is an autosomal recessive neuromuscular disorder characterized by hypotonia, muscle weakness, and muscle wasting that begins in infancy. 10 MDC1A is caused by mutations in the LAMA2 gene, which encodes laminin-2. 11 Laminins are proteins of the extracellular matrix that are essential components of the basement membrane. 11,12
DM1 is the most prevalent adult-onset form of MD. 13 Patients may also develop intellectual impairment, respiratory insufficiency, and cardiac conduction abnormalities, in addition to muscle weakness and stiffness. 14,15 DM1 is caused by a microsatellite expansion of cytosine, thymine, and guanine (CTG) triplet repeats in the 3′ untranslated region (UTR) of the dystrophia myotonic protein kinase (DMPK) gene. 13,15 As a result, these mutated DMPK transcripts generate nuclear foci, which cause aberrant splicing across a broad spectrum of pre-mRNAs. 14
Various treatments for MDs are currently approved or are in clinical testing. Included in this category are antisense oligonucleotides (ASOs), gene therapies, and stop codon read-through drugs. 1 Although effective in treating other neuromuscular disorders such as spinal muscular atrophy (SMA), unfortunately, with regard to MDs, their relatively poor performance and transient nature limit their clinical applications. 1 An effective treatment must target and, ideally, permanently repair the genetic cause of MDs. Interventions based on CRISPR/Cas9 are extremely promising because of their unrivalled utility and accuracy in performing targeted genome editing.
CRISPR/Cas 9 GENOME EDITING STRATEGIES TO TREAT MDs
Exon skipping and reframing
Exon skipping using CRISPR/Cas9 endonucleases can restore the open reading frame (ORF) of an out-of-frame gene and lead to the restoration of a functional protein. Exon skipping has been achieved in DMD patients with the use of ASOs, which omit the out-of-frame exon(s) from the final transcript. 16 Eteplirsen, Golodirsen, and Viltolarsen are examples of such drugs that use ASOs to target out-of-frame exons in the DMD gene, excluding them from the final transcript and restoring the reading frame. 17 –21 Unfortunately, ASOs typically result in low uptake in cardiac tissue, limiting their clinical relevance regarding the treatment of MDs with a cardiac pathology. 20,21
As a result, CRISPR/Cas9 is currently under investigation as an alternative to mediating exon skipping and reframing. CRISPR/Cas9-mediated exon-skipping would be a one-time therapeutic option that results in permanent correction and restoration of a functional gene product (a summary of such studies is listed in Table 1). Three main exon-skipping and reframing approaches (Fig. 1A) have been tested in preclinical studies and are described next.
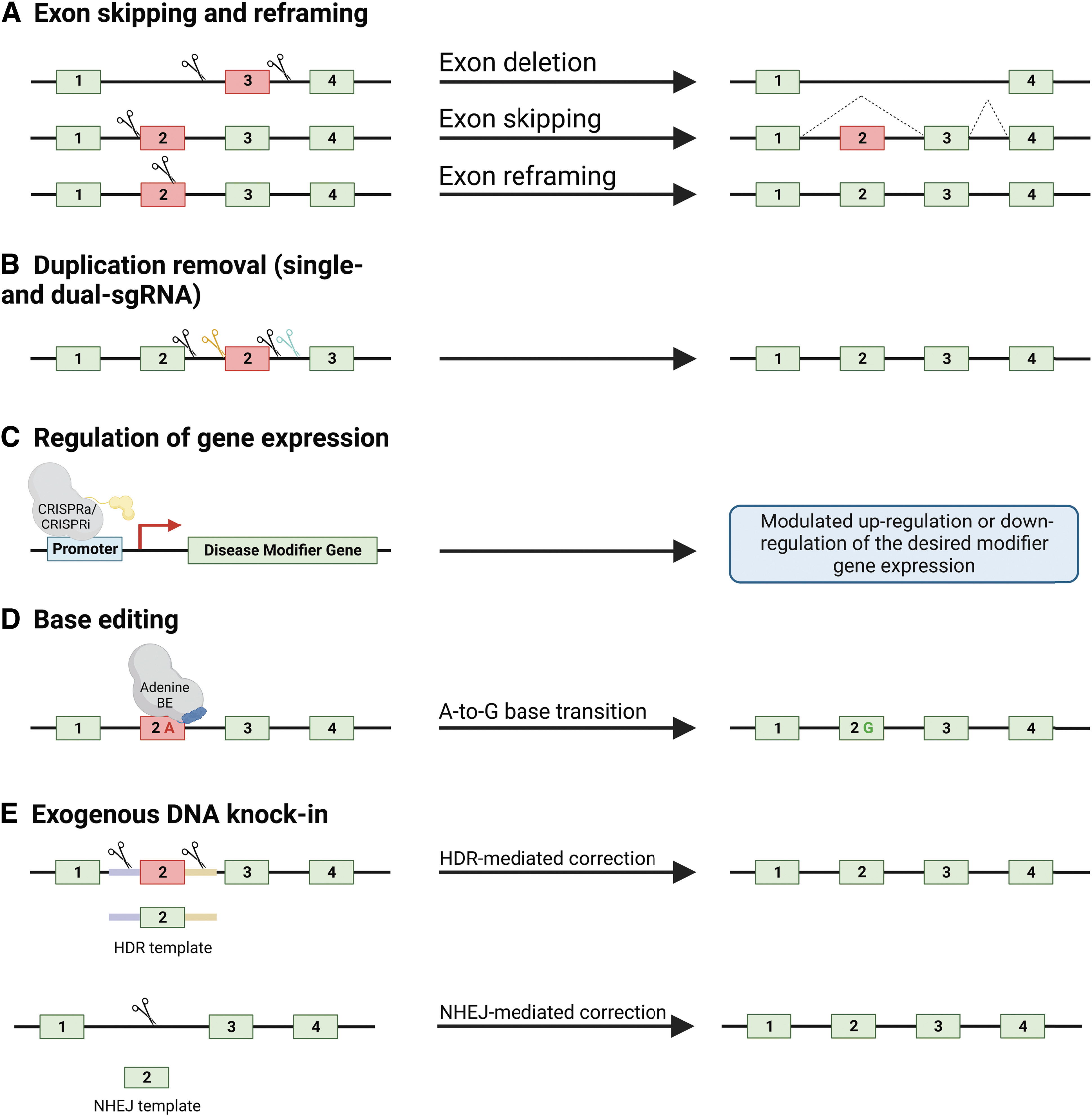
Summary of in vivo validated CRISPR/Cas9 genome editing strategies for the treatment of MDs. On the left are gene stretches containing a mutated exon, highlighted in red, as well as respective CRISPR/Cas9 strategies, leading to the desired outcomes on the right.
Summary of in vivo CRISPR studies for the treatment of muscular dystrophies
AAV, adeno-associated virus; ASO, antisense oligonucleotide; Cas9, CRISPR-associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; CTG, cytosine, thymine, and guanine; DAPC, dystrophin-associated protein complex; dCas9, dead Cas9; DM1, dystrophy type 1; DMD, Duchenne muscular dystrophy; GRMD, golden-retriever model of DMD; HDR, homology-directed repair; HITI, homology-independent targeted integration; LRMD, Labrador retriever muscular dystrophy; MD, muscular dystrophy; NHEJ, non-homologous end-joining; RNP, ribonucleoprotein; sgRNA, single-guide RNA; tsAAV, trans-splicing adeno-associated virus; WCMD, Welsh corgi muscular dystrophy.
Exon removal
This approach aims at restoring the ORF of a gene by excising the out-of-frame exon(s), resulting in the expression of a truncated protein. This strategy has been assessed pre-clinically in several DMD models, including mice, pigs, and dogs. 22 –25 Studies in mice focused on several exons of the Dmd gene, including exons 23 and 52–53. 23,25 These studies show restoration of a functional dystrophin protein and improved disease phenotypes. Studies in pigs and dogs provided proof-of-principle that CRISPR/Cas9-mediated exon removal is a viable therapeutic option in larger animal models, paving the way for its translation to the clinical trials. 22,24
Exon skipping and inclusion through splice site manipulation
The next strategy designed to mediate exon skipping relies on manipulation of splice sites using CRISPR/Cas9 systems. Here, a single-guide RNA (sgRNA) is used to disrupt splice sites through the error-prone non-homologous end-joining (NHEJ) repair pathway, which subsequently allows for the skipping of the desired exon(s). 26 The NHEJ-mediated introduction of random deletions and insertions (indels) renders the splice site no longer identifiable by the spliceosome. 27,28
The disruption results in the exclusion of the exon from the final transcript, restoring the ORF and expressing a truncated gene product. This strategy has been tested pre-clinically in mouse and canine DMD models and resulted in restoration of a functional dystrophin protein in skeletal (including the diaphragm) and cardiac tissues. 22,26
In an MDC1A mouse model, Kemaladewi et al. used a pair of sgRNAs to remove a mutated splice site in the LAMA2 gene and allowed NHEJ to restore a functional splice donor site. As a result, full-length laminin-2 was restored in muscles and the sciatic nerve. Treated mice exhibited reduced fibrosis, increased muscle fiber size, and functional improvements comparable to wild-type mice. 29
Indel-mediated reframing
An alternative reframing approach consists of restoring the ORF by introducing small indels in the coding sequence of an out-of-frame exon. The Olson group has tested this strategy by using the engineered KKH variant of SaCas9 in patient-derived induced pluripotent stem cells (iPSCs) to induce a single cut that results in the introduction of indels that are able to reframe exon 51 of the DMD gene. 30 This approach resulted in a high frequency of indels introducing a two-nucleotide deletion and, thus, the restoration of the reading frame.
The group subsequently tested this approach in vivo in mice with a deletion of Dmd exon 50. Results demonstrated that this single-cut strategy restored dystrophin expression to 30–50% in skeletal muscle and 50% in the heart, along with improved muscle integrity and function. The authors note that using SpCas9 tends to favor NHEJ-mediated insertion of one base pair, which is not the case with SaCas9. To address this challenge, the authors screened for guide RNAs that can induce double-strand breaks (DSBs) in regions of microhomology, which tend to produce predictable deletions.
Min et al. also tested single-cut genome editing strategies in mouse and cell models of DMD. 31 They compared both reframing and exon skipping for three mutations using SaCas9. From their results, they concluded that +1 reframing has comparable editing efficiencies to −1 reframing depending on a chosen guide RNA. They also note that the indel type observed in iPSC-derived cardiomyocytes can be used to predict the levels of dystrophin restoration in vivo.
Advantages, limitations, and future directions of CRISPR/Cas9-mediated exon skipping and reframing
CRISPR/Cas9-mediated exon skipping and reframing has several advantages compared with ASOs, including the ability to potentially achieve a permanent correction using a single treatment, which significantly improves the cost-benefit profile of this approach, especially as the cost of clinical-grade AAV production continues to decrease. 32 Exon skipping and reframing have been tested in vitro and in vivo, including in large animal models, and have led to significant and widespread restoration of dystrophin. The major limitation of these strategies is that exon skipping and reframing typically lead to the production of a truncated gene product. Depending on which exons are skipped or lost, this may lead to varying clinical improvements. 33 To address this, focus should be placed on studying the impact of such correction strategies on patients as we approach clinical trials.
Duplication removal
Large genomic duplications typically encompass one or more exons and result in reading frame disruption or impairment of protein function through the addition of these exons to the transcript. In DMD, these duplications represent 10–15% of patient mutations and occur at low but appreciable frequencies for several other MDs, such as MDC1A. 34 –37 Therapeutics specific for treating MD duplication mutations have been mostly neglected, leading to a significant unmet need for a sizeable patient population. Presently, applications of CRISPR/Cas9 toward treating large duplications have only been published for DMD, though the principles behind these studies should be broadly applicable to other MDs.
Duplication removal using single- and dual-sgRNA approaches
Large duplications can be corrected in one of two ways using CRISPR/Cas9 (Fig. 1B). The first employs two sgRNAs that flank the duplicated region and, through coordinated cleavage, excises the intervening duplication, with subsequent ligation restoring the wild-type sequence. The second relies on a solitary sgRNA targeting within the duplicated sequence. As a result, the original and duplicated sequences are cut in the same location with the intervening sequence being excised, also restoring the wild-type sequence.
There are several key advantages to the latter approach that make it the preferred option for correcting most large duplications. Using one sgRNA rather than two notably reduces the complexity of delivery. Minimizing the essential CRISPR components is critical, as AAVs will likely remain the delivery vehicle of choice for clinical trials for the foreseeable future. The single-sgRNA approach enables packaging of all components into one AAV. 38 Using the same sgRNA for both target sites may improve coordinated cutting at both sites by reducing the potential for asynchronous cleavage and thus enhance excision of the duplication. In addition, the entire duplicated region is made available for guide selection, which enables the assessment of numerous sgRNAs to identify the most efficacious candidates. It should be noted that to minimize potential disruption to the coding sequence, sgRNA design should be limited to introns.
In DMD patients, large duplications typically manifest in tandem, which enables the use of the single-sgRNA approach. It is worth noting that non-contiguous DMD duplications have been reported, whereby a dual sgRNA strategy may be the only option. 39,40 Several groups have demonstrated the successful restoration of full-length dystrophin in DMD patient-derived myoblasts harboring large duplications using the solitary sgRNA strategy. Wojtal et al. were the first in 2016, correcting a duplication of exons 18–30. 41
Studies soon followed, correcting DMD patient duplications of exon 2, exons 55–59, exons 18–25, and exons 3–16. 42 –45 We were unable to identify any published study utilizing the dual sgRNA strategy for correcting a duplication mutation, likely owing to its notable disadvantages and the relatively early reporting of the successful use of a solitary sgRNA. Unfortunately, in vivo application of CRISPR/Cas9 for correcting DMD duplications is limited to a single 2021 study by Maino et al.
There, a novel DMD mouse model harboring a 130 kb exon 18–30 duplication was generated and subsequently treated with the single-sgRNA approach using SaCas9 and delivered systemically by AAV9s. Significant full-length dystrophin restoration, averaging ∼17%, was observed in muscle tissues, most notably in the heart, resulting in significant improvements in muscle histology and function. 38
Advantages, limitations, and future directions of CRISPR/Cas9-mediated duplication removal
In vivo studies on large duplications are extremely limited owing to the immense difficulty in generating appropriate animal models relative to large deletions and small insertions or duplications. In addition to the exon 18–30 duplication mouse, only one other model has been described, which possesses an exon 2 duplication, the most common duplication observed in DMD patients. 39,46 However, this model was obtained by inserting exon 2 and only small portions of its flanking intronic sequences into a duplication hotspot in intron 2. 46
As a result, the full genomic architecture of a typical large tandem duplication is lost, limiting investigations of duplication removal strategies. There does remain immense utility for this model, as demonstrated by the successful evaluation of an AAV-packaged ASO, which is currently under investigation in a clinical trial. 47 –49 Most recently, a family of Labrador retrievers possessing a tandem 400 kb duplication of exons 2–7 in the DMD gene has been described and may prove to be an important large animal model for pre-clinical research into duplication-specific CRISPR/Cas9 therapies. 50
Although ASOs demonstrate some potential use for single exon duplications, care must be taken as different exons respond variably to ASOs, with non-productive skipping of both exons having been observed. 40,47,51 Multi-exonic duplications magnify the difficulty proportional to their size and remain extremely problematic to treat with ASO. 51 CRISPR/Cas9 provides a solution for treating duplications, particularly tandem ones, using single- and dual-sgRNA approaches. Maino et al. have importantly demonstrated that generating mouse models with large multi-exonic duplications is feasible. 38
Looking ahead, focus should be shifted toward developing novel duplication animal models of various MDs that faithfully reflect patient genomic architecture, including humanized mouse models. These will be essential for enabling translational CRISPR/Cas9 preclinical research. Although studies on the topic remain sparse, there is a growing appreciation for large duplications in MD and how CRISPR/Cas9 can be applied to correct them.
Transcriptional modulation of disease modifiers in MDs
CRISPR/Cas9 technology can be harnessed to modulate the expression of target genes of interest (Fig. 1C). In this approach, it is typical to target an inactivated or “dead” Cas9 (dCas9) protein, fused to a transcriptional activator or repressor, to the promoter region of a target gene. 52 CRISPR activation (CRISPRa) enables overexpression of target genes via recruitment of transcriptional machinery or relaxation of surrounding chromatin. 53
CRISPR interference (CRISPRi) is a similar technique that enables the suppression of target gene expression by interfering with transcriptional machinery to transiently inhibit gene expression. 54 The technique works by targeting a dCas9 fused to a transcriptional repressor, to the promoter region of the gene of interest. Alternatively, dCas9 can be fused to a variety of epigenetic enzymes that perform biochemical modifications to condense local chromatin structure and/or methylate promoters, masking the gene of interest from transcriptional machinery. For a more detailed description of CRISPR/Cas9-mediated transcriptional regulation systems, please refer to the following review. 55
CRISPR/Cas9-mediated transcriptional regulation of MD disease modifiers
Disease modifier genes are those, separate from the mutated gene, that can affect the severity and progression of a disorder. Restoring full-length dystrophin via gene therapy remains challenging due to its large size. 1 Expression of alternative homologs capable of performing the functions of dystrophin would be an ideal mutation-independent approach, applicable to all patients. 56,57 Utrophin is a cytoskeletal protein, highly similar in structure and function to dystrophin, encoded by the autosomal UTRN gene. 58 –61
Utrophin is primarily expressed in fetal skeletal muscle and is largely replaced by dystrophin in adult tissues. 58 Interestingly, patients with DMD have endogenously increased levels of utrophin distributed in damaged muscle, and several early studies demonstrated that utrophin overexpression could rescue the DMD phenotype in mice. 62,63 In fact, double knockout mice of Dmd and Utrn possess a far more severe phenotype than Dmd null mice. 64,65 Although several small molecule-based approaches are in development to promote utrophin expression and/or decrease the rate of UTRN transcript downregulation, 59,66,67
CRISPRa has been demonstrated to be effective as a highly specific and efficacious method for upregulation of utrophin. Wojtal et al. demonstrated that a dCas9-VP160 fusion targeting either the UTRN A or B promoters could effectively upregulate utrophin protein expression in patient-derived myoblasts. 41 Although these results were recently validated in DMD patient-derived stem cells, 57 this approach requires assessment of efficacy, longevity of utrophin expression, and characterization of its effects on the dystrophic phenotype in vivo.
A similar CRISPRa approach has been used to treat MDC1A via disease modifier upregulation. There are over 350 known pathogenic nonsense, missense, splice site, and deletion mutations in LAMA2; an efficacious disease modifier would provide a therapeutic alternative to gene-editing that is applicable to all MDC1A patients. 68 LAMA1, which encodes the structurally similar laminin-a1 protein, was demonstrated via overexpression studies to be a suitable candidate to substitute the function of laminin-a2 in vivo.
Kemaladewi et al. were able to upregulate Lama1 in skeletal muscle via systemic delivery of a dSaCas9-VP64 protein in mice harboring a splice site mutation in the Lama2 gene. 56 This strategy led to significant expression of the laminin-a1 protein and was correlated with improvements in the histological hallmarks of MDC1A, including significantly reduced fibrosis. Importantly, systemic treatment of MDC1A mice led to a remarkable rescue of the advanced hind-limb paralysis phenotype, with significant improvements in ambulation as well as increases in specific tetanic force and conduction velocity in the sciatic nerve.
It is predicted that this approach may also provide new opportunities for treatment of DMD, as overexpression of Lama1 has been shown to stabilize the sarcolemma in dystrophic myofibers from mice with dystrophinopathy, further expanding the utility of this approach. 69,70 Ultimately, these results demonstrate the utility of disease modifiers as legitimate therapeutic strategies for the treatment of MDs.
The modular nature of the CRISPR/dCas9 system means it can be used not only to upregulate but also to downregulate the expression of a target gene. Pinto et al. treated the transgenic HSALR mouse, which carries a fragment of the human skeletal actin (HSA) gene with 250 CTG repeats in the 3′ UTR, with dCas9 targeting the CTG repeats and preventing their inclusion in HSA transcripts. 71 This resulted in a significant decrease in repeat transcription and an improvement in phenotype. Applications involving a combinatorial approach with concurrent upregulation of protective disease-modifier genes and downregulation of detrimental genes could represent a new mutation-independent approach for ameliorating disease phenotype.
Advantages, limitations, and future directions of CRISPR/Cas9-mediated transcriptional regulation of disease modifiers
Modulation of gene expression by CRISPR has many advantages over traditional nuclease-based approaches. First, as CRISPRa/CRISPRi is based on fusions of effector domains linked to a catalytically inactivated Cas9, there is negligible risk for the introduction of double-stranded DNA breaks at the target site and the induction of severe chromosomal damage. 72,73
Although it is still possible for a dCas9-fusion to localize effector modules to an off-target location in the genome, it is unlikely that this region is proximal to the promoter or enhancer motifs, further reducing the chance of modulating the transcriptional output of an off-target gene. 74 Transcriptomic studies from the dCas9-VP160-mediated Lama1 upregulation study largely suggest this concern is negligible. 56 Second, modulation of disease modifiers via CRISPRa/CRISPRi offers a mutation-independent approach to treating genetic diseases.
Combining CRISPR transcriptional modulation with traditional CRISPR-mediated gene editing strategies can generate novel therapeutic strategies to treat MDs. These would involve using CRISPRa to increase the expression of a CRISPR/Cas9-corrected gene. We believe the primary advantage of this combinatorial strategy may be especially relevant to diseases where a small number of corrected nuclei can rescue a phenotype by supplying sufficient gene product for the entire tissue. MDs can also benefit from such a strategy since in many neuromuscular disorders, a single edited nucleus can complement the entire myofiber syncytium with a functional gene product. 75
Base-editing in MDs
The introduction of base-editing was the first iteration of true gene-editors capable of producing targeted modifications to specified regions of DNA (Fig. 1D). 76 Base-editing directly generates precise point mutations in genomic DNA without double-stranded breaks, a DNA donor template, or cellular homology-directed repair (HDR). 76 –78 These editors comprised a fusion between a catalytically impaired Cas9 (D10A) nickase and a base-modifying enzyme that operates on transient single-stranded DNA intermediates. 76,79
Two classes of DNA base editors have been described to date: cytosine base editors (CBEs), which convert a C-G base pair into a T-A base pair, and adenine base editors (ABEs), which convert an A-T base pair into a G-C base pair. 76,80 Collectively, CBEs and ABEs can mediate all four possible transition mutations (C-to-T, A-to-G, T-to-C, and G-to-A). More information can be found on the development of base editors in this review by Rees and Liu. 81
Use of base-editors in the therapeutic context for MDs
Therapeutic applications of base editing for neuromuscular disease are a promising strategy due to its highly efficient nature and precise editing of pathogenic point mutations in post-mitotic cells, including neurons and myofibers. 82 Specifically, base editors have the potential to be used as therapeutic interventions in ∼16% of DMD patients who harbor a pathogenic single nucleotide polymorphism that could be corrected by this technology alone. 83 Base editors have been utilized as such in two publications, which highlight their potential for correcting nonsense mutations and manipulating splice sites in exon-skipping strategies in vivo.
Ryu et al. used a trans-splicing adeno-associated virus vector system to deliver an ABE7.10 base editor that was programmed to target a point mutation in Dmd exon 20, a causative mutation that introduces a premature termination codon and subsequently produces a DMD phenotype. 84 Intramuscular delivery of the ABE7.10 via AAV9s to the tibialis anterior (TA) was able to successfully perform single-nucleotide substitution via A-T to G-C deamination to restructure the premature termination codon at a frequency of 3.3%, 8 weeks post-injection.
Notably, this genomic correction was accompanied by a 17% restoration of dystrophin-positive myofibers compared to control. Base editors can also be used as a modified approach for exon-skipping in DMD, as described earlier. Chemello et al. utilized a dual AAV9 split-intein trans-splicing ABEmax-SpCas9-NG system to accomplish exon-skipping via A-T to G-C deamination of the splice donor site to interrupt it and exclude exon 50 from the mature Dmd transcript in a Dmd exon 51 deletion mouse. 85
Intramuscular injection into the TA achieved an on-target genomic editing efficiency of 35%, and Sanger sequencing of transcripts illustrated scarless splicing of Dmd exon 49 to exon 52 as expected. Immunoblotting for full-length dystrophin demonstrated 54% protein restoration compared with wild-type control. Histological findings suggested a substantial reduction in fibrosis and 96.5% restoration of dystrophin-positive fibers in treated animals. However, the authors note that the titer of intramuscular AAV9s delivered in this study would extrapolate to a systemic intravenous dosage corresponding to 1.5E16 viral genomes per milliliter, significantly higher than an acceptable threshold for therapeutic usage.
In a recent study, the Liu lab used an ABE to treat an SMA mouse model (Δ7SMA). 86 Conveniently, SMN1 has a paralogous gene in humans known as SMN2, which mainly differs from SMN1 by a C-G-to-T-A transition in exon 7, resulting in the skipping of exon 7 in most SMN2 transcripts and the production of only low levels of SMN. Following intracerebroventricular injection of a split-intein ABE-Cas9 packaged in an AAV9 vector in neonatal mice, the authors could detect ∼40% A-to-G editing on average in bulk cortical tissue. The treatment also increased the lifespan of the Δ7SMA mice by ∼33%, although the authors note that the short lifespan of these animals makes phenotypic analysis challenging.
Therapeutic advantages, limitations, and future directions of base-editing in MDs
Although the technological framework has been rapidly developed to date, there exist several key challenges to the translation of base-editing into the clinic, some of which are currently being resolved. First, the original base-editing platforms are too large for packaging into traditional AAV vectors in clinical use today. 87 Currently, most preclinical base-editing work is being performed using a dual-vector system, whereby the base editor is assembled post-translationally in a co-transduced cell via a split-intein mechanism. 85,88
The nature of dual-vector systems necessitates increased titers of viral vehicles, which possess immunological and toxicological concerns. In addition, dual vector systems are more expensive to produce. Nonetheless, the Liu lab has recently developed an optimized suite of ABEs that can be constructed into a single, all-in-one vector with the added benefit of increased efficiency of transgene expression, suggesting potentially lower titer requirements. 89
This was achieved by fusing a single evolved TadA deaminase domain to smaller nickase variants of Cas9 (SaCas9, SaKKH Cas9, SauriCas9, and NmeCas9), which has the additional benefit of creating a toolbox of base editor enzymes with alternative or expanded protospacer adjacent motif or PAM, theoretically permitting base-editing at ∼80% of sites within the genome by the combination of these four smaller nickases. In addition, the group has utilized directed evolution to generate highly active cytosine deaminase enzymes from the previously evolved and smaller adenosine deaminase, as well as hybrid deaminase enzymes capable of both adenosine and cytosine deamination. These novel base editor variants should lead to the development of CBEs that are small enough for single AAV packaging in the near future.
CRISPR/Cas9-mediated exogenous DNA knock-in to restore wild-type proteins
To permanently correct MDs, restoration of the wild-type protein at physiologically relevant levels is required. Multi-exonic deletions constitute the majority of DMD patient mutations (∼60%). For precise CRISPR/Cas9 correction of deletions, two DNA knock-in strategies have been pursued: HDR and NHEJ (Fig. 1E).
HDR-mediated exogenous DNA knock-in
Although HDR is not an efficient pathway in muscle cells due to their post-mitotic nature, 90,91 several investigations have attempted to correct a variety of mutations using this strategy in vivo. In a golden-retriever model of DMD, Mata López et al. demonstrated correction of a splice acceptor mutation by delivering plasmids carrying the Cas9 endonuclease and an HDR donor template to the tibiotarsal flexor muscle, restoring 2–16% of wild-type dystrophin levels. 92
In another study, local delivery of AAV9s carrying CRISPR/Cas9 and an HDR template to the TA successfully replaced exon 53 with an efficiency of 0.18%. 25 Finally, Lee et al. obtained an HDR frequency of 0.8% in mdx mice via local delivery using gold nanoparticles (CRISPR-Gold). 93 HDR efficiency was raised to 5.4% when mdx mice were simultaneously treated with cardiotoxin, which activates the proliferation of muscle stem cells. 93,94 Although several potential HDR-based strategies have been developed to repair mutations in other forms of MDs, no in vivo studies have been conducted to date. 95
The inability of HDR-based techniques to restore clinically significant quantities of protein in muscle cells prevents their use as a treatment for MD. These findings demonstrate the limitations of HDR and the need for alternative strategies to improve its effectiveness in muscle.
NHEJ-mediated exogenous DNA knock-in
NHEJ is the predominant DNA repair pathway in muscle cells that corrects DSBs. Unlike HDR, NHEJ does not require a homologous repair template. Homology-independent targeted integration (HITI) relies on CRISPR/Cas9 and NHEJ to achieve precise DNA knock-ins. Pickar-Oliver et al. used HITI to knock-in exon 52 of DMD in a humanized mouse model (hDMDΔ52/mdx) both locally and systemically. 96 Localized delivery to the TA using AAV9s resulted in knock-in rates of 0.24–0.67%. In addition to reintroducing a single exon, a novel strategy using a “superexon” donor was investigated.
This knock-in template includes DMD exons 52–79 and a synthetic poly-A signal, which has the potential to correct many DMD mutations located downstream of exon 51. Delivery of the superexon to TAs resulted in knock-in rates of 0.17%. This treatment was also administered systemically and resulted in knock-in efficiencies of 0.15–3.05% across multiple tissues, including both skeletal and cardiac muscles. Although generally more efficient at introducing exogenous DNA sequences to the genome of muscle cells, NHEJ-based knock-in strategies are still too inefficient to warrant translation to clinical trials. Further investigation is required to improve the efficiency of this approach in skeletal and cardiac tissue.
Advantages, limitations, and future directions of CRISPR/Cas9-mediated exogenous DNA knock-Ins
Low editing efficiencies in muscle are a major hurdle slowing the development of DNA knock-in strategies to treat MDs. Effective integration of exogenous DNA would enable a range of new CRISPR applications, including those unrelated to MDs. Due to the post-mitotic nature of myotubes and satellite cells, alternative strategies to HDR must be developed to allow for efficient editing in these cells. 97,98 HITI, homology-mediated end joining (HMEJ), and prime editing are three emerging techniques that could address this challenge. 96,99,100
It is proposed that HMEJ occurs via single-strand annealing using a modified DNA donor template that contains sgRNA target sites for its excision from a delivery vector. AAV9 delivery into the mouse cortex resulted in a transduced neuron population with a knock-in rate of 50%. An alternative in vitro study on human-derived IPSCs and myoblasts demonstrated knock-in rates of 6–30% when inserting the full DMD transcript. 101 HMEJ is a promising strategy for the correction of a multitude of MD mutations, although further investigation is required regarding its efficacy in vivo.
Cas9 nickase and reverse transcriptase proteins are fused to create prime editors. 100 Utilizing a 3′-extended pegRNA encoding the required template, comparable in vitro knock-in rates to HDR have been achieved. Chemello et al. rescued a deletion of DMD exon 51 by delivering a prime editor to patient-derived IPSCs. 85 This strategy aimed at restoring the expression of the gene by reframing exon 52 through a +2 insertion and achieved a correction rate of 20.2%. Prime editing, however, is highly inefficient at introducing larger sequences of exogenous DNA, such as those the size of exons. 100,102
Novel strategies such as twin-PE allow for the introduction of large sequences into the genome using prime editors and integrases together. 102 This creates the possibility of designing systems that can integrate entire gene transcripts, including engineered promoter regions to express proteins of interest at physiologically relevant levels. For in vivo MD applications, however, downsizing of the technology and validation in muscle cells are still required. It, nevertheless, presents a promising avenue to pursue.
IN VIVO CRISPR/Cas 9 DELIVERY SYSTEMS
Progress and current challenges with adeno-associated viral vectors
Adeno-associated vectors have emerged as a promising tool for gene therapy in MDs. 103,104 One of the most promising AAV serotypes for MD gene therapy is AAV9, which has shown remarkable efficacy in preclinical studies. 105 –108 AAV9 has a broad tropism for various cell types, including muscle cells, and can efficiently transduce both skeletal and cardiac tissues. 109 However, AAVs also have limitations and disadvantages that must be considered. One of the main disadvantages is their limited cargo capacity. 109
In addition, AAVs can elicit an immune response, particularly on repeated administration or due to pre-existing immunity in some patients. 110 –113 Further, the production and purification of AAV vectors is currently complex and expensive, leading to high manufacturing and treatment costs for patients. 32 Despite these challenges, the use of AAVs for gene therapy is a promising approach, and ongoing research is focused on overcoming these limitations and further advancing the clinical application of AAV-mediated gene therapies for MDs.
In 2020, the Grimm group described a novel AAV capsid variant termed AAVMYO, which consistently outperformed AAV9s in skeletal and cardiac tissue when analyzing the level of transcribed cargo. 114 Another recent advance is the generation of a novel muscle-specific serotype termed MyoAAV1. 115 With a novel engineered capsid, MyoAAV1 promises higher specificity for muscle tissue and reduced tropism for the liver. Increasing the efficiency of cargo delivery to skeletal and cardiac cells while reducing the potential for side effects is associated with off-target transduction. Further research is needed to fully evaluate the safety and efficacy of AAVMYO and MyoAAV1 for the treatment of MDs.
DISCUSSION
Long-term considerations for the use of in vivo CRISPR gene editing
It is essential to understand the long-term consequences and effects of utilizing CRISPR-mediated editing as a therapeutic strategy. The stability of genomic manipulation in muscle cells is still poorly understood within the context of MD patients, with a key concern being the rate of muscle turnover. It is our opinion that without the correction of satellite cell populations, it is unlikely that the life-long benefits of a single, virally mediated treatment will be realized. This issue is especially pertinent for MDs, where the optimal therapeutic window is early childhood, before the progression of more severe symptoms. 1,86 Rapid muscle growth at this age may quickly render CRISPR therapies that target non-satellite cells ineffective through the loss or dilution of corrected nuclei.
Although AAVs are considered to be a relatively efficient vector for use in humans, adverse effects have been documented, which may limit their clinical utility. Immunogenicity against the virus and the Cas9 machinery, as well as integration of the AAV into the genome, further exacerbate immunological and genotoxic concerns. 113,116,117 In a study by the Gersbach group, a humoral and cellular immune response was observed in adult mdx mice when treated by AAV8/9s carrying an SaCas9, which was mostly avoided when the same treatment was given to P2 neonates. 117
AAV-based delivery is the current gold standard for FDA-approved systemic treatments, and the dose-dependent adverse effects of AAV, such as temporary thrombocytopenia and transaminitis, can be effectively managed. 112,118 The Duan group evaluated intramuscular and intravenous Cas9 and sgRNA delivery in three distinct canine models of DMD to measure the effect of pre-existing Cas9 immunity. 119 They discovered that AAV-mediated expression of CRISPR reagents not only promoted editing of the DMD gene but also induced a B and T cell-based immune response to Cas9, which reduced the extent and duration of dystrophin restoration.
Expression of a novel protein that was absent during the removal of self-reactive lymphocytes can stimulate immunological responses. 113 Anti-dystrophin antibodies and immunological rejections have been observed in DMD animals who have received remedial treatments. 120 Several studies have also demonstrated significantly higher rates of AAV integration into the genome than previously thought. 113,116,117 AAV genomes can spontaneously integrate into the AAVS1 safe harbor site on chromosome 19 and were previously thought to rarely integrate into DSBs created in the genome by Cas9. 121
Across two studies, the Gersbach group reports between ∼1 and ∼5% of editing outcomes, resulting in AAV genome integration in both skeletal and cardiac tissue when Cas9 nucleases were delivered systemically to mice. 96,117 AAV integration events can pose serious immunogenic concerns when paired with pre-existing Cas9 immunity. Hence, along with pharmacological options such as immunosuppressants to counter immune reactions, extensive safety monitoring will be required during clinical trials to reduce immune response in patients.
Conclusions and future directions for the MD gene editing field
Current innovations in the application of CRISPR/Cas9 have created various new treatment prospects for MDs. There is tangible excitement in this field, but there continues to be a lot of necessary work ahead as we begin to consider clinical trials for these therapeutic avenues. Here, we lay out four areas that require the most focus: efficient targeting of satellite cells, improved editing efficiency in muscle tissue, vector enhancements, and immunogenic response (Fig. 2).
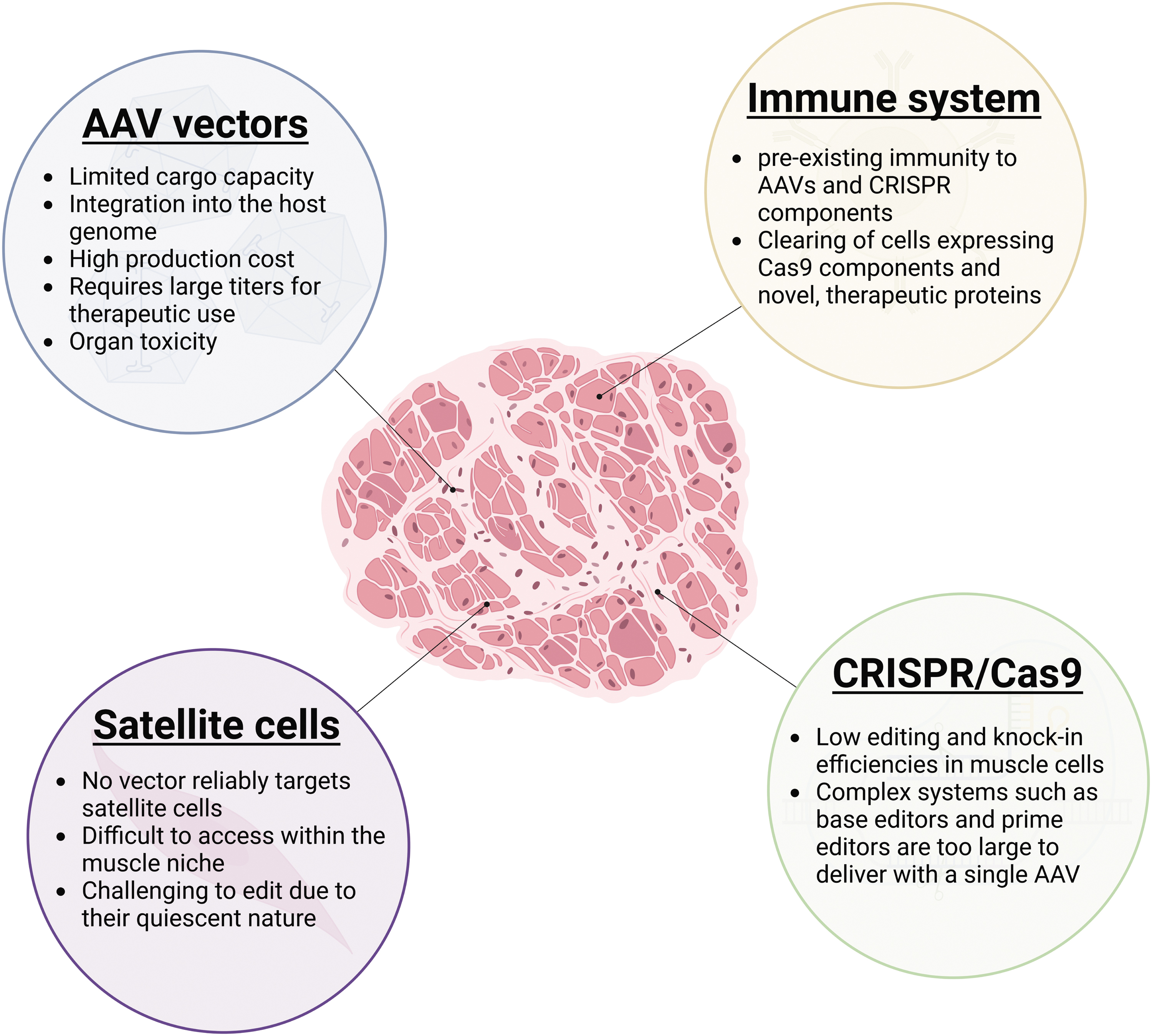
Current challenges with in vivo CRISPR/Cas9 genome editing for the treatment of MDs. An overview of the technical difficulties associated with the use of AAVs for MD therapies, delivery of CRISPR/Cas9 components to satellite cells, immune responses to targeted or edited cells, and CRISPR/Cas9 editing efficiencies in muscle. AAVs, adeno-associated viruses.
An ideal MD therapy would target satellite cells, and so it is imperative to determine if currently available serotypes can target these cells efficiently. If not, further work needs to be done to design or evolve a novel serotype that can efficiently deliver gene editing components to these targets. Other systems, such as virus-like particles or nanoparticles, are also potential candidates for this role but require further in vivo testing regarding efficacy and safety.
CRISPR/Cas9 editing efficiencies are lower than desired when targeting muscle tissue. Although less than ideal rates of correction in muscle are partly due to the challenges associated with delivery of editing machinery to cells, there is a need to further develop more efficient systems and strategies. Tools such as base and prime editors are promising but require further development and in vivo validation. Knock-in strategies allow for restoration of the wild-type proteins; however, current approaches such as HDR are too inefficient. Development of systems reliant on HMEJ and integrases could open new opportunities to develop MD therapies based on DNA knock-ins.
AAV9s will likely be used in the first generation of CRISPR therapeutics for MDs, given their track record in in vivo investigations and clinical trials. AAV9s may be the best option available, but future therapies will certainly require an improved delivery mechanism. It may be in the form of nanoparticles, as they are highly modular and can carry a range of transiently expressed cargo. 122,123 By re-administering nanoparticles to counteract the consequences of muscle turnover, musculoskeletal dystrophies could be cured.
The scarcity of published studies reveals that systemic muscle delivery by nanoparticles remains a formidable obstacle, most likely due to the large quantity of muscle in the human body and the necessity to target muscles within the thoracic cavity. Future research should prioritize the development of an effective, muscle-specific nanoparticle that can be delivered through the circulatory system.
The immune system poses a significant, multi-dimensional challenge for gene therapy strategies. Pre-existing immunity toward AAVs and Cas9 can pose a health risk to patients. Clearing of edited cells by the immune system due to the expression of Cas9 or the therapeutically relevant protein can reduce the effectiveness of any strategy. The use of immunosuppressants is a potential strategy to address the immune response elicited by CRISPR gene therapies, particularly in the case of repeated administrations. 124 –126 However, immunosuppressants increase the risk of infections, cancer, and potential organ damage. Further work is required to investigate innate and humoral responses to CRISPR/Cas9 gene editing strategies in vivo and potential solutions to these challenges.
Although there are challenges to bringing therapeutic genome editing for MDs closer to the clinic, many exciting advances have been made to date in the field of gene-editing in MDs. Ongoing research focusing on improving delivery vehicles and new formulations of CRISPR components such as mRNA and ribonucleoproteins for transient and tissue-specific delivery will undoubtedly bring this technology to the forefront of clinical translation.
Footnotes
ACKNOWLEDGMENTS
The Cohn and Ivakine lab members are gratefully acknowledged for their input in this work.
AUTHORs' CONTRIBUTIONS
S.F.: Conceptualization, Data curation, Writing—Original draft, Writing—Review and editing, Visualization.
R.M.M.: Conceptualization, Data curation, Writing—Review and editing, Visualization.
M.J.R.: Conceptualization, Data curation, Writing—Review and editing.
L.P.: Conceptualization, Data curation, Writing—Review and editing.
E.A.I.: Conceptualization, Writing—Review and editing, Supervision.
R.D.C.: Conceptualization, Writing—Review and editing, Supervision.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was funded by the Canadian Institutes of Health Research (Ronald D. Cohn and Evgueni A. Ivakine), the McArthur family (Ronald D. Cohn), Jesse's Journey (Ronald D. Cohn), and the Michael Hyatt Foundation (Ronald D. Cohn).