
Abstract
Autosomal dominant Alzheimer's disease (ADAD) is a rare early-onset form of Alzheimer's disease, caused by dominant mutations in one of three genes: presenilin 1, presenilin 2, and amyloid β precursor protein (APP). Mutations in the presenilin 1 gene (PSEN1) account for the majority of cases, and individuals who inherit a single-mutant PSEN1 allele go on to develop early-onset dementia, ultimately leading to death. The presenilin 1 protein (PS1) is the catalytic subunit of the γ-secretase protease, a tetrameric protease responsible for cleavage of numerous transmembrane proteins, including Notch and the APP. Inclusion of a mutant PS1 subunit in the γ-secretase complex leads to a loss of enzyme function and a preferential reduction of shorter forms of Aβ peptides over longer forms, an established biomarker of ADAD progression in human patients. In this study, we describe the development of a gene therapy vector expressing a wild-type (WT) copy of human PSEN1 to ameliorate the loss of function associated with PSEN1 mutations. We have carried out studies in mouse models using a recombinant AAV9 vector to deliver the PSEN1 gene directly into the central nervous system (CNS) and shown that we can normalize γ-secretase function and slow neurodegeneration in both PSEN1 conditional knockout and PSEN1 mutant knockin models. We have also carried out biodistribution studies in nonhuman primates (NHPs) and demonstrated the ability to achieve broad PS1 protein expression throughout the cortex and the hippocampus, two regions known to be critically involved in ADAD progression. These studies demonstrate preclinical proof of concept that expression of a WT human PSEN1 gene in cells harboring a dominant PSEN1 mutation can correct the γ-secretase dysfunction. In addition, direct administration of the recombinant AAV9 into the NHP brain can achieve broad expression at levels predicted to provide efficacy in the clinic.
INTRODUCTION
Alzheimer's disease (AD)
For most cases of EOAD, the etiology is unknown, but ∼15% of EOAD is driven by mutations in one of three genes—presenilin 1 (PSEN1), presenilin 2 (PSEN2), or amyloid β-precursor protein (APP). This rare genetic form of AD, termed Autosomal dominant Alzheimer's disease (ADAD), accounts for <1% of all AD but its monogenic etiology provides an opportunity to pursue a gene therapy approach to slow or halt disease progression. 2 Given that mutations in PSEN1 account for ∼60–70% of all ADAD, the majority of these patients can benefit from a gene therapy approach.
The presenilin 1 protein (PS1) is the catalytic subunit of a tetrameric aspartyl protease known as γ-secretase, which is responsible for proteolysis of several substrates, including APP, the precursor of Aβ peptides. More than 200 different pathogenic mutations have been identified in PSEN1, with most mutations resulting in a loss of γ-secretase function (LOF), decreased production of total Aβ, and disruption of several neuronal homeostatic processes. 3 –8 Although total Aβ production is reduced, shorter forms of the peptide, including Aβ40, are preferentially decreased, leading to an increase in the Aβ42:40 ratio. 6,7 The relative increase in the more amyloidogenic Aβ42 relative to Aβ40 is associated with amyloid deposition, which is characteristically observed in the brains of ADAD patients.
The role of amyloid deposition in AD progression remains controversial, but the causative role of PSEN1 mutations in ADAD is unequivocal. Although the underlying PSEN1 mutations, which cause ADAD are not present in LOAD, pathological biomarkers and clinical symptoms are very similar between the early and late-onset forms of the disease. 9 Thus, the extensive body of work that has been carried out to understand the natural history of LOAD is relevant to the less-prevalent ADAD and can be leveraged in preclinical and clinical development of a therapeutic.
While amyloid plaques and neurofibrillary tangles are diagnostic for LOAD, the lack of a single genetic target has made gene therapy more challenging in the sporadic disease population. In contrast, the monogenic etiology and well-understood biochemical mechanisms causative in ADAD provide a clear path for the development of a targeted gene therapy. Herein we describe a gene replacement approach targeting ADAD patients with mutations in PSEN1. We have used the adeno-associated virus AAV9, to deliver a wild-type (WT) human PSEN1 into the central nervous system (CNS) of mouse models of ADAD and have demonstrated rescue of biochemical deficits and slowing of neurodegeneration. We have also demonstrated CNS biodistribution to disease-relevant regions in a nonhuman primate (NHP) brain following several routes of administration (ROAs). These results are encouraging for the continued development of a gene therapy that can effectively treat ADAD patients harboring PSEN1 mutations.
MATERIALS AND METHODS
AAV vector
For murine studies AAV9-PS1 was manufactured in adherent HEK293 cells by calcium phosphate triple transfection with the gene of interest plasmid (p45-U1.5), the adenovirus helper plasmid (pHelper), and the Rep/Cap plasmid (pRC9) (manufacturing carried out by SAB Tech, Philadelphia, PA, USA). The p45-U1.5 plasmid contains a human influenza hemagglutinin (HA) tag appended to the N-terminus of the human PSEN1 transgene to allow the exogenous human protein to be distinguished from the endogenous mouse PSEN1 protein. The HA-PSEN1 transgene was placed under the transcriptional control of the ubiquitin C (UBC) promoter. AAV9-PS1 was purified by double cesium chloride gradient ultracentrifugation. Vector purity was assessed by silver staining and titered by ddPCR. Measured endotoxin levels were below-detectable limits (<0.1 EU/mL). AAV9-PS1 vector was resuspended for dosing in phosphate-buffered saline (PBS) containing 0.001% Pluronic F-68.
For NHP studies, AAV9-PS1 was manufactured by triple transfection of HEK293 suspension cultures using p45-U1.5, pHelper, and pRC9 plasmids as described above (manufacturing carried out at CRL, Rockville, MD, USA). Viral purification was achieved by AAV9 affinity chromatography with enrichment for full capsids by iodixanol step gradient ultracentrifugation. The resulting vector was assessed for purity by silver staining, empty:full capsid ratio by analytical ultracentrifugation (>80%), sterility, endotoxin burden, host cell contaminants, and the presence of mycoplasma. The final AAV9-PS1 preparation was suspended in 1.058 mM KH2PO4, 154 mM NaCl, 5.6 mM Na2HPO4, 5% Sorbitol, and 0.001%F-68 at pH 7.3. The vector was titered by ddPCR with a final concentration of 9.5 × 1013 vg/mL.
Administration of AAV9-PS1 to mouse models
Generation of conditional double KO (cDKO) and Psen1 L435F knockin mice has been described previously. 6,7,10 Experiments were conducted at Charles River Laboratories (South San Francisco, CA, USA) in accordance with protocols approved by the Institutional Animal Care and Use Committee. Mice received bilateral intraparenchymal infusions of AAV9-PS1 (1 × 1013 vg/mL), or formulation buffer (vehicle), in the dorsal hippocampus (A/P: −1.5, M/L: ±1.5, D/V: −1.4) and lateral cortex (A/P: −2.0, M/L: ±2.8, D/V: −1.4), in a volume of 1.5 μL/injection at a rate of 0.3 μL/min (6 × 1010 total dose/animal). Each infusion was followed by a 5-min rest period.
Before surgery, mice were anesthetized using isoflurane. Bupivacaine was used for local analgesia and carprofen for peri-/postoperative analgesia. The animals were placed in a stereotaxic frame (Kopf Instruments, USA). Coordinates were zeroed on Bregma, and skull flatness was measured. A Hamilton syringe (model no. 80308; 10-μL syringe with corresponding 30-gauge blunt tip needle) and a stereotactic micromanipulator were used, and the location of the burr holes were designated and drilled. The drill was only used to penetrate the bone. Dura mater was gently removed with a needle and the infusion needle was slowly lowered into the brain to the depth of the desired location over ∼1 min. The injections were controlled by an ultra-micropump III with microcontroller (World Precision Instruments, or similar). The needle was lowered to D/V −0.1 below coordinates, and allowed to equilibrate for 1 min, before being drawn back to the correct position. When delivery was completed, there was a wait period of 5 min before the needle was withdrawn over ∼1 min.
Once all infusions were completed, animals were sutured and allowed to recover the righting reflex, then moved back to their colony room. Animal health was monitored closely until terminal procedure day.
Eight weeks postinfusion, animals were deeply anesthetized through isoflurane followed by transcardial perfusion with a volume of PBS corresponding to 1 mL/g of body weight. The brains were extracted from the skull and divided into two hemispheres with a midsagittal cut. The right hemibrain was immersion fixed in fresh 4% paraformaldehyde in PBS for 2 days at 4°C, transferred into PBS, and reserved for sectioning and immunohistochemistry (IHC). The left hemibrain was placed into a brain matrix to allow for evenly spaced sectioning and further dissected to collect 10 total brain samples, including: olfactory bulb, hindbrain (posterior to the cerebral cortex), and 8 evenly spaced ∼1 mm sections. Terminal cerebrospinal fluid (CSF) was collected through the cisterna magna and flash frozen.
Administration of AAV9-PS1 to NHPs—Route of Administration Selection Study
This study was performed at Virscio, Inc., using adult African green monkeys (Chlorocebus aethiops sabaeus, 4–6 kg). The non-GLP study was conducted in a Virscio exam and procedure room located on the St. Kitts Biomedical Research Foundation campus, Lower Bourryeau Estate, St. Kitts, West Indies. Study animals were housed in a Virscio primate enclosure at the facility. Procedures were conducted according to facility Standard Operating Procedures (SOPs) and the study protocol and associated amendments.
Prescreening of animals involved physical exams, clinical chemistries, and complete blood counts, and serum samples were collected for analysis of anti-AAV9 capsid/neutralizing antibody (NAb). Animals in good health and with low NAb titers (no activity at 1:5 serum dilution in AAV9 luciferase assay) were recruited into one of three groups receiving test article through intracerebroventricular (ICV) (n = 3), intrathalamic (n = 3), or intrahippocampal (n = 3) routes. In addition, three animals received vehicle treatment through each of the three routes mentioned above (n = 1/route). Before the study, animals received methylprednisolone acetate (10 mg/kg) for immunosuppression.
Monkeys were fasted for 12–16 h and received atropine sulfate (0.02–0.04 mg/kg IM) and penicillin-G benzathine (60,000 IU/kg IM), before anesthesia. Animals were then sedated with ketamine (8 mg/kg, IM) to allow a peripheral IV line and endotracheal tube to be placed, after which animals were maintained on isoflurane anesthesia. Buprenorphine (0.005–0.010 mg/kg, IM) and meloxicam (0.2 mg/kg, SC) were administered for prophylactic analgesia. The surgical area was shaved and prepped with chlorhexidine scrub before sterile draping and application of a Betadine impregnated barrier. The head was fixed into a primate stereotaxic unit, using the external meatus of the ear and the inferior orbital ridges as the fixation points, and checked to ensure orientation in the correct plane and the exact midline. Stereotaxic coordinates were confirmed for each target by reference to baseline MRI images and stereotaxic atlases. A midline incision was performed on the scalp and the temporalis muscle on each side was retracted using a periosteal elevator sufficient to expose the stereotaxic target for the procedure.
Using a stereotaxic guided drill, one craniotomy (∼2.0 mm) was performed on each side for bilateral dosing of the appropriate target. Once the drill penetrated the skull and the dura was exposed and resected, a Kopf stereotaxic apparatus was used to allow stable advance and positioning of the cannula tip within the specific injection site as described for each site below. The cannula was lowered to the target coordinates within a few seconds. During the insertion, the microinjector syringe pump infused at a rate of 3 μL/min as the cannula descended to avoid clogging of tissue within the cannula tip. After the initial dosing on the right side, the left side was similarly prepared and dosed.
ICV route
A 500-μL Hamilton syringe was connected to a Clearpoint Neuro SmartFlow cannula (NGS-NC-01), both previously filled with AAV9-PS1 for delivery (2.5 × 1013 vg/hemisphere in 500 μL). The syringe was mounted to a stereotaxic syringe pump and the cannula was attached to a stereotaxic needle carrier. The target coordinates were determined by presurgical MRI. The target coordinates for the lateral ventricles were ear bar zero (EBZ) +15.0 mm anterior and 3.0 mm right or left of midline, respectively. The depth was 15.0 mm. The cannula was swiftly lowered to the target coordinates with test article infusing at 3 μL/min as described above. Once the cannula reached the target depth, the infusion rate was changed to 5 μL/min for 20 min and increased to 10 μL/min for 40 min for a total infusion time of 60 min. After delivery, with the pump off, the cannula was maintained in place for 5 min to allow pressures to equilibrate, then slowly withdrawn at a rate of 2 mm/min. The test article syringe was refilled, ensuring no air bubble formed in the cannula, and the procedure was repeated on the opposite side to complete bilateral dosing.
Intrathalamic route
A 500-μL Hamilton syringe was connected to a Clearpoint Neuro SmartFlow cannula (NGS-NC-01), both previously filled with the test article for delivery (3.5 × 1012 vg/hemisphere in 400 μL). The syringe was mounted to a stereotaxic syringe pump and the cannula was attached to a stereotaxic needle carrier. The target coordinates for the thalamus were: EBZ +12.0 mm anterior and 5.0 mm right or left of midline, respectively. The depth was EBZ +18.0 mm. The cannula was swiftly lowered to the target coordinates with test article infusing at 3 μL/min as described above. Once the cannula reached the target depth, the infusion began at 1 μL/min for 10 min and increased to 3 μL/min for another 30 min. At that time the infusion rate was increased to 5 μL/min for 60 min. The total infusion time was 100 min per hemisphere. After delivery, with the pump off, the cannula was maintained in place for 5 min to allow pressures to equilibrate. The cannula was withdrawn at a rate of ∼2 mm/min. The procedure was repeated on the opposite side to complete dosing.
Intrahippocampal dosing
A 500-μL Hamilton syringe was connected to a ClearPoint Neuro SmartFlow cannula (NGS-NC-01), both previously filled with the test article for delivery (2.5 × 1012 vg/hemisphere in 160 μL). The syringe was mounted to a stereotaxic syringe pump and the cannula was attached to a stereotaxic needle carrier. The target coordinates for the hippocampus were: EBZ +12.9 mm anterior and 5.0 mm right or left of midline, respectively. The depth was EBZ +5.0 mm. Once the cannula reached the target depth, the infusion began at 1 μL/min for 10 min and increased to 3 μL/min for another 10 min. At that time the infusion rate increased to 5 μL/min for 24 min. The total infusion time was 44 min for each hemisphere. After delivery, with the pump off, the cannula was maintained in place for 5 min to allow pressures to equilibrate, then the cannula was withdrawn at a rate of ∼2 mm/min, which allowed tissue to equilibrate. The 500-μL Hamilton syringe contained sufficient volume for two infusions, and the procedure was repeated on the opposite side to complete dosing.
Following each route of administration, the temporalis muscles were then repositioned and sutured across the midline with 2-0 absorbable sutures, followed by closure of the fascia with a 3-0 absorbable suture. The skin was closed with a 3-0 absorbable suture in a subcuticular pattern. Bupivacaine (Nocita, 1 mg/kg) was infused intradermally into the incision site to minimize local pain and discomfort. Antiseptic was applied to the closed skin incision and tissue adhesive was applied to the incision site. Penicillin (60,000 UI/kg IM QD × 5 days) was given postoperatively for antibiotic prophylaxis. Meloxicam (0.2 mg/kg, SC) was administered every 8–12 h for 72 h. Buprenorphine (0.015 mg/kg, IM) was administered 8–12 h for 48 h.
Necropsy
At the scheduled time for euthanasia, monkeys were sedated with ketamine (8–10 mg/kg, IM) and euthanized by an overdose of sodium pentobarbital (100 mg/kg, IV to effect). Upon loss of corneal reflex, transcardial perfusion was performed with cold heparinized saline for ∼15 min at 100 mL/min until the escaping fluid ran clear before tissue collection. Intact left-brain hemispheres were immersion-post-fixed for 72 h in one liter of 4% paraformaldehyde in 0.1 M PBS, pH 7.4 at room temperature. After ∼72 h, the left hemispheres were transferred to PBS +0.05% sodium azide for further sectioning and IHC. Spinal cord, dorsal root ganglia (DRG), heart, liver, spleen, and kidney tissues were removed and flash frozen for vector genome (vg) quantification. Right brain hemispheres were cut into ∼4 mm slabs using a brain matrix, laying the sections on a cold surface for further dissection. Brain tissue samples were punched with a 2 mm diameter tissue punch and flash frozen for vg quantification.
Administration of AAV9-PS1 to NHPs—Dose selection study
The second NHP study was performed by Northern Biomedical Research (NBR, Norton Shores, MI, USA) using cynomolgus monkeys (Macaca fascicularis, 1.5–3 kg, 2 years of age). The study was performed according to the Protocol, Amendments, and SOPs of NBR. Animals were enrolled and prepared for surgery in a similar manner to that described above. The study included nine animals in four groups that received intrathalamic injections of either a high dose (3.5 × 1012 vg/hemisphere, n = 3), mid dose (1.2 × 1012 vg/hemisphere, n = 2), low dose (3.5 × 1011 vg/hemisphere, n = 3), or vehicle (artificial CSF, n = 1).
Each hemisphere was injected with a volume of 200 μL containing 2 mM gadoteridol to enable visualization of the infusate using intraoperative MRI. Animals were placed in an MRI-compatible frame. A coronal incision was made over the calvarium, and a craniotomy made over the thalamus (bilateral). MRI scans were performed using a 3T Philips MRI and MRI-compatible frame. An anatomical MRI scan was performed to identify the infusion site. T1- and T2-weighted MRI scans were performed before cannula insertion. Following cannula insertion and through cannula removal, repeated T1-weighted MRI scans were performed to monitor distribution of the dosing solution within the brain. Upon identification of the target, a cannula guide was aligned to the target infusion site. Trajectories and target depth were confirmed by MRI. A small opening in the dura was made by passing a pointed ceramic lancet through the cannula guide to penetrate the dura.
A 16-gauge, 10-foot SmartFlow® Neuro Ventricular Cannula (Clearpoint Neuro) primed with the test article had depth stops attached at the target depth and was carefully inserted through the cannula guide to the target depth. The dose was administered through a calibrated MRI-compatible infusion pump (Harvard Apparatus Pump Model: PHD 2000 Infuse/Withdraw). The syringe containing dosing solution was attached directly to the cannula. The cannula was swiftly lowered to the target coordinates with test article infusing at 5 μL/min. Once inserted to the target, the rate was reduced to 1 μL/min. The rate was increased to 2, then 3, and finally 5 μL/min following visualization of infusate by MRI. The cannula remained in the target site for a minimum of 5 min following infusion completion. Necropsy was performed in a similar manner to that described above.
Immunohistochemistry
NHP and murine hemispheres were paraffin embedded and freeze sectioned at 40 μm thickness in coronal planes through the entire length of the specimen. IHC and counterstaining with Neutral Red was performed through the entire brain hemisphere on every 24th section spaced at 960 μm intervals. Anti-HA antibody recognizing the six amino acid HA-tag was used to detect exogenous HA-PS1 protein.
Morphological and histological analysis
To quantify mouse brain cortical thickness, Hematoxylin and Eosin-stained coronal sections with consistent morphology across all animals corresponding to figures 45–50 of the atlas of Franklin and Paxinos were selected in a blinded manner independently by two scientists. 11 Linear measurements were performed in parallel using the Concentriq digital pathology software package and three to six measurements were taken in each cortical area and averaged to yield mean thickness measures.
Amino cupric silver stain percent area measurements were obtained in an automated, unbiased manner using FIJI–ImageJ. 12 Whole coronal sections corresponding to figures 45–50 of the atlas of Franklin and Paxinos 11 were imaged at 20 × magnification. Coronal sections were processed to minimize background. Positive signal was identified using an Otsu threshold. Positively identified pixels were then normalized to the entire area of the coronal section as described previously. 13
Biochemical analysis
De novo Aβ production
Mouse tissue was homogenized in a bead mill using homogenization buffer (20 mM PIPES, 140 mM KCl, 250 mM Sucrose, 5 mM ethylene glycol tetraacetic acid [EGTA], pH 7.3). The homogenate was centrifuged at 1,000 rcf for 10 min at 4°C. The supernatant was then ultracentrifuged at 100,000 rcf for 30 min at 4°C. The supernatant was then removed, and the pellet was resuspended in CHAPSO buffer (50 mM PIPES, 250 mM Sucrose, 1 mM EGTA, 10 mg/mL CHAPSO) and incubated on ice for an hour. This solubilized membrane preparation was then ultracentrifuged for 30 min at 100,000 rcf at 4°C. Protein content of the supernatant was measured using a micro bicinchoninic acid (BCA) assay. The membrane preparation (15 μg) was incubated with C99 substrate for 2 h in assay buffer consisting of 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 300 mM NaCl, 10 mM EDTA, 20 μM phosphoramidon salt, 10 mM 1,10 phenanthroline, and 100 μg phosphatidylcholine. After 2 h, the reaction was quenched with 1 × Diluent 35 (MSD). Quantification of Aβ species was performed following the manufacturer's protocol (Meso Scale Diagnostics, Rockville, MD, USA).
Endogenous Aβ detection
Brain tissue was homogenized using 0.2% diethanolamine with 50 mM NaCl using a bead mill. The homogenate was then incubated on ice for 3 h. The sample was then ultracentrifuged at 100,000 rcf for 30 min at 4°C. The supernatant contains soluble Aβ. The pellet was resuspended in 5M guanidine-HCl, 50 mM tris-HCl, at pH 8.0. Both the soluble fraction and the resuspended insoluble fraction were then run on a 60 mg Oasis HLB LP 96-well plate using the manufacturer's protocol. The eluted fraction was vacuum centrifuged at 300 rpm at 60°C for 2 h. Once the sample was completely dried, it was resuspended in Diluent 35 and Aβ species were quantified following the manufacturer's protocol (Waters Corporation).
Vector genome assay
Brain tissue was homogenized using a bead mill in PBS with protease inhibitor. Genomic DNA was isolated using the Qiagen DNA Blood and Tissue Kit (Qiagen, Venlo, the Netherlands). For vg quantification, known plasmid was used for the standard curve. Genomic DNA (50 ηg in 2 μL) was loaded per well in a 384-well dish along with 5 μL of 2 × Reaction Master Mix, 0.5 μL of 20 × primer, and 2.5 μL H2O. The quantitative polymerase chain reaction (qPCR) was performed in ThermoFisher QuantStudio 6 using a standard protocol. Vector genomes/diploid cell was calculated using the absolute number of DNA molecules based on a standard curve and dividing by diploid genomes per nanogram of DNA.
Western blot
Brain tissue homogenate was incubated with RIPA buffer containing protease inhibitor at 1:1 ratio of homogenate:radioimmunoprecipitation assay (RIPA) buffer for 1 h on ice. A BCA protein assay (Thermo Fisher) was performed on the sample and 25 μg of protein was incubated with 4 × loading dye on ice for 10 min before loading on a gel for the PS1 N-terminal fragment (PS1-NTF) or at 90°C for 10 min for amyloid precursor protein C-terminal fragment (APP-CTF). The primary antibody used for APP-CTF was RbαAPP-CTF (Y188; Abcam, Cambridge, United Kingdom). The primary antibody used for PS1-NTF was MsαPS1 (APS 11; Abcam) and the primary antibody for HA-PS1 was MsαHA (2-2.2.14; Thermo Fisher). The secondary antibodies used for detection were from LI-COR (IRDye 800CW GtαMS and GtαRb). All western blot experiments were conducted blinded and blot images were analyzed by a different scientist using FIJI–ImageJ.
RESULTS
Distribution of HA-PS1 in the mouse brain
We designed a recombinant AAV9 vector (AAV9-PS1) containing a codon-optimized human PSEN1 cDNA, under the transcriptional control of the UBC promoter, which is known to provide strong expression in most CNS cell types. 14 To distinguish vector-derived PS1 from endogenous PS1, we appended a human influenza HA tag to the N-terminus of the PSEN1 sequence (HA-PS1), in a manner previously shown not to impair the function of the PS1 protein. 5
To assess the ability of the AAV9-PS1 vector to drive expression of a functional HA-PS1 protein in vivo, we initiated target-engagement studies with the Psen1/2 cDKO mouse. 10 This model has a germline KO of PSEN2 in all cells and conditionally lacks PSEN1 in postnatal forebrain neurons. These mice display a loss of presenilin function and γ-secretase activity in excitatory forebrain neurons, leading to biochemical and functional deficits, including neurodegeneration. 10,15
We first sought to demonstrate distribution and expression of AAV9-PS1 vector throughout the cDKO mouse brain using stereotaxic injection into the CA1 region of the hippocampus and the lateral cortex (SBF1). Bilateral, intraparenchymal administration of 1.5 × 1010 vector genomes (vg)/site (6.0 × 1010 vg total dose) resulted in robust expression of HA-PS1 protein throughout the dorsal cortex and hippocampus as shown by IHC 8 weeks following administration (Fig. 1A). This expression was overwhelmingly observed in neurons (>99%, data not shown) that express the neuronal marker NeuN (Fig. 1B), although some glial fibrillary acid protein (GFAP) + astrocytes proximal to the infusion site also expressed HA-PS1 protein (Fig. 1C).

Expression of HA-PS1 in Psen1/2 cDKO mouse brain tissue.
Presenilin is proteolytically processed into amino-terminal and carboxy-terminal fragments (NTF and CTF, respectively) during maturation of the γ-secretase complex. 16 To assess whether administration of AAV9-PS1 produced a functional HA-PS1 protein, we measured the levels of HA-PS1-NTF detectable in cDKO mouse brain following administration. HA-PS1-NTF was detectable by immunoblot using probes against either the HA-tag or PS1 (Fig. 1D), indicating that HA-PS1 is both expressed and incorporated into the γ-secretase complex.
Normalization of γ-secretase function in the cDKO mouse model by AAV9-PS1
To determine the ability of exogenous HA-PS1 to restore γ-secretase activity in the cDKO model, we delivered AAV9-PS1 intraparenchymally to cDKO animals at 2 months of age as described above. Eight weeks postadministration, we assessed the impact of AAV9-PS1 on biochemical and functional deficits associated with the neuronal loss of PSEN1.
cDKO animals that were administered a vehicle control (n = 5) showed a marked loss of γ-secretase activity as quantified by dramatically reduced de novo enzymatic production of Aβ peptides in plasma membrane preparations (Fig. 2A) and accumulation of the endogenous substrate APP-CTF in the brain tissue (Fig. 2B). In contrast, animals that were administered AAV9-PS1 (n = 5) showed restoration of γ-secretase function in brain lysates to levels observed in WT littermate control animals (Fig. 2A, B). Animals that received AAV9-PS1 also showed an increase in Aβ40 and Aβ42 peptides in the CSF as compared with animals who received vehicle treatment (Fig. 2C). In a subsequent dose-ranging study, we confirmed that AAV9-PS1 can normalize de novo production of Aβ peptides and reduce accumulation of the endogenous substrate APP-CTF in the brain in a dose-dependent manner (Supplementary Fig. S1A, B).

Restoration of γ-secretase function in the Psen1/2 cDKO mouse model.
Previous studies have established that cDKO animals exhibit progressive cortical thinning and neurodegeneration starting at ∼4 months of age. 10,17 Remarkably, 8 weeks following administration of AAV9-PS1 to 2-month-old cDKO mice we observed a statistically significant increase in cortical thickness compared with vehicle-treated controls (p < 0.0001) with no significant difference from age-matched WT control mice (p = 0.99) (Fig. 2D). This normalization of cortical thickness is likely due to prevention of neuronal loss that has been previously shown to occur in the absence of PS1. 10
To measure the effect of AAV9-PS1 on neuronal degeneration, we used amino-cupric-silver staining, which binds to degenerating neurons, and employed unbiased quantitative image analysis to identify the percent of positive staining present across coronal sections of cDKO animals treated with AAV9-PS1 or vehicle. We observed a significant reduction in the percent positivity in AAV9-PS1-treated animals compared with vehicle treatment (p < 0.0001), and no significant difference between AAV9-PS1-treated mice and age-matched WT control animals (p = 0.83) (Fig. 2E).
These data demonstrate that administration of AAV9-PS1 is sufficient to restore the biochemical and morphological deficits associated with loss of PS1 in cDKO animals.
To better define the minimum amount of virus required for efficacy in the cDKO model, we leveraged the diminishing levels of AAV9-PS1 present in the brain as distance from the parenchymal infusion site increased. Brain tissue from cDKO animals was sectioned in eight 1 mm increments along the rostral–caudal axis from olfactory bulb to hindbrain with the fourth section containing the infusion site. We then quantified both vector genomes/diploid cell (vg/dc) and the amount of γ-secretase activity (de novo Aβ peptide production) from each section (Fig. 2F). Using this approach, we determined that 0.5 vg/dc (Fig. 2F, section 2) is the minimum amount of virus needed to significantly rescue γ-secretase function in this model.
Normalization of γ-secretase function in the Psen1 L435F model of ADAD by AAV9-PS1
While the cDKO model is an excellent system to assess the distribution, expression, and function of AAV9-PS1 in vivo, the Psen1 L435F model more closely reflects ADAD as it harbors a pathogenic PSEN1 mutation that is known to cause ADAD in human patients, knocked into the endogenous mouse Psen1 locus. 7 Heterozygous Psen1 L435F animals exhibit decreased production of Aβ peptides, with greater reduction in shorter isoforms such as Aβ40, and more modest decreases in longer isoforms like Aβ42. This preferential reduction in the shorter forms of Aβ results in an increased ratio of Aβ42:40, a well-established biomarker of the pathogenesis of ADAD. 18
We administered AAV9-PS1 to 18-month-old Psen1 L435F mice as described above. At 8 weeks postadministration, Psen1 L435F animals showed robust expression of HA-PS1 protein proximal to the infusion sites as evidenced by IHC (Fig. 3A, arrows). Interestingly, animals treated with AAV9-PS1 (n = 9) showed a significant increase in shorter species of endogenous Aβ (Aβ38, Aβ40) compared with vehicle-treated controls (p < 0.001), with levels statistically indistinguishable from WT littermates (p = 0.061 and p = 0.17 for Aβ40 and Aβ38, respectively) (Fig. 3B).

Normalization of γ-secretase function in the heterozygous Psen1
L435F mouse model.
However, there was no significant effect of AAV9-PS1 treatment on Aβ42 levels (p = 0.079). As a consequence of the above changes in individual Aβ peptide levels, the Aβ42:40 ratio, which is a biomarker of ADAD, was significantly reduced in AAV9-PS1-treated mice versus vehicle controls, although not fully returning to WT littermate control levels (Fig. 3B). The lack of an effect of AAV9-PS1 treatment on Aβ42 levels was expected given that there is no reduction in this longer species in the vehicle-treated heterozygous Psen1 L435F mutant mouse (n = 7, Fig. 3B lower left panel). Therefore, treatment with AAV9-PS1 would not be expected to elevate Aβ42 above normal WT levels. These data demonstrate that vector-derived WT PS1 can outcompete mutant PS1 to normalize γ-secretase function.
Distribution of AAV9-PS1 in NHPs
Upon establishing the efficacy of AAV9-PS1 in two mouse models, we next focused on the challenge of delivering the virus to those brain regions that are early sites of ADAD pathology, including the cerebral cortex and the hippocampus. We carried out these studies in NHPs, given the greater similarity in brain size and neuroanatomy to the human brain. We assessed three bilateral ROAs in African green monkeys, including (1) parenchymal administration to the hippocampus (2.5 × 1012 vg/hemisphere, n = 3), (2) parenchymal administration to the thalamus (3.5 × 1012 vg/hemisphere, n = 3), and (3) ICV administration to the frontal horn of the lateral ventricle (2.5 × 1013 vg/hemisphere, n = 3). The animals were euthanized 4 weeks postadministration and CNS and peripheral tissues were isolated and analyzed by both qPCR to quantify AAV9-PS1 vector genomes (Fig. 4A–C), and IHC using an anti-HA antibody to assess protein expression throughout the brain (Fig. 5).
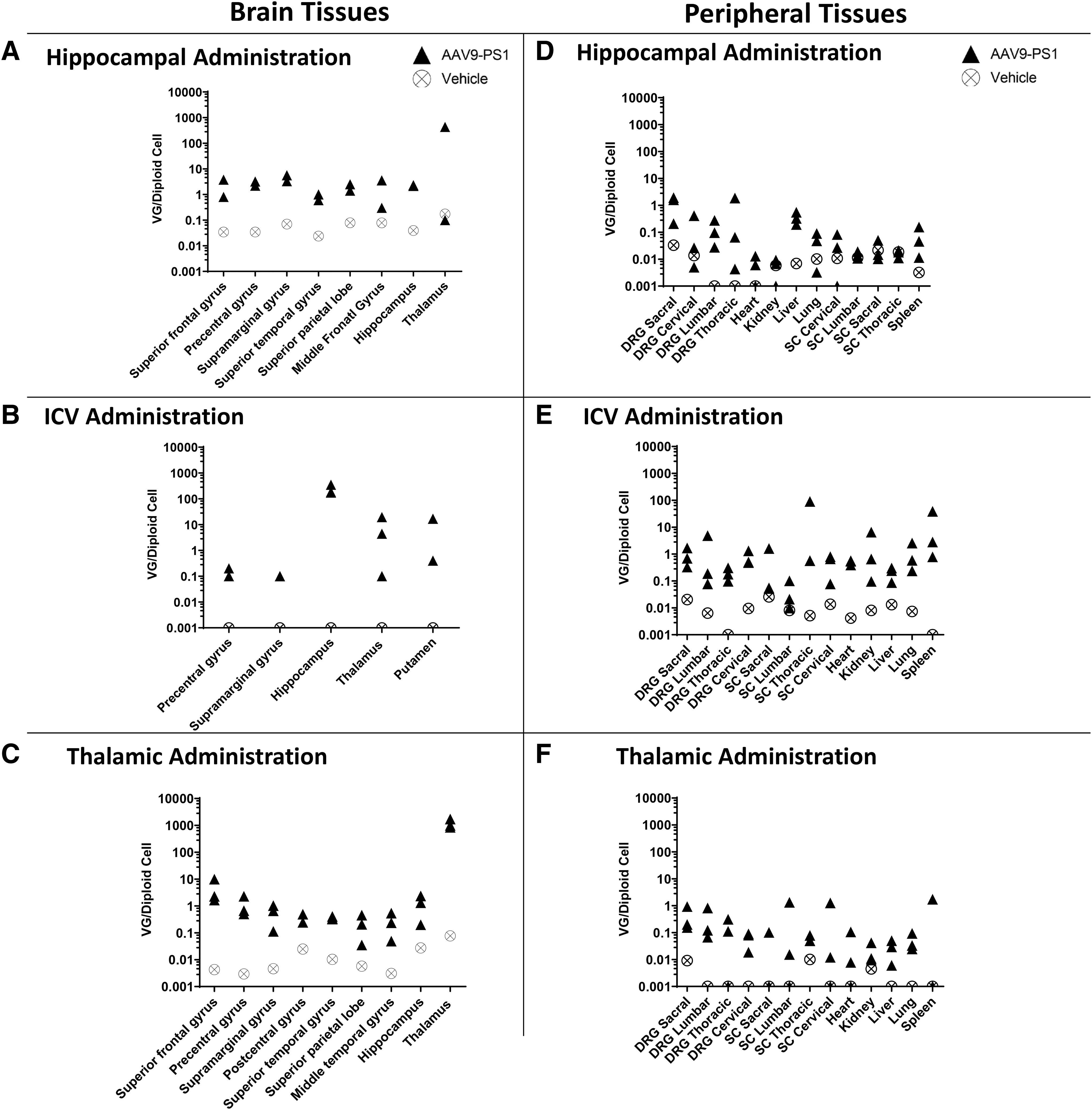
Vector genome quantification of AAV9-PS1 from NHP tissues following three different ROAs. AAV9-PS1 vector genomes were quantified by qPCR from tissue punches isolated from select cortical and subcortical brain regions of NHPs at 4 weeks following bilateral administration of AAV9-PS1 through

Expression of HA-PS1 protein in NHP brain tissue following three different ROAs. HA-PS1 expression measured by IHC at 4 weeks following administration of AAV9-PS1 by hippocampal
When AAV9-PS1 was administered into the hippocampus, high levels of AAV9-PS1 vector genomes (Fig. 4A) and robust expression of HA-PS1 (Fig. 5A) were observed within this structure with lower levels detectable outside of the hippocampus. ICV administration resulted in extensive distribution of AAV9-PS1 (Fig. 4B) and expression of HA-PS1 throughout the cortex, with limited expression in subcortical structures (Fig. 5B). Thalamic administration also resulted in broad cortical distribution of AAV9-PS1 (Fig. 4C) and expression of HA-PS1 throughout the cortex in addition to a wide range of subcortical structures, including the thalamus and hippocampus (Fig. 5C). Unsurprisingly, animals that received thalamic administration of AAV9-PS1 had very high levels of vector genomes and HA-PS1 protein expression in the thalamus. Notably, the pattern of HA-PS1 in the cortex differed between ICV and intrathalamic routes. ICV administration exhibited a more punctate pattern consistent with viral spread within the perivascular space (Fig. 5B).
In contrast, intrathalamic administration resulted in a more even distribution throughout the cortex (Fig. 5C) thought to be driven by anterograde transport along thalamocortical projection neurons as previously reported. 19 ICV administration resulted in cortical expression of HA-PS1 broadly along the rostral–caudal axis, while intrathalamic administration resulted in distribution from mid brain through the frontal cortex (Fig. 5D). Although the ICV and thalamic routes were chosen to achieve broad cortical expression, we also observed hippocampal expression of HA-PS1 in animals that were treated through these ROAs (Fig. 5D, note circled regions).
To maximize therapeutic efficacy and minimize toxicity, it is important to achieve biodistribution of AAV9-PS1 in the brain while reducing distribution in off-target tissues, such as the spinal cord, DRG, and peripheral organs. To assess the peripheral spread associated with each ROA, we quantified AAV9-PS1 vector genomes present in the spinal cord and DRG from the sacral, lumbar, thoracic, and cervical regions, and in the heart, kidney, liver, lung, and spleen (Fig. 4D–F). We observed a large difference between animals that received ICV administration versus those that were treated through a parenchymal route, with ICV animals showing higher levels in the spinal cord, DRG, and peripheral tissues when compared with either hippocampal or thalamic administration.
We selected intrathalamic administration for a second dose-ranging study based on the broad expression of HA-PS1 observed throughout the cortex and several subcortical structures with this route. In this study cynomolgus macaques received intrathalamic infusion of either a high (3.5 × 1012 vg/hemisphere, n = 3), medium (1.2 × 1012 vg/hemisphere, n = 2), or low dose (3.5 × 1011 vg/hemisphere, n = 3), seeking to identify the minimum dose required to reach the efficacy threshold as defined in the earlier mouse model study (Fig. 2F). The animals were euthanized 4 weeks postadministration and brains were processed for HA-PS1 expression by IHC using an anti-HA antibody and quantification of AAV9-PS1 by qPCR from tissue captured from 32 brain regions. Expression of HA-PS1 showed a clear dose–response by IHC evaluation (Fig. 6A). In addition, all three doses resulted in distribution of AAV9-PS1 that reached or exceeded our previously defined efficacy threshold of 0.5 vg/dc for most brain regions analyzed (Fig. 6B, data shown for high- and low-dose groups only).
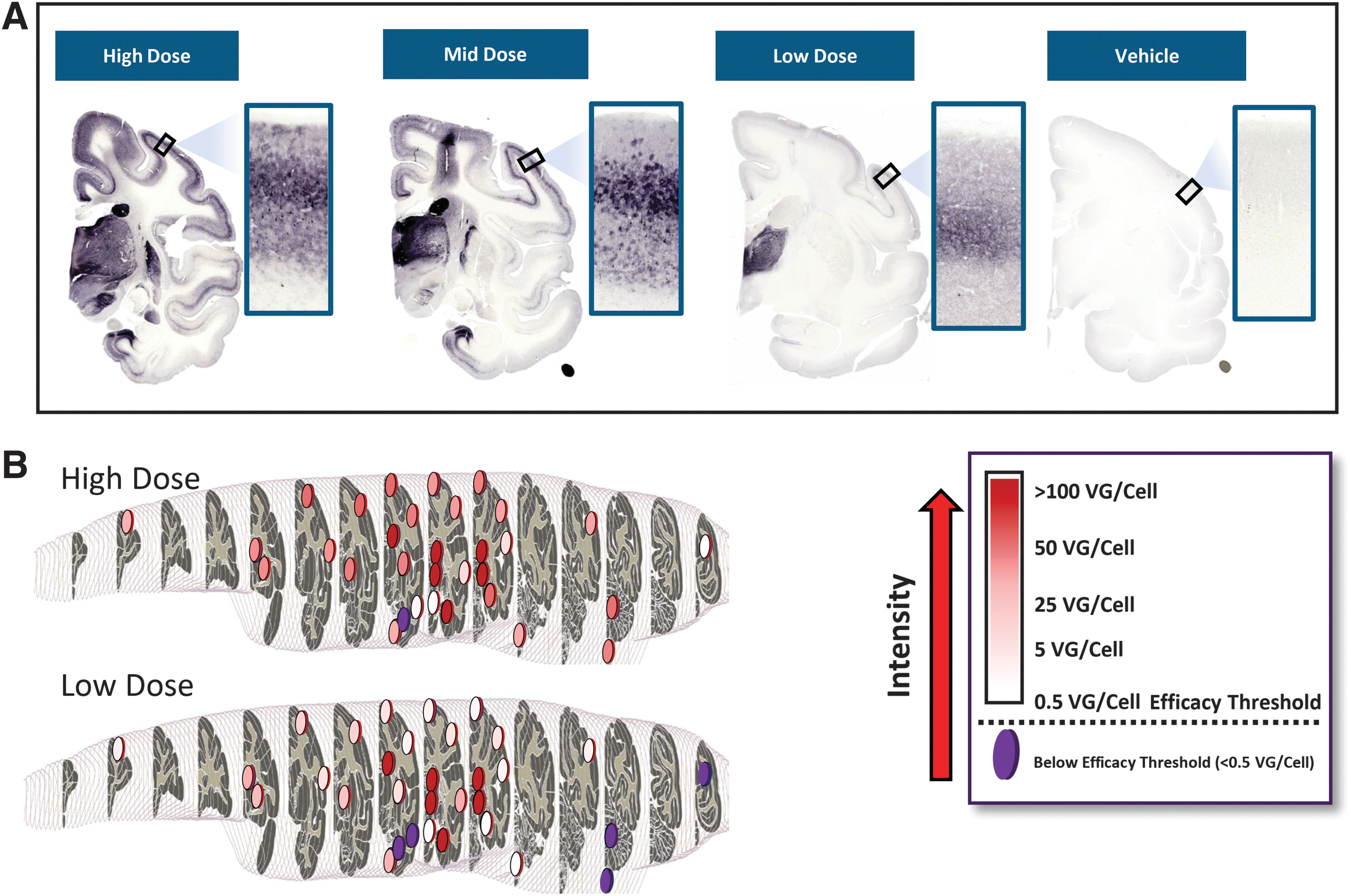
Dose-dependent biodistribution of AAV9-PS1 and HA-PS1 expression in NHP brain following thalamic administration.
DISCUSSION
While several genes have been linked to an increased risk of developing LOAD, 20 only three genes have been identified which are causative for the development of ADAD. Two of these genes (PSEN1 and PSEN2) encode the catalytic subunit of the γ-secretase protease, while the third (APP) encodes a γ-secretase substrate. 2 While this confluence of human genetic findings has been adduced as support for the amyloid hypothesis, which posits a central role for amyloid in the pathophysiology of AD, several lines of evidence have emerged over the years which have challenged this hypothesis. 3 –8 While recent regulatory approvals of plaque-clearing antibodies have provided additional support for the role of amyloid, even robust clearing of plaques leads to only a 20–30% slowing in cognitive decline 21,22 suggesting that plaque accumulation alone is unlikely to fully explain AD pathogenesis.
With the above in mind, it is useful to highlight the distinction between our PSEN1 gene replacement approach and the amyloid-centric approaches that have been the focus of most late-stage clinical trials. The anti-amyloid antibody therapies rely on the assumption that amyloid deposits are a key driver in AD. In contrast, the gene therapy approach we describe in this study is targeting the single causative genetic driver in the ADAD patient population and is agnostic with respect to the role of amyloid. While the ability of AAV9-PS1 to normalize Aβ peptide production is expected to slow the accumulation of new amyloid deposits, it is important to note that this therapy will also be impacting a range of other dysregulated neuronal functions that occur downstream of mutant PSEN1, potentially including both γ-secretase-dependent and -independent activities. 23 –25
Some of the early work investigating the effect of PSEN1 mutations on γ-secretase activity suggested that these mutations led to a gain of enzyme function, increasing the amount of Aβ42 produced by the mutant γ-secretase enzyme. However, work carried out over the past two decades has clearly demonstrated that pathogenic PSEN1 mutations result in a LOF, which leads in turn to a preferential reduction in shorter forms of Aβ (e.g., Aβ38 and 40). 4 –8 This specific enzyme dysfunction explains the increase in the Aβ42:40 ratio that is typically observed across most PSEN1 mutations. 27 The current study demonstrates that overexpression of a WT copy of human PSEN1 in cells that harbor a mutant gene can effectively replace the mutant PS1 protein and thereby restore normal γ-secretase activity. Our findings further show that correcting PS1 function and γ-secretase activity in the brain can normalize Aβ production and rescue neurodegeneration caused by a pathogenic PSEN1 mutation.
We initially characterized the expression of HA-PS1 following injection of AAV9-PS1 into the lateral cortex and hippocampus of cDKO mice. Eight weeks post-treatment we observed HA-PS1 protein expression throughout the dorsal and lateral cortex and hippocampus of treated animals (Fig. 1A). We also observed formation of the HA-PS1-NTF, indicative of incorporation of the exogenously expressed protein into a functional γ-secretase complex (Fig. 1B). In addition, using double-labeling immunofluorescence, we showed that most cells expressing HA-PS1 are neurons based on their costaining with a NeuN antibody (Fig. 1C).
To study the therapeutic potential of our gene replacement approach, we assessed γ-secretase function in brain homogenates of cDKO mice 8 weeks following intraparenchymal administration of AAV9-PS1. Treated animals showed restoration of de novo Aβ peptide production in the brain to WT levels (Fig. 2A), a reduction in the accumulated APP-CTF substrate (Fig. 2B), and increases in endogenous levels of Aβ peptides in the CSF (Fig. 2C). AAV9-PS1-treated cDKO mice also showed a reduction in cortical thinning (Fig. 2D) and significantly reduced neuronal degeneration (Fig. 2E). In addition, we quantified the amount of virus that was required to restore normal γ-secretase function, allowing us to establish an efficacy threshold of ∼0.5 vg/dc in this model (Fig. 2F).
The second animal model which was employed was the Psen1 L435F knockin mouse. These mice carry a pathogenic L435F “knockin” mutation introduced into one of the endogenous Psen1 alleles. 6,7 This model recapitulates the genetic makeup of ADAD patients with an L435F PSEN1 mutation, and like the cDKO model, shows a neurodegenerative phenotype. Importantly, these mice also show a LOF, characterized by a preferential decrease in shorter forms of Aβ peptides and a corresponding increase in the Aβ42:40 ratio, as observed in patients. 7 Like our results in the cDKO model, when the AAV9-PS1 virus was administered into the brains of these animals, we observed a normalization of γ-secretase function, demonstrating that expression of the WT human PS1 protein can effectively out-compete the mutant protein to form a functionally normal γ-secretase complex (Fig. 3).
The above mouse models were critical in demonstrating that (1) introduction of a WT copy of PSEN1 into the mouse brain leads to expression and incorporation into a functional γ-secretase complex, and (2) that the exogenous WT PSEN1 can overcome the effect of the dominant PSEN1 mutation, which together support the biological rationale for this approach. 3,5
However, for this approach to be clinically feasible it is necessary to demonstrate that introduction of WT PSEN1 into a larger primate brain leads to expression of the WT PS1 protein in disease-relevant brain regions. To address this challenge, we have carried out studies in NHPs, exploring specific ROAs intended to achieve broad coverage throughout the cortex and hippocampus, two regions that are critically affected in ADAD. 27 We chose two ROAs, intrathalamic and ICV, seeking to distribute the virus broadly throughout the cortex, although through different mechanisms. Intrathalamic administration was expected to achieve cortical distribution through transport of AAV9 along thalamocortical projection neurons, while ICV administration should allow virus to access cortical regions by introducing the virus into the CSF compartment. 19 Our results have largely supported previous work, and indeed, show robust viral spread and protein expression throughout most cortical regions (Figs. 4 and 5).
The third ROA involved introduction of AAV9-PS1 directly into the hippocampus, a subcortical region known to be critically involved in AD pathogenesis. The results following hippocampal administration were consistent with previous reports, showing very high expression, largely restricted to this region (Figs. 4 and 5). 28 It is notable that AAV9-PS1 vector genomes and HA-PS1 protein expression were also observed in the hippocampus of animals that were injected through the thalamic and ICV routes (Figs. 4 and 5), raising the possibility that direct hippocampal injection may not be required to achieve expression in this brain region.
Several reports have described DRG pathology following administration of high doses of AAV into the CSF compartment. 29 We therefore measured viral levels in peripheral nervous tissue, including spinal cord and DRGs, as well as several peripheral organs. Consistent with previous work, the ICV-administered group showed significant levels of AAV9-PS1 in DRG, spinal cord, and peripheral organs (Fig. 4E). In contrast, lower peripheral exposure was seen in those animals that received direct intraparenchymal injection (Fig. 4D, F). While clinical safety issues have not been clearly linked with CSF administration thus far, this remains an area of concern and needs to be closely monitored in both preclinical and clinical safety studies that involve CSF administration of recombinant AAV.
Considering the higher peripheral exposure and increased risk of DRG toxicity associated with the ICV ROA, and the attractive cortical expression associated with the intrathalamic route, we chose to further explore the intrathalamic ROA in a second NHP study. In this dose-ranging study we evaluated three doses in an effort to determine the minimum dose required to reach our previously defined efficacy threshold (Fig. 2F). Four weeks following intrathalamic administration of a high, medium, or low dose of AAV9-PS1, animals were euthanized, and brain tissue analyzed for vector genome levels and protein expression. Immunohistochemical analysis showed dose-responsive protein expression throughout the cortex, with expression patterns similar to what was observed in the initial study (Fig. 6A). We demonstrated that viral levels as measured in multiple cortical and subcortical tissue punches, generally reached or exceeded levels that were associated with biochemical rescue in the mouse model, even in the low-dose group (Fig. 6B).
We believe the above studies indicate that sufficient biodistribution to most cortical regions should be achievable in the clinic utilizing either intrathalamic or ICV ROAs. Further studies will be required to assess safety and tolerability more thoroughly.
While CNS administration of gene therapy either directly into brain parenchyma or into the CSF compartment can be advantageous in terms of limiting peripheral exposure to the virus, these are relatively invasive procedures, which may limit the patient population that would undergo treatment. Therefore, in the future, it will be important to evaluate novel AAV capsids that have more recently been described, which have the ability to achieve robust CNS exposure following peripheral administration. 30 While transduction of peripheral organs, such as the liver, may increase following intravenous administration, the enhanced safety and convenience of peripheral administration is anticipated to increase patient and physician acceptance, and encourage treatment in younger presymptomatic patients, where gene therapy is expected to show the greatest benefit.
Footnotes
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Jie Shen for the conceptualization of using Presenilin based gene therapy to treat FAD and for providing intellectual and technical guidance throughout the study.
AUTHORs' CONTRIBUTIONS
B.M.: Investigation (lead); supervision (lead); writing—original draft (equal); and writing—review and editing (equal); A.S.: Investigation (equal); methodology (lead); and writing original draft (supporting); M.G. and C.P.: Investigation (equal); T.S.: Investigation (equal); and methodology (equal); R.H.: Supervision; E.S.: Supervision (lead); writing—original draft (equal); and writing—review and editing (equal); R.J.K.: Conceptualization; resources; supervision; writing—review and editing; project administration; funding acquisition.
AUTHOR DISCLOSURE
Authors listed as affiliated with Paros Bio were full-time employees of the company at the time the work was carried out.
FUNDING INFORMATION
This work was funded by private investment from Mass General Brigham Ventures, Arkin Holdings, Qiming Venture Partners, and Dolby Ventures. R.J.K. was supported by NIH awards 5K24NS093185 and 5R01NS075346.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.