
Abstract
Interleukin 2 (IL-2) plays a crucial role in T cell growth and survival, enhancing the cytotoxic activity of natural killer and cytotoxic T cells and thus functioning as a versatile master proinflammatory anticancer cytokine. The FDA has approved IL-2 cytokine therapy for the treatment of metastatic melanoma and metastatic renal cell carcinoma. However, IL-2 therapy has significant constraints, including a short serum half-life, low tumor accumulation, and life-threatening toxicities associated with high doses. Oncolytic viruses (OVs) offer a promising option for cancer immunotherapy, selectively targeting and destroying cancer cells while sparing healthy cells. Numerous studies have demonstrated the successful delivery of IL-2 to the tumor microenvironment without compromising safety using OVs such as vaccinia, Sendai, parvo, Newcastle disease, tanapox, and adenoviruses. Additionally, by engineering OVs to coexpress IL-2 with other anticancer transgenes, the immune properties of IL-2 can be further enhanced. Preclinical and clinical studies have shown promising antitumor effects of IL-2-expressing viral vectors, either alone or in combination with other anticancer therapies. This review summarizes the therapeutic potential of IL-2-expressing viral vectors and their antitumor mechanisms of action.
INTRODUCTION
Interleukin 2 (IL-2)

Proinflammatory effects of IL-2 and its mechanisms of action. (1) High-affinity IL-2 signaling is mainly produced in response to antigen stimulation. First, through trans presentation, IL-2 derived from dendritic cells binds to CD25 (a.k.a. IL2Rα) expressed on activated dendritic cells. Then, the ligand–receptor complex will bind to IL2Rβ and IL2Rγ receptors on adjacent T cells to trigger downstream mechanisms. (2) Upon cis presentation of high-affinity IL-2 to the trimeric IL-2R receptor (i.e., IL-2Rα, IL2-Rβ, and IL-2Rγ), downstream signaling cascade is triggered. (3) IL-2 binds with low–moderate affinity to the dimeric IL-2R to induce downstream mechanisms. Notably, the monomeric IL-2 receptor (not pictured) does not result in downstream signaling. The three major IL-2 downstream mechanisms include activation of the PI3K-AKT pathway, JAK-STAT pathway through STAT5 dimerization, and the MAPK pathway. All the listed pathways will result in transcription of target genes downstream. While IL-2 is mainly associated with antigen-stimulated CD4+ T cells, which can differentiate into multiple subtypes (i.e., Th1, Th2, Th17, and Tregs), IL-2 is also associated with CD8+ T cells, NK cells, and cytotoxicity. IL-2, interleukin 2; MAPK, mitogen-activated protein kinase; NK, natural killer; PI3K, phosphoinositide 3-kinase; Treg, regulatory T-cell.
IL-2 cytokine therapy for cancer presents several significant constraints, including a short serum half-life 3 and low accumulation within tumors. 5 Consequently, frequent administration of high doses of IL-2 is necessary, which may result in adverse events, including life-threatening toxicities. 6 For instance, administration of a high dose of IL-2 may lead to various common adverse effects, such as hypotension, gastrointestinal symptoms (nausea, vomiting, and diarrhea), grade 2 or 3 central nervous system toxicity, malaise, and pancytopenia. High dose of IL-2 can also trigger capillary leak syndrome, a life-threatening condition resembling septic shock. Clinical manifestations of capillary leak syndrome include pulmonary edema, respiratory distress, hypotension, cardiac insufficiency, cerebral edema, and reduced kidney perfusion. 7 Renal failure is a major concern for high-dose IL-2 in patients with renal cell carcinoma, as they may face an increased risk due to having one kidney after undergoing nephrectomy. 8 Even with high doses, the concentration achieved in tumors can be too low for efficacy—this can be reversed by local production of IL-2. 3,9
Conversely, low doses may compromise the anticancer efficacy of IL-26 and stimulate the expansion of Tregs, 10 an undesirable outcome in cancer immunotherapy. Despite efforts to improve safety through subcutaneous and intraperitoneal delivery techniques, alternative dosing and schedules, modification of binding affinity, and combination with fusion proteins, the safety of IL-2 cytokine therapy has only seen slight improvements. 11 –13 Thus, a fundamentally better approach is required to effectively deliver IL-2 to tumors to maximize antitumor immunity while promoting patient safety.
Oncolytic viruses (OVs), whether naturally occurring or genetically modified, preferentially target and lyse cancer cells while sparing healthy cells. 14 –17 OVs have emerged as a promising option for cancer immunotherapy as they can deliver cytokine transgenes directly to tumors, mitigating the harmful effects of systemic cytokine therapies. 14 –26 Numerous studies have demonstrated the effective delivery of IL-2 to the tumor microenvironment using viral vectors, without compromising safety (Table 1). Furthermore, by engineering viral vectors to coexpress IL-2 with other anticancer transgenes, the immune properties of IL-2 can be further enhanced (Table 2). Preclinical studies and clinical trials have shown promising antitumor effects of IL-2-expressing OVs either alone or in combination with other anticancer therapies (Fig. 2). This review summarizes the therapeutic potential of IL-2-expressing viral vectors and their antitumor mechanisms of action.
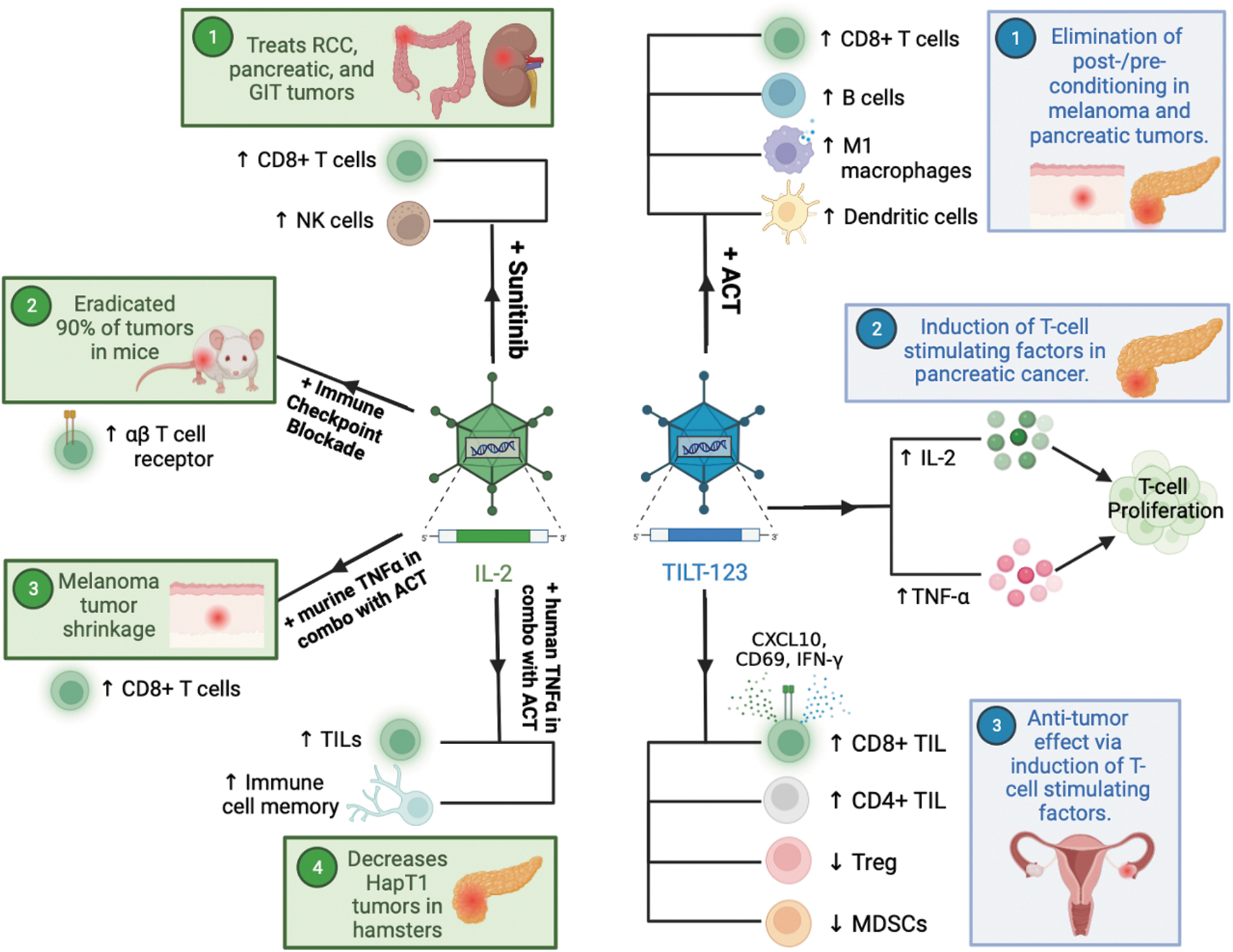
Summary of IL-2-expressing viruses as potential cancer therapeutics. Referring to IL-2-expressing virus (depicted in green), there are multiple combination therapies to treat cancer demonstrating promising activity in preclinical models. (1) IL-2 in combination with sunitinib demonstrated therapeutic activity in RCC, pancreatic neuroendocrine tumors, and GIT stromal tumors through an increase in CD8+ T cells and NK cells; 61 –64 (2) IL-2 in addition to an immune checkpoint blockade induced tumor eradication in 90% of mice through modulation of the αβ T cell receptor; 28 (3) IL-2 in addition to murine TNF-α in combination with ACT will increase CD8+ T cells and is known to hinder the growth of melanoma tumors; and, lastly; 56 (4) IL-2 in addition to human TNF-α in combination with ACT will increase TILs and immunogenic memory, which will ultimately decrease HapT1 tumor growth. 58,65 TILT-123 in an oncolytic adenovirus encoding IL-2 and TNF-α (depicted in blue), (1) TILT-123 in combination with ACT eliminates postconditioning and preconditioning in melanoma and pancreatic tumors through high CD8+ T cell levels, elevated B cells, an increase in M1-like macrophages, and proliferation of dendritic cells, which ultimately results in high antitumor effect and low toxicity profile; 58,65 (2) TILT-123 in pancreatic cancer induced T cell-stimulating factors that promote T cell proliferation; 58 (3) TILT-123 in ovarian cancer increased overall TIL activation as evidenced by increased CD69, CXCL10, and IFN-γ. TILT-123 in ovarian cancer is also associated with a decrease in functional signatures of infiltrating Tregs and MDSCs. 56,58,59 ACT, adoptive cell therapy; GIT, gastrointestinal; MDSCs, myeloid-derived suppressor cells; RCC, renal cell carcinoma; TIL, tumor-infiltrating lymphocyte; Tregs, regulatory T cells.
Viral vectors expressing interleukin 2 as a single cytokine
CTL, cytotoxic T-lymphocyte; HVJ-E, hemagglutinating virus of Japan envelope; I.C., intracranial; IL-2, interleukin 2; I.P., intraperitoneal; I.T., intratumoral; I.V., intravenous; mIL-2, murine interleukin-2; mIL2v, mouse IL2 variant; MOI, multiplicity of infection; n/a, not applicable; NDV, Newcastle disease virus; PBS, phosphate-buffered saline; pfu, plaque-forming units; rNDV, recombinant Newcastle disease virus; RoA, route of administration; SCC, squamous cell carcinoma; SeV, Sendai virus; TIL, tumor-infiltrating lymphocyte; TNBC, triple-negative breast cancer; TPV, tanapoxvirus; Treg, regulatory T cell; VP, viral particle; VV, vaccinia virus; VV-IL2, vaccinia virus expressing membrane-bound IL-2; wtTPV, wild-type TPV.
List of viral vectors to coexpress interleukin 2 with other anticancer transgenes
HNSCC, head and neck squamous cell carcinoma; ICI, immune checkpoint inhibitor; IL-12, interleukin 12; IP-10, inducible protein-10; MDSCs, myeloid-derived suppressor cells; mPD-L1, murine programmed death ligand 1; NK, natural killer; PD-L1, programmed death ligand 1; SCID, severe combined immunodeficiency; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
VIRAL EXPRESSION OF IL-2 AS SINGLE CYTOKINE
IL-2-expressing vaccinia viruses
Vaccinia virus (vv or VVs), a member of the orthopoxvirus, contains a linear double-stranded DNA genome of ∼190 kb in length. The large genome size allows manipulation of viral genes and insertion of large therapeutic anticancer transgenes up to 40 kb in size. Extensive research has been conducted on VV vectors as an oncolytic agent in preclinical tumor models and through multiple clinical trials for treating patients diagnosed with advanced-stage solid cancers. 27
VV expressing membrane-bound IL-2 versus secretory IL-2 in colorectal cancer
As discussed earlier, viral expression of IL-2 is typically confined to tumors and devoid of systemic toxicities associated with high-dose IL-2 treatment. However, an increased replication of virus expressing secretory IL-2 can result in the release of IL-2 into the peripheral circulation, potentially leading to IL-2-mediated systemic toxicities. 28 To overcome this scenario involving secretory IL-2, Liu et al. developed oncolytic VVs that express membrane-bound IL-2, such as vvDD-IL-2-FG, where a flexible linker is fused to GPI (glycosylphosphatidylinositol), and vvDD-IL-2-RG, where a rigid linker is fused to GPI. 28
When tumor cells were infected with vvDD-IL-2-FG or vvDD-IL-2-RG, IL-2 expression was limited on the surface of the infected tumor cells, thereby preventing systemic release of IL-2 and its associated toxicities. The membrane-bound IL-2 retains its biological activity, as demonstrated by the proliferation of CTLL-2 cells, whose growth is dependent on IL-2 stimulation. This proliferation occurs when CTLL-2 cells were cocultured with a stable IL-2-RG-expressing cell line (MC38-IL-2-RG) or vvDD-IL-2-RG-infected MC38 cells.
The addition of an IL-2-neutralizing antibody to the culture media reverses the proliferative activity of CTLL-2 cells. 28
Liu et al. conducted a comparison of the antitumor effects and IL-2 release profiles of three oncolytic VVs: vvDD-IL-2, which expresses the secreted form of IL-2, and vvDD-IL-2-FG and vvDD-IL-2-RG, which encode membrane-bound IL-2. 28 In vitro, both membrane-bound IL-2 constructs (vvDD-IL-2-FG and vvDD-IL-2-RG) and the secreted IL-2 construct (vvDD-IL-2) demonstrated similar cytotoxic potential in tumor cells. However, vvDD-IL-2 infection resulted in a significantly higher level of IL-2 in the cell culture supernatant compared with the membrane-bound IL-2 constructs. 28 When injected intraperitoneally at a dose of 2 × 108 plaque-forming unit (pfu)/virus, all three viruses (vvDD-IL-2, vvDD-IL-2-FG, and vvDD-IL2-RG) exhibited strong therapeutic activity. Among the three, vvDD-IL-2-RG demonstrated the most favorable survival outcomes in mice with early-stage peritoneal MC38-luc colon cancer. 28
All long-term survivors from the vvDD-IL-2, vvDD-IL2-FG, or vvDD-IL2-RG groups rejected MC38 rechallenge but favored the growth of tumors derived from antigenically distinct Lewis lung cancer cells. This finding indicates treatment-induced tumor-specific memory protection in the long-term survivors. However, intriguingly, some mice receiving vvDD-IL-2 (expressing secretory IL-2) succumbed to tumors within 7 days after virus injection. This interesting finding was correlated with a staggeringly 100 times higher level of serum IL-2 in mice receiving treatment with VV expressing secretory IL-2 compared with those receiving treatment with VV expressing membrane-bound IL-2. 28 These findings reiterate that VVs expressing secretory IL-2 cause easy release of IL-2 into the serum, which may have resulted in death of those animals. Furthermore, the toxic effects of VV expressing secretory IL-2 (vvDD-IL-2) were even more pronounced in the late-stage tumor model, indicating a positive correlation between tumor burden, virus replication, IL-2 production, and associated toxicities. 28
In contrast, VV encoding membrane-bound IL-2 (i.e., vvDD-IL2-RG) significantly prolonged the survival of mice compared with control vvDD, without compromising safety.
The superior antitumor efficacy of the vvDD-IL2-RG virus compared with other treatments, such as phosphate-buffered saline (PBS), vvDD, or vvDD-IL-2-FG, is associated with several factors. These include: (1) an increase in the percentage of CD8+ T cells and a reduction in exhausted tumor-infiltrating CD8+ T cells, such as PD1+CTLA-4+CD8+, PD1+Tim-3+CD8+, PD1+TIGIT+CD8+, and PD1+LAG-3+CD8+ T cells; (2) an increase in the number of tumoral CD4+FOXP3− and CD8+IFN-γ+ T cells, and CD8+CD44hi memory T and CD3−NK1.1+ cells in both tumors and spleens; (3) an increase in the CD8+/Treg ratio, although the absolute number and percentage of CD4+FOXP3+ cells were increased in tumors; and (4) an increased level of IFN-γ, Granzyme B, perforin, Th1-type chemokine CXCL9, and a decrease in the level of TGF-β and angiogenesis markers such as CD105 and VEGF in tumors. 28 Furthermore, immune cell depletion studies demonstrate that the antitumor efficacy of vvDD-IL2-RG was dependent on CD8+ T cells and IFN-γ, with CD4+ T cells not playing any role.
Although the depletion of natural killer (NK) cells did not impair the treatment efficacy of the vvDD-IL2-RG virus, a slightly better survival benefit was observed when NK1.1+ cells were depleted after vvDD-IL-2-RG treatment compared with vvDD-IL-2-RG treatment alone. 28 This suggests that NK cells may impede virotherapy. 29
Overall, this study suggests that viral expression of membrane-bound IL-2 is safer for localized IL-2 production and more effective in treating tumors than viral expression of the secreted form of IL-2.
VV encoding membrane-bound IL-2 in malignant pleural disease
Ekeke et al. recently conducted a study using a mouse model of malignant pleural disease (MPD), where they administered Lewis lung carcinoma cells in the pleural cavity to develop MPD. Seven days later, mice bearing MPD received intrapleural treatment with vaccinia virus expressing membrane-bound IL-2 (VV-IL2) with a dose of 2 × 108 pfu/mouse. 30 VV-IL2 treatment significantly reduced tumor burden and increased median survival (23 days) compared with control VV virus (17 days) or PBS (16 days). This survival benefit of VV-IL2 was attributed to a significantly higher tumor infiltration of CD3+ and CD8+ T cells (vs. control VV), indicating the crucial role of IL-2 expression in inducing antitumor immunity. 30
Additionally, a side-by-side comparison between intrapleural (local) and intraperitoneal (systemic) VV-IL2 treatments showed that local VV-IL2 was significantly superior in inhibiting tumor growth and extending survival compared with systemic VV-IL2. The increased antitumor effects of local VV-IL2 treatment were also confirmed by a significantly increased infiltration of both CD4+ and CD8+ T cells (vs. control VV vector). It is important to point out that only CD3+ T cells were increased with systemic VV-IL2 delivery. 30 This study reinforces the idea that local delivery of OV may be a superior choice in cancer immunotherapy than systemic delivery.
Antitumor effects of tumor-reactive tumor-infiltrating lymphocytes induced by VV expressing IL-2
It is generally accepted that OVs expressing cytokines, such as IL-2, induce a strong antitumor response, primarily by increasing the intratumoral accumulation of tumor-infiltrating lymphocytes (TILs). 28,30 Feist et al. came up with the idea of isolating TILs from OV-IL2-treated tumors and using these TILs for adoptive cell therapy in the treatment of solid cancer. In this context, Feist et al. injected vvDD-IL-228 (along with respective controls) with a dose of 1 × 108 pfu/tumor directly into subcutaneous MC38 tumors to induce tumor-reactive TILs. 31 Intratumoral injection of vvDD-IL-2 resulted in a significantly increased infiltration of tumor-reactive CD8+ TILs in MC38 tumor lesions compared with other cytokine or chemokine-expressing VVs such as vvDD-CCL5, vvDD-CXC11, and vvDD-IL15Rα. 31
TILs were then isolated from vvDD-IL2-treated tumors, expanded ex vivo, and adoptively transferred at a dose of 5.0 × 106 TILs (naive TILs included as controls) into sublethally irradiated mice bearing intraperitoneal MC38-luc tumors. After the adoptive transfer of TILs, the treated mice also received exogenous cytokine support of IL-2 intraperitoneally at a dose of 100,000 IU/mouse every 12 h for 3 days. 31 The mice treated with vvDD-IL-2-induced TILs had the highest reduction in tumor burden and longer median survival (61 days) compared with mice treated with TILs from PBS-treated tumors (27 days) or control T cells from naive spleens (29 days), indicating virally produced IL-2 enhances the antitumor effects of TILs. 31
IL-2 expressing Sendai viruses
Sendai virus, also known as murine parainfluenza virus type 1, is an enveloped RNA virus used an oncolytic vector as the virus does not integrate into the host genome. An inactive replication-defective Sendai viral vector, also called hemagglutinating virus of Japan envelope (HVJ-E), can be used for delivering IL-2 cytokine directly into tumors. 32 The Sendai viral vector HVJ-E is known to augment antitumor immunity by activating cytotoxic T lymphocytes and NK cells and by inhibiting the activity of Tregs. 33 Due to the immunomodulatory effects of HVJ-E and IL-2's versatile antitumor properties, HVJ-E containing pVAX-mIL-2 (a plasmid expressing IL-2; HVJ-E/IL-2) was tested in various tumor models. 32,34
Antitumor efficacy of HVJ-E/IL-2 in intradermal glioma
Matsuda et al. constructed a number of HVJ-E Sendai viral vectors carrying pVAX-mGM-CSF (HVJ-E/mGM-CSF), pVAX-mIFN-β (HVJ-E/mIFN-β), pVAX-mIL-12 (HVJ-E/mIL12), or pVAX-Luc (HVJ-E expressing luciferase [HVJ-E/Luc]) and compared their antitumor effects with HVJ-E containing pVAX-mIL-2 (HVJ-E/IL-2) in intradermal RSV-M mouse glioma tumor model. 32 For the intradermal model, mice bearing established RSV-M tumors received three injections of PBS or HVJ-E Sendai virus particles (6 × 109 particles/injection/virus) on days 5, 8, and 11 post-tumor implantations, and animals were monitored for tumor growth. It was found that HVJ-E/IL-2 treatment resulted in the eradication of RSV-M intradermal tumors in 70% of mice, compared with 0%, 20%, 30%, and 40% tumor eradication in the HVJ-E/Luc, HVJ-E/mIFN-β, HVJ-E/mGM-CSF, and HVJ-E/mIL12 treatment groups, respectively.
This study suggests that, for immunogene therapy, expressing IL-2 using the Sendai virus is a more favorable option than expressing IFN-β, GM-CSF, or interleukin 12 (IL-12) in the RSV-M intradermal tumor model. 32 However, it is yet to be established whether this observation can be replicated in other cancer models using IL-2 expressing Sendai virus or other viral vectors, such as IL-2 expressing VV.
Antitumor efficacy of HVJ-E/IL-2 in intracranial glioma
Matsuda et al. also evaluated the antitumor effects of single intratumoral injections of HVJ-E/IL-2, HVJ-E/Luc, or PBS in an intracranially implanted RSV-M mouse glioma tumor model. 32 Seven days postvirus injection, mice from all treatment groups were euthanized, and their brains and spleens were harvested to evaluate tumor volume and measure RSV-M-specific CD8+ T cell response by enzyme-linked immunosorbent spot assay, respectively. Similar to the findings from intradermal RSV-M tumors, a single intracranial injection of HVJ-E/mIL2 resulted in a significant extension of survival compared with those that received single injections of HVJ-E/Luc or PBS. This survival benefit of HVJ-E/IL-2 (vs. HVJ-E/Luc or PBS) coincided with a significant reduction in tumor volume and the generation of a tumor-specific IFN-γ response. 32
In addition, the authors also concluded that a single intracavitary administration of HVJ-E/IL-2 vector following surgical resection of the primary tumor resulted in the successful delivery of the IL-2 gene into postoperative residual tumor cells and a significant inhibition of the expansion of Tregs compared with murine stem cell virus (MSCV) retroviral vector-mediated delivery of the IL-2 gene. All these factors contributed to the improvement in survival. HVJ-E-mediated IL-2 gene therapy and its associated survival benefit were also correlated with a robust tumoral infiltration of CD4+ and CD8+ T cells into tumors compared with mice treated with PBS or the control vector HVJ-E/Luc. The superior survival and antitumor immunity due to HVJ-E/IL-2 treatment over HVJ-E/Luc or PBS treatments highlight the therapeutic potential of IL-2 expression within central nervous system tumors. 32
Antitumor efficacy of HVJ-E/IL-2 in angiosarcoma
In a separate study, Takehara et al. evaluated the therapeutic effect of inactivated Sendai virus particles carrying an IL-2 complementary DNA (cDNA; HVJ-E/IL-2) in the ISOS-1 angiosarcoma model in BALB/c mice. 34 They demonstrated that intratumoral injection of HVJ-E/IL-2 on days 12, 16, and 20 posttumor implantations more efficiently suppressed the growth of ISOS-I angiosarcoma compared with treatment with non-IL-2-expressing Sendai virus (HVJ-E), resulting in complete tumor eradication in 35.7% of mice in the HVJ-E/IL-2 treatment group, and they remained tumor free for up to ∼130 days from the initial implantation. Importantly, 57% of these tumor-free mice rejected lethal ISOS-1 rechallenge at a distant site and remained tumor free for an additional 50 days, indicating HVJ-E/IL-2 induced long-term memory protection. 34
Immunologically, although HVJ-E/IL-2 treatment resulted in an increased infiltration of CD8+ T cells and NK cells in tumors and a reduced number of Tregs (CD3+CD25+FoxP3+) in regional lymph nodes, these changes were not statistically different compared with HVJ-E treatment (i.e., no IL-2 expression). However, interestingly, CD8+ T cells isolated from the HVJ-E/IL-2 treatment group produced a strikingly high level of ISOS-1 tumor cell-specific IFN-γ responses compared with CD8+ T cells isolated from the HVJ-E and other control treatment groups. 34
IL-2 expressing parvoviruses
The rat parvovirus, H-1PV, is an established cancer-killing agent and possesses preferential oncotropic and oncolytic properties. 35 The safety and antitumor properties of H-1PV have been established through clinical studies in glioma and pancreatic ductal adenocarcinoma patients. 35 Similar to other OVs, H-1PV induces immunogenic cell death and enhances antitumor immunity. 36 To further enhance H-1PV-mediated antitumor immunity, Dempe et al. genetically modified H-1PV to express IL-2, resulting in a modified version called Chi-H1/IL2. 37 The lytic and immune potential of Chi-H1/IL2 was tested both in vitro and in vivo, as discussed below.
In vitro effects of Chi-H1/IL2
Infection of pancreatic Panc-1 cells with the IL-2-expressing H-1PV virus, Chi-H1/IL2, resulted in robust release of IL-2 in the cell culture supernatant. The authors then investigated the biological activity of IL-2 released from H-1PV-infected Panc-1 cells in activating NK cells isolated from healthy donor peripheral blood mononuclear cells. These NK cells were stimulated for 5 days with recombinant IL-2 cytokine (100 IU/mL) or cell-free conditioned media collected from mock-treated, Chi-H1/Δ800-infected, or Chi-H1/IL2-infected Panc-1 cells. 37 The stimulated NK cells were cocultured with 51 Cr-labeled target K562 cells, responsive to NK killing activity in vitro, and 51 Cr release was measured. Compared with NK cells stimulated by supernatants from mock- or Chi-H1/Δ800-infected Panc-1 cells, the NK cells stimulated by the Chi-H1/IL2-infected Panc-1 supernatant showed robust killing activity (increased by ∼30%) of target K562 cells, which was comparable to that observed with NK cells stimulated with recombinant IL-2. 37
In a separate in vitro experiment, the authors evaluated the ability of virally released IL-2 to affect the chemotactic property of NK cells. Naive NK cells were separated by a porous membrane from cell-free conditioned media harvested from mock-, Chi-H1/Δ800-, or Chi-H1/IL2-infected Panc-1 cells. There was an increase in the migratory ability of NK cells toward IL-2-expressing H-1PV (Chi-H1/IL2)-infected Panc-1 supernatant compared with those with control vector Chi-H1/Δ800- or mock-infected Panc-1 supernatants. 37 The increased migratory ability toward Chi-H1/IL2-infected Panc-1 supernatant suggests that virally released IL-2 has chemoattractant properties, which are likely due to IL-2-mediated expression of the chemokine receptor CCR2 on NK cells. 38
In vivo effects of Chi-H1/IL2
To evaluate the antitumor activity of H-1PV constructs, including Chi-H1/IL2 in vivo, Panc-1, or MiaPaCa-2 cells were treated with buffer (mock) or infected with wild-type H1-PV, Chi-H1/Δ800, or Chi-H1/IL2 at an multiplicity of infection (MOI) of 1.5 and 2.0 RU/cell, respectively. The buffer- or virus-treated Panc-1 or MiaPaCa-2 cells were then subcutaneously implanted in BALB/c nude mice, and the animals were monitored for tumor growth. Compared with all three control groups, IL-2 expression from Chi-H1/IL2 resulted in profound antitumor effects, leading to 100% tumor-free mice in both Panc-1 and MiaPaCa-2 pancreatic tumor models. 37 The robust antitumor effect of Chi-H1/IL2 is associated with increased infiltration of NKG2D-positve NK cells in Panc-1 tumors and marked expression of NK cell cytotoxic factors such as granzyme B, perforin, inducible nitric oxide synthase, and IFN-γ when compared with the control vector. 37
IL-2 expressing Newcastle disease viruses
Newcastle disease virus (NDV) is an enveloped, single-stranded RNA avian paramyxovirus. Native NDV preferentially replicates and lyses human neoplastic cells making it an attractive virus for OV development. 39
Antitumor effects of NDV/Anh-IL-2
Wu et al. utilized the Anhinga strain of NDV as a backbone to construct a recombinant (r)NDV-expressing IL-2 (NDV/Anh-IL-2) and tested its antitumor efficacy in a hepatocellular H22 cancer model. 40 Mice were treated with intratumoral injections of 107 pfu virus every other day for a total of four times. NDV/Anh-IL-2 treatment resulted in tumor eradication in 50% of the treated mice within 2–3 weeks of treatment initiation (which survived long-term), compared with none in the control non-IL-2-expressing NDV (NDV/Anh) or PBS groups. 40 The long-term survivors due to NDV/Anh-IL-2 therapy developed memory protection as evidenced by rejection of lethal tumor rechallenge with the same tumor cells. The treatment efficacy with NDV/Anh-IL-2 was linked to a significant increase in tumor infiltration of both CD4+ and CD8+ T cells compared with the NDV/Anh or PBS groups. 40
Antitumor effects of rNDV/IL-2
Zhao et al. evaluated the ability of rNDV/IL-2, an NDV encoding human IL-2, to deliver and express IL-2 in four different human cell lines: MCF-7 and MCF-10 (breast cell lines), HT29 (colon-adenocarcinoma cell line), and Jurkat (acute T cell leukemia cell line). 41 The MCF-7 cells had the highest expression of IL-2 compared with other three cell types. The tumor cells produced biologically active IL-2 after infection with rNDV/IL-2, which led to an increase in the proliferation of IL-2-dependent CTLL-2 cells, a subclone of T cells derived from C57BL/6 mice. 41 Despite these promising in vitro results, rNDV/IL-2 has not yet been tested in vivo.
Antitumor effects of rLaSota/IL-2
Bai et al. evaluated the antitumor effects of rLaSota/IL-2, an NDV (derived from the LaSota strain) expressing human IL-2, in syngeneic mice bearing H22 or B16-F10 tumors. 42 Similar to the rNDV/IL-2 virus, 41 the rLaSota/IL-2 treatment significantly increased CTLL2 cell proliferation in vitro (p < 0.01) compared with the unarmed rLaSota NDV virus.
In vivo, rLaSota/IL-2 efficiently replicated and produced IL-2 in the tumor microenvironment. The intratumoral delivery of rLaSota/IL-2 resulted in significant inhibition of tumor growth, leading to long-term survivors (75% in the H22 model and 62.5% in the B16-F10 model), compared with non-IL2-expressing NDV or control. Some long-term survivors due to rLaSota/IL-2 treatment in both models rejected tumor rechallenge, indicating the development of a memory response. 42 The superior antitumor efficacy of rLaSota/IL-2 treatment was associated with a robust tumor-specific IFN-γ response and enhanced tumor infiltration of both CD4+ and CD8+ T cells when compared with control NDV or mock therapies. No observable IL-2-related systemic toxicities were reported. 42
IL-2 expressing tanapoxviruses
Tanapoxvirus (TPV), classified under the Yatapoxvirus genus, is responsible for inducing a self-limiting febrile illness characterized by localized skin lesions. The virus was initially documented during two outbreaks in 1957 and 1962 in Kenya, primarily impacting communities residing in the floodplain of the Tana River. 43,44 TPV lacks the capability to transmit from person to person, 45 making it an advantageous attribute for the safe utilization of oncolytic virotherapy. Poxviruses are large double-stranded DNA viruses. The large genome size of TPV, ∼145 kb, allows the deletion of genes such as thymidine kinase (TK) to achieve specificity for cancer cells. Moreover, it also allows for the insertion of therapeutic transgenes such as IL-2 to promote the induction of effective antitumor immunity. 46 TPV/Δ66R/mIL-2 is a modified form of TPV where the TK(66R) gene was deleted and a murine IL-2 cytokine gene was inserted. 46 The oncolytic potential and antitumor efficacy of an TPV, named TPV/Δ66R/mIL-2, were assessed in both in vitro and in vivo models. 46,47
In vitro oncolytic activity of TPV/Δ66R/mIL-2
In the in vitro experiments, the replication potential and oncolytic activity of TPV/Δ66R/mIL-2 were evaluated in MDA-MB-231, MBA-MB-436, and MBA-MB-157 triple-negative breast cancer (TNBC) cells. Non-IL-2-expressing TPV (TPV/Δ66R) and wild-type TPV (wtTPV) were included as controls. The results showed that TPV/Δ66R/mIL-2 replicated efficiently in all TNBC cells, similar to the control TPV/Δ66R. However, the overall replication efficiency of both TPVs was lower than wtTPV, indicating that the genetic modifications had some impact on the replication potential of TPV recombinants. Nevertheless, both wtTPV and TPV recombinants demonstrated efficient oncolytic activity against MDA-MB-231 TNBC cells in a dose-dependent manner. 46
In vivo antitumor efficacy of TPV/Δ66R/mIL-2
In vivo, TPV/Δ66R/mIL-2 virotherapy demonstrated significant effectiveness in controlling the growth of subcutaneous MDA-MB-231 xenografted tumors in athymic nude-Foxn1nu mice, in comparison to mock treatment. This antitumor effect was sustained throughout the course of virotherapy. The sustained treatment efficacy was associated with tumor necrosis, varying degrees of fibrosis in the tumor capsule, and infiltration of inflammatory cells deep into the tumor tissues in the TPV/Δ66R/mIL-2 treatment group. 46
The antitumor efficacy of TPV/Δ66R/mIL-2 was also evaluated in a subcutaneously xenografted SKEML-3 human melanoma tumor model. 47 In this study, expression of IL-2 resulted in significant regression of SKEML-3 tumor growth when compared with controls (wtTPV or TPV/Δ66R without IL-2 expression). 47 Similar to the findings in the MDA-MB-231 TNBC model, the antitumor effects of TPV/Δ66R/mIL-2 treatment in the SKEML-3 melanoma model were also associated with a remarkable level of tumor cell degeneration, along with significant peritumoral infiltration of mononuclear cells, when compared with control treatments (wtTPV or TPV/Δ66R). These findings highlight the importance of IL-2 expression in the TPV platform. 47
The in vitro oncolysis and in vivo suppression of tumor growth, along with the recruitment of innate inflammatory immune cells in both TNBC and melanoma models, suggest that TPV/Δ66R/mIL-2 could be a potential candidate for the treatment of TNBC and melanoma.
Other viruses have been used to express IL-2, including VV, adenovirus, and herpes simplex virus type 1. A list of viral vectors used in cancer therapy is summarized in Table 1.
VIRAL COEXPRESSION OF IL-2 WITH OTHER TRANSGENES
Viral coexpression of IL-2 plus tumor necrosis factor-related apoptosis-inducing ligand (rNDV-IL2-TRAIL)
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), also known as CD253, selectively induces apoptosis in cancer cells in a p53-independent manner. 48 Bai et al. aimed to determine if coexpression of IL-2 and TRAIL by a viral vector would yield superior anticancer effects compared with the expression of a single transgene, such as IL-2 or TRAIL alone. 49 For this, they genetically modified the parental LaSota strain of NDV and constructed recombinants expressing no cytokine (rNDV), IL-2 (rNDV-IL2), TRAIL (rNDV-TRAIL), or IL-2 plus TRAIL (rNDV-IL2-TRAIL). These were then evaluated in both in vitro and in vivo models of murine H22 hepatic carcinoma and murine B16-F10 malignant melanoma. 49
In vitro replication and transgene production by rNDV-IL2-TRAIL
The genetic insertion of IL-2, TRAIL, or IL-2 plus TRAIL caused a slight delay in the onset of viral replication. However, by 72 h postinfection, the NDVs regained viral titers comparable to those of the parental LaSota NDV strain. This confirms that the genetic modification and insertion of foreign transgenes did not have a negative impact on viral replication in this case. Infection of H22 or B16-F10 cancer cells with rNDV-IL2, rNDV-TRAIL, or rNDV-IL2-TRAIL at a MOI of 0.01 resulted in efficient messenger RNA (mRNA) expression (determined by reverse transcriptase polymerase chain reaction) and the presence of the desired gene products, IL-2, TRAIL, or both, respectively, in the harvested cell culture supernatant (determined by enzyme-linked immunosorbent assay [ELISA]). In contrast, these cDNA products were not detected in H22 or B16-F10 cells infected with the control NDV (rNDV). 49
IL-2 produced from rNDV-IL2-TRAIL infection is biologically active
To assess the biological activity of IL-2 derived from rNDV-IL2 or rNDV-IL2-TRAIL-infected cells, DF1 chicken embryo fibroblast cells were infected with rNDV, rNDV-TRAIL, rNDV-IL2, or rNDV-IL2-TRAIL at an MOI of 0.1 for 48 h. The infected cell culture supernatant was harvested, IL-2 was purified, and 20 ng of the purified IL-2 was added to IL-2-dependent CTLL2 T cells for 48 h, and the proliferation index of CTLL2 cells was measured. As expected, IL-2 harvested from rNDV-IL2- or rNDV-IL2-TRAIL-infected supernatants stimulated the proliferation of CTLL2 cells, while no proliferation was observed with the other groups. 49
To further evaluate IL-2's biological activity, virally released IL-2 was used to generate lymphokine-activated killer (LAK) cells, which were then tested for their cytotoxic activity against H22 or B16-F10 cancer cells. Fresh splenocytes were stimulated with cytokine-free control medium or purified IL-2 obtained from rNDV-IL2-infected cancer cell supernatants for 3 days. The IL-2-stimulated splenocytes were considered LAK cells and cocultured with H22 or B16-F10 target tumor cells for 3 days. The purified IL-2 from rNDV-IL2- or rNDV-IL2-TRAIL-infected DF1 cell supernatants effectively stimulated splenocytes to differentiate into LAK cells, which exhibited significant cytotoxic activity against B16-F10 or H22 cancer cells compared with the controls. 49 These in vitro studies confirmed the biological activity of IL-2 produced from rNDV-IL2- or rNDV-IL2-TRAIL-infected cells.
In vivo antitumor efficacy of rNDV-IL2-TRAIL
Treatment of tumors with rNDV-IL2-TRAIL, compared with controls and single transgene recombinants, resulted in significant antitumor activity, with a significant extension of median survival in both murine H22 hepatic carcinoma and malignant B16-F10 melanoma models. This was associated with an increase in mRNA expression of apoptosis-related genes in rNDV-IL2-TRAIL-treated tumors compared with rNDV or rNDV-IL2 treatments. Additionally, both rNDV-IL2 and rNDV-IL2-TRAIL treatment resulted in a significantly increased accumulation of CD4+ and CD8+ T cells within tumors and an enhanced H22- or B16-F10-specific IFN-γ response, compared with treatments with rNDV-TRAIL, rNDV, or PBS. Although IL-2 was detected in the serum of mice receiving rNDV-IL2- or rNDV-IL2-TRAIL treatments, no toxicities were reported in vivo throughout the entire study period. 49
Overall, this study indicates that the antitumor immunity and anticancer effects of viral expression of IL-2 can be further enhanced by coexpression of a second transgene, such as TRAIL.
Viral coexpression of IL-2 plus interleukin 12 (rClone30-IL12-IL2)
Like IL-2, IL-12 is a promising heterodimeric anticancer cytokine that is naturally produced by antigen-presenting cells such as dendritic cells, macrophages, neutrophils, and B cells following antigenic stimulation. 50 It produces multifaceted antitumor immune effects: (1) stimulates T cells (both CD4+ and CD8+ T cells) and NK cells to produce immune effectors such as TNF-α and IFN-γ; (2) induces differentiation of naive T cells toward a Th1 phenotype; and (3) inhibits tumor angiogenesis through the IFN-γ-inducible protein-10 (IP-10) pathway. 51 Due to these diverse immune effects, IL-12 has been considered a master proinflammatory anticancer cytokine and has been tested in various cancers. 52 While early clinical trials of recombinant IL-12 suggested there was clinical activity, development was halted due to significant IL12-related adverse events, including fatal hepatotoxicity and neutropenia. 53
Since both IL-2 and IL-12 stimulate T and NK cells, 1 –3,51,52 Ren et al. hypothesized that viral coexpression of both IL-2 and IL-12 would induce superior antitumor effects compared with viral expression of IL-2 or IL-12 alone. 54 To test this hypothesis, they genetically modified the NDV lentogenic strain and generated multiple rNDV variants: rClone30-IL2 (NDV expressing IL-2), rClone30-IL12 (NDV expressing IL-12), rClone30-IL12-IL2 (NDV expressing IL-12 plus IL-2), and rClone30 (no cytokine expression), and tested their antitumor efficacy both in vitro and in vivo. 54
In vitro studies with rClone30-IL12-IL2
In vitro, all four recombinant viruses (rClone30, rClone30-IL2, rClone30-IL12, and rClone30-IL12-IL2) replicated in Vero cells with similar replication kinetics. Infection of tumor cells (HepG2 and U251 tumor cell lines) with cytokine-expressing rNDVs and their subsequent replication resulted in the efficient release of IL-2 or IL-12 cytokines in the cell culture supernatant, while no cytokines were found with rClone30. Similarly, all four recombinant viruses exhibited similar cytotoxic effects in different tumor cell lines, including U251, HepG2, HeLa, and A549. These in vitro studies suggest that dual transgene expression had no impact on viral replication or virus-induced cytotoxic effects. 54
In vivo studies with rClone30-IL12-IL2
OV-mediated cytokine release usually remains confined within the tumor microenvironment, thus limiting cytokine-related systemic toxicity. 14 –26 In a mouse H22 hepatocarcinoma model, Ren et al. demonstrated that after intratumoral injection of 1 × 107 rNDVs, the release of IL-2 and/or IL-12 remained confined within the tumors, with almost negligible levels of IL-2 and/or IL-12 found in the sera of treated mice. Regarding IFN-γ and IP-10, tumor homogenates treated with rClone30-IL12-IL2 had higher levels of IFN-γ and IP-10 than those treated with rClone30-IL2 or rClone30-IL12. 54
In terms of controlling H22 tumor growth in vivo, rClone30 treatment significantly reduced tumor burden compared with PBS treatment, which was further reduced due to IL-2 and/or IL-12 expression. Although no obvious difference in reducing tumor volume was observed between single and dual cytokine expression, rClone30-IL12-IL2 treatment resulted in 80% long-term survivors compared with 69%, 63%, 44%, and 0% in the rClone30-IL12, rClone30-IL2, rClone30, and PBS treatment groups, respectively. 54 This study suggests that when IL-2 is coexpressed with IL-12, it is more effective, but not significantly, than the expression of IL-2 alone.
Viral coexpression of IL-2 plus tumor necrosis factor alpha
Antitumor effects of the combination of Ad5-CMV-mTNFα plus Ad5-CMV-mIL2 viruses (defined as Ad5-CMV-mTNFα/mIL2)
Like IL-2, 10 the proinflammatory cytokine TNF-α plays a dual role in cancer. On one hand, it can inhibit tumor growth, while on the other hand, endogenous TNF-α can also support tumor growth and metastasis. 55 Havunen et al. explored whether adenoviral expression of both TNF-α and IL-2 within the tumor microenvironment induces a potent antitumor immune response locally and systemically and controls the growth of both local and untreated distant tumors. 56
To investigate this, they developed bilateral mouse B16-OVA melanoma tumors and treated one tumor intratumorally with PBS or replication-deficient adenoviruses, such as Ad5-Luc1 (expression of luciferase), Ad5-CMV-mIL2 (expression of murine IL-2), Ad5-CMV-mTNFα (expression of murine TNF-α), or a combination of Ad5-CMV-mTNFα plus Ad5-CMV-mIL2 (referred to as Ad5-CMV-mTNFα/mIL2). 19,57 The treatments were administered on days 11 and 14 post-tumor implantation, with a dose of 1 × 109 viral particles (VPs) for single viruses or 0.5 × 109 VPs each when two viruses were combined, while the other tumors were left untreated. 56
They observed that cytokine-armed viruses were effective in controlling the growth of both treated and untreated tumors compared with the control, although no significant differences were observed in tumor sizes between the groups receiving treatments with adenoviruses expressing single versus dual cytokine expression. 56 Importantly, only armed viruses were able to control the growth of noninjected tumors, not the unarmed viruses. As expected, immune analysis revealed that treatment with cytokine-armed viruses resulted in increased intratumoral infiltration of natural killer cells (CD3−CD11b+NK1.1+), mature dendritic cells (CD11c+CD86+; especially in tumors receiving IL-2 cytokine expression), and macrophages (CD11b+F4/80+) in both injected and noninjected tumors. The aforementioned viruses were also tested in combination with adoptive T cell therapy or TILs, which are discussed in the IL-2 Expressing Viruses in Combination with Nonviral Therapies section. 56
Antitumor effects of Ad5/3-E2F-d24-hTNFα-IRES-hIL2 (also known as TILT-123)
Havunen et al. engineered replication-competent oncolytic adenoviruses, such as Ad5/3-E2F-d24-hTNFα-IRES-hIL2 (adenoviral coexpression of human TNF-α and human IL-2, also known as TILT-123), and its respective controls, including Ad5/3-E2F-d24-hIL2 (adenoviral expression of human IL-2), Ad5/3-E2F-d24-hTNFα (adenoviral expression of human TNF-α), and Ad5/3-E2F-d24 (no cytokine expression), 58 and tested their efficacy in various tumor models (Fig. 2).
It was demonstrated that following treatment of HapT1 tumors with TILT-123, IL-2 production was localized within HapT1 tumors, with almost no detectable levels of IL-2 found in the serum. 58 In vivo, in severe combined immunodeficiency (SCID) mice bearing intraperitoneal SKOV3-Luc human ovarian carcinoma, all four viruses, including TILT-123, demonstrated similar efficacy against SKOV3-Luc tumors when compared with mock (PBS) treatment. 58 No statistical differences were observed between single and dual cytokine expression in controlling the growth of ovarian tumors, which could be due to the use of SCID mice that lack an immune system, thus cytokine expression could not produce any additional immunostimulatory effect over control Ad5/3-E2F-d24 (no cytokine expression). However, in an immunocompetent HapT1 pancreatic cancer model in Syrian hamsters, immune analysis of tumors revealed that viral expression of IL-2 was critical in recruiting CD4+ and CD8b+ T cells into tumors and augmenting the proliferation of splenocytes ex vivo. 58
Santos et al. utilized TILT-123 to reshape the immunosuppressive ovarian tumor microenvironment in ex vivo patient-derived tumor tissue samples. Single-cell suspensions of ovarian tumor cultures were inoculated with cytokine-armed viruses (TILT-123) or unarmed viruses (Ad5/3-E2F-d24) at an MOI of 100. TILT-123 treatment of ex vivo tumor cultures resulted in an increased release of proinflammatory effector molecules such as IFN-γ, TNF-α, and IL-2, as well as activation of CD4+ and CD8+ TILs, as demonstrated by their expression of CD69, compared with treatments with Ad5/3-E2F-d24 or the vehicle. This underscores the importance of cytokine expression in modulating antitumor effector responses. 59 The same group also tested these human cytokine-armed viruses in combination with TILs, which we discuss in the IL-2 Expressing Viruses in Combination with Nonviral Therapies section.
Viral coexpression of IL-2 plus antiprogrammed death ligand 1
In addition to the construction of vaccinia viruses expressing membrane-bound IL-2 (vvDD-IL-2-FG and vvDD-IL-2-RG), Liu et al. also engineered a VV (vvDD-IL-2-FPTM) that expresses transmembrane-conjugated IL-2 fused with a murine programmed death ligand 1 (PD-L1) transmembrane domain and a (G4S)3 flexible linker. 28 However, unlike the survival benefits observed with vvDD-IL-2-FG or vvDD-IL-2-RG in the intraperitoneal MC38-luc murine colon cancer model, treatment with vvDD-IL-2-FPTM did not provide any additional survival benefit compared with treatment with the control vvDD virus. The failure to produce therapeutic benefits with vvDD-IL-2-FPTM treatment is likely associated with the low levels of IL-2 expressed on the cell membrane following virus infection. 28
A list of viral vectors to coexpress IL-2 with other anticancer transgenes is summarized in Table 2.
IL-2 EXPRESSING VIRUSES IN COMBINATION WITH NONVIRAL THERAPIES
Viral expression of IL-2 in combination with Sunitinib
Sunitinib, a multitargeted tyrosine kinase inhibitor, inhibits tumor angiogenesis by interacting with platelet-derived growth factor receptor (PDGFR), vascular endothelial-derived growth factor receptor (VEGFR), c-kit (CD117), and FMS-like tyrosine kinase-3 receptor (FLT3). 60 It is approved for patients with metastatic renal cell carcinoma, 61,62 pancreatic neuroendocrine tumors, and gastrointestinal stromal tumors. 63,64 In the ISOS-1 angiosarcoma model in BALB/c mice, Takehara et al. evaluated the therapeutic effect of sunitinib in combination with an inactivated Sendai virus carrying an IL-2 cDNA (HVJ-E/IL-2). 34
The combined use of sunitinib plus HVJ-E/IL-2 resulted in superior inhibition (statistically nonsignificant) of ISOS-1 angiosarcoma growth, resulting in 75% tumor-free mice compared with only 35.7% in the HVJ-E/IL-2 treatment group. 34 Immune analysis revealed that sunitinib prevented infiltration of CD8+ T and NK cells into the tumors, and this inhibitory effect was counteracted by the viral expression of IL-2 (HVJ-E/IL-2). Similar findings were observed by Saha et al. in an immunocompetent brain tumor model, where axitinib, another tyrosine kinase inhibitor, reduced the infiltration of CD3+ T cells in the tumor, and this immune inhibitory effect of axitinib was overcome by viral expression of IL-2-like cytokine such as IL-12 in the tumor microenvironment. 23
Viral expression of IL-2 in combination with immune checkpoint blockade
Although vvDD-IL-2-RG (expressing membrane-bound IL-2) treatment induced a systemic antitumor immune response in the intraperitoneal MC38-luc tumor model, it failed to completely eradicate tumors, possibly due to elevated expression of PD-1, PD-L1, and CTLA-4 within the tumors. 28 Consequently, Liu et al. assessed vvDD-IL-2-RG in combination with immune checkpoint inhibitors, such as anti-PD-1/PD-L1 or anti-CTLA-4 antibodies, in a late-stage established MC38-luc tumor model. The combination of unarmed vaccinia virus (vvDD) with immune checkpoint inhibitors (anti-PD-1, anti-PD-L1, or anti-CTLA-4) did not confer any survival benefits compared with vvDD monotherapy. However, the combination of IL-2 expression (vvDD-IL-2-RG) plus anti-PD-1 or anti-PD-L1 (but not anti-CTLA-4) treatment resulted in tumor eradication in 90% of mice, compared with only 10% in the vvDD-IL-2-RG treatment group. 28
The survival benefits observed in the combination studies with armed (vvDD-IL-2-RG) viruses plus immune checkpoint blockade indicate a role for IL-2 expression in enhancing the efficacy of T cell-modulating immunotherapies against cancer.
As mentioned above, Ekeke et al. evaluated the antitumor effects of a VV-IL2 in a murine model of MPD. 30 In the same study, the authors examined the antitumor effects of combination therapy (VV-IL2 plus anti-PD-1) in MPD mice. Unfortunately, no additional survival benefit was observed when the combination was compared with VV-IL2 treatment alone. 30
Viral expression of IL-2 in combination with adoptive cell therapy
Santos et al. developed a 5/3 chimeric oncolytic adenovirus encoding human IL-2, designated as Ad5/3-E2F-d24-hIL2, and examined its antitumor efficacy in combination with adoptive TIL therapy. 65 They demonstrated that intratumoral administration of Ad5/3-E2F-d24-hIL2 in HapT1 tumors in immunocompetent Syrian hamsters resulted in localized production of IL-2, replacing the systemic use of exogenous recombinant IL-2, which is known to cause severe toxicities in patients. Notably, the combination of Ad5/3-E2F-d24-hIL2 and TILs exerted superior control over HapT1 tumor growth compared with the combination of recombinant human IL-2 and TILs. The enhanced antitumor efficacy of Ad5/3-E2F-d24-hIL2 plus TILs was associated with increased infiltration of CD8b+ T cells in the tumor. 65 These findings suggest that local production of IL-2 supports T cell immunotherapies while ensuring safety.
Viral coexpression of murine IL-2 and murine TNF-α in combination with adoptive cell therapy
Havunen et al. investigated the combination of Ad5-CMV-mTNFα/mIL2 with OVA-specific OT-1 T cells in bilateral B16-OVA melanoma tumors, as in Antitumor Effects of the Combination of Ad5-CMV-mTNFα Plus Ad5-CMV-mIL2 Viruses (Defined as Ad5-CMV-mTNFα/mIL2) section. 56 On one side, tumors received intratumoral injections of Ad5-CMV-mTNFα/mIL2 (0.5 × 109 VPs each), Ad5-Luc1 (1 × 109 VPs/injection), or a mock treatment on days 11 and 14 after tumor implantation, while tumors on the other side remained untreated. On day 11, all three groups of mice also received a single intraperitoneal injection of CD8-enriched OVA-specific OT-1 cells at a dose of 1.4 × 106 cells per animal. Intratumoral expression of mTNFα/mIL2 + OT-1 treatment did not impact the growth of the injected tumors but did delay the growth of the noninjected tumors compared with the mock treatment. The antitumor effect at distant sites was not attributed to virus spread since Ad5-CMV-mTNFα/mIL2 and Ad5-Luc1 are replication deficient, and no virus was detected in the noninjected tumors. 56
Viral coexpression of human IL-2 plus human TNF-α in combination with adoptive cell therapy
In the Antitumor Effects of Ad5/3-E2F-d24-hTNFα-IRES-hIL2 (also known as TILT-123) section, we discussed the antitumor effects of TILT-123 (Ad5/3-E2F-d24-hTNFα-IRES-hIL2) and its respective controls (Ad5/3-E2F-d24-hIL2, Ad5/3-E2F-d24-hTNFα, or Ad5/3-E2F-d24) as monotherapeutic agents. 58 Furthermore, Havunen et al. conducted additional tests to evaluate the antitumor effects of TILT-123 in combination with TILs both ex vivo and in vivo using a HapT1 pancreatic cancer model in Syrian hamsters. 58 To conduct the ex vivo studies, the authors established subcutaneous HapT1 tumors, harvested the tumors, and minced them on day 10 posttumor implantation. They then cultured the harvested tumor fragments in the presence of human IL-2 and extracted stimulated TILs, which were used for ex vivo and in vivo studies. 58
Combination of TILT-123 and TILs ex vivo
For the ex vivo studies, HapT1 cells were infected with 5 × 103 VPs of TILT-123 (or respective controls) for 72 h. They added TILs isolated from HapT1 tumors to the virus-infected cells and cocultured HapT1/TILs for 24 h. The cytotoxicity was assessed using an MTS assay. The data demonstrated that the addition of TILs significantly increased the cell-killing effect of viruses, including IL-2-expressing viruses, compared with the killing effects observed with viruses alone (without added TILs). 58
Combination of TILT-123 and TILs in vivo
In an immunocompetent HapT1 Syrian hamster model, treatment with Ad5/3-E2F-d24-hIL2 or Ad5/3-E2F-d24-hTNFα-IRES-hIL2 (TILT-123) exhibited robust antitumor efficacy, which was further enhanced in mice receiving the combination therapy of virus plus TILs. Importantly, combination therapy with TILT-123 plus TILs resulted in tumor remission in all mice, leading to 100% long-term survivors or cure. The cured mice from the cytokine-armed virus-treated groups rejected tumor rechallenge with the same HapT1 tumor cells and supported the growth of tumors resulting from the challenge with antigenically different DDT1-MF2 hamster leiomyosarcoma cells. In contrast, the cured mice from the unarmed virus-treated group did not reject the HapT1 rechallenge but slowed the growth of HapT1 tumors. 58 This rechallenge study indicates that TILT-123 treatment induces tumor (HapT1)-specific immunological memory.
Combination of TILT-123 and autologous TILs ex vivo
Santos et al. utilized TILT-123 in combination with autologous TILs to reshape the immunosuppressive ovarian tumor microenvironment in ex vivo patient-derived tumor tissue samples. 59 For this study, TILs were isolated from the tumor specimens and expanded in vitro, resulting in autologous TILs. Single-cell suspensions of ovarian tumor cultures, depleted of CD3+ cells, were then inoculated with cytokine-armed viruses (TILT-123) or unarmed viruses (Ad5/3-E2F-d24) at a MOI of 100. After virus treatment, 0.85 × 106 autologous expanded TILs were added to the culture. The supernatants and cells from this coculture study were subsequently collected and evaluated for cytokines and T cell markers, respectively. Autologous TILs produced a significantly higher level of IFN-γ in response to treatment of ex vivo ovarian tumor cultures with TILT-123 compared with treatments with Ad5/3-E2F-d24 or vehicle. Importantly, this increased level of IFN-γ release from autologous TILs was independent of PD-L1 expression in the ovarian cancer cells. 59
TILT-123 eliminates the necessity for lymphodepletion chemotherapy while enhancing the antitumor efficacy of TILs
Lymphodepleting chemotherapy, such as cyclophosphamide plus fludarabine, is commonly utilized to enhance the effectiveness of adoptive T cell therapy, despite the severe toxicity associated with these agents. 66 Santos et al. demonstrated that TILT-123 therapy could enhance the efficacy of adoptively transferred TILs, thus eliminating the need for a lymphodepletion chemotherapy regimen. For instance, the combination of intratumoral TILT-123 with intravenous TILs yielded comparable antitumor efficacy against HapT1 tumors as the combination of lymphodepleting chemotherapy with TILs or the combination of lymphodepleting chemotherapy with intratumoral TILT-123 followed by intravenous TILs. 66
CONCLUSIONS
The experimental studies presented in this article showcase the versatility of IL-2 as an important factor in promoting T cell growth, enhancing cytolytic activity, and eliciting antitumor immune responses. However, the clinical application of recombinant IL-2 cytokine therapy for cancer treatment encounters significant challenges due to its potential multiorgan systemic toxicities. Fortunately, OVs have emerged as a promising avenue for delivering IL-2 directly to tumors while minimizing systemic side effects. The studies presented herein have demonstrated the delivery of IL-2 by OVs, either alone or in combination with other anticancer transgenes, can result in promising antitumor effects in murine tumor models. These IL-2-expressing OVs possess tremendous potential as targeted immunotherapy strategies for cancer. Further validations in clinical trials are needed to determine the full therapeutic potential of this approach.
Among the IL-2-expressing OVs discussed above, TILT-123 is the furthest along in the clinic. Based on the aforementioned findings, TILT-123 can be regarded as an interesting treatment strategy for combination with adoptive T cell therapy or other forms of immunotherapies in clinical settings. In fact, there are currently multiple ongoing clinical trials with TILT-123, either as a monotherapy or in combination with TILs, anti-PD-1, or anti-PD-L1 therapies (NCT05271318, NCT04695327, NCT05222932, and NCT04217473) and we await their results. Nevertheless, further research is warranted to fully elucidate their therapeutic potential and underlying mechanisms of action. In summary, the development of IL-2-expressing OVs represents a promising approach for enhancing the delivery, efficacy, and safety of IL-2 in cancer immunotherapy.
Footnotes
AUTHORs' CONTRIBUTIONS
H.W.: prepared tables and figures, wrote/edited the article; M.B.: prepared figures; A.H. and S.B.: edited and critically reviewed the article; N.S.: literature review; H.L.K. and S.D.R.: wrote/edited the article; D.S.: conceptualization, wrote the article text, methodology, edited tables/figures, and funding acquisition. All authors contributed to the article and approved the submitted version.
AUTHOR DISCLOSURE
S.D.R. is a co-inventor on patents relating to oncolytic herpes simplex viruses, owned and managed by Georgetown University and Massachusetts General Hospital, which have received royalties from Amgen and ActiVec, Inc., and acted as a consultant and received honoraria from Replimune, Cellinta, and Greenfire Bio, and honoraria and equity from EG 427. H.L.K. is an employee of Ankyra Therapeutics and has received honoraria for participating on advisory boards for Castle Biosciences, Midatech Pharma, Marengo Therapeutics, and Virogin. A.H. is a shareholder in Targovax ASA and an employee and a shareholder of TILT Biotherapeutics Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FUNDING INFORMATION
D.S. was supported in part by a fund from the DOD (W81XWH-20-1-0702) and S.D.R. was supported in part by a grant from NIH (R01 CA160762) and the Thomas A. Pappas chair in Neurosciences.