
Abstract
Interleukin 7 Receptor alpha Severe Combined Immunodeficiency (IL7R-SCID) is a life-threatening disorder caused by homozygous mutations in the IL7RA gene. Defective IL7R expression in humans hampers T cell precursors' proliferation and differentiation during lymphopoiesis resulting in the absence of T cells in newborns, who succumb to severe infections and death early after birth. Previous attempts to tackle IL7R-SCID by viral gene therapy have shown that unregulated IL7R expression predisposes to leukemia, suggesting the application of targeted gene editing to insert a correct copy of the IL7RA gene in its genomic locus and mediate its physiological expression as a more feasible therapeutic approach. To this aim, we have first developed a CRISPR/Cas9-based IL7R-SCID disease modeling system that recapitulates the disease phenotype in primary human T cells and hematopoietic stem and progenitor cells (HSPCs). Then, we have designed a knockin strategy that targets IL7RA exon 1 and introduces through homology-directed repair a corrective, promoterless IL7RA cDNA followed by a reporter cassette through AAV6 transduction. Targeted integration of the corrective cassette in primary T cells restored IL7R expression and rescued functional downstream IL7R signaling. When applied to HSPCs further induced to differentiate into T cells in an Artificial Thymic Organoid system, our gene editing strategy overcame the T cell developmental block observed in IL7R-SCID patients, while promoting full maturation of T cells with physiological and developmentally regulated IL7R expression. Finally, genotoxicity assessment of the CRISPR/Cas9 platform in HSPCs using biased and unbiased technologies confirmed the safety of the strategy, paving the way for a new, efficient, and safe therapeutic option for IL7R-SCID patients.
INTRODUCTION
The group of severe combined immunodeficiencies (SCID) represents the most serious form of primary immunodeficiency diseases, affecting approximately one infant in every 50,000 live births. SCID is characterized by a block in T cell development or function, variably associated with defects in B or natural killer (NK) lymphocytes. IL7R deficiency causes ∼10% of SCID cases, and the majority of T-B+NK+ cases. 1 IL7R-SCID is caused by biallelic loss-of-function mutations in the IL7RA gene, which encodes for the α chain of the interleukin-7 receptor (IL7R).
The interaction of IL-7 with IL7R leads to the recruitment of intracellular signaling molecules and to activation of multiple downstream pathways, which are important for transcriptional activation of genes involved in T cell differentiation, survival, maturation, and TCR rearrangement. IL7R expression is restricted to T lymphocytes in humans and it is tightly regulated during T cell development to ensure correct cell maturation. 2 In the human thymus, αβ T cell thymopoiesis proceeds from hematopoietic stem and progenitor cells (HSPCs) through common lymphoid progenitors (CLPs), which commit to the T-lineage following Notch signaling. CLPs differentiate into double-negative (DN1, DN2, DN3; CD4–CD8–), then double-positive (DP; CD4+CD8+) thymocytes, which are positively selected for TCR functionality and become single-positive (SP) CD4+ or CD8+ mature T cells. 3 Only CLPs express IL7R without requiring it, allowing human IL7RA-deficient CLPs to develop into normal B cells. 4 From the DN2 stage, BCL-2 expression, essential for protecting thymocytes from apoptosis, becomes IL7 dependent.
IL7R also promotes V(D)J recombination in DN2–3 cells and T cell receptor γ (TCRγ) rearrangement and is thus essential for γδ-T cell development. 5 IL7R expression is then downregulated at the DN4 stage and disappears in DP thymocytes, when TCR-αβ becomes expressed and takes over the antiapoptotic role (reviewed in Mazzucchelli and Durum2). As such, disruption of IL-7 signaling arrests T cell development at the DN2-3 stage in IL7RA-knockout mice, 5,6 preventing productive TCR rearrangement and leading to T cell lymphopenia. The absence of T lymphocytes results in profound failure of both humoral and cellular immunity, with severe and opportunistic infections and failure to thrive, leading to fatal outcome within the early years of life of IL7R-SCID children.
Hematopoietic stem cell transplantation (HSCT) from matched sibling donors is the leading therapy for patients with SCID, 7 however, it is available to less than 20% of patients. While matched HSCT often successfully reconstitutes T cells in IL7R-SCID patients and raises their low immunoglobulin levels, 8 mismatched donor transplantation is associated with aggressive graft-versus-host disease, long-term hepatic complications of myeloablative conditioning, insufficient immune reconstitution, and mortality, especially if infection occurs. 9 Consequently, HSCT does not represent a viable therapeutic option for the remaining patients lacking a suitable donor. Genetic correction of autologous patient HSPCs and subsequent transplantation eliminates the risk of alloreactivity associated with HSCT and facilitates the use of submyeloablative conditioning. Successful gene therapy approaches for ADA-SCID and SCID-X1 have demonstrated the applicability of this technology to treat rare genetic diseases that affect the hematopoietic system. 10
However, preclinical gene therapy studies using a retroviral vector to introduce a correct copy of IL7RA in HSPCs derived from IL7R-deficient mice showed that constitutive, unregulated, and ectopic expression of the protein can promote nonlymphoid cell proliferation, causing preleukemic neutrophil expansion and splenomegaly. 11 Evidence also suggests that forced IL7R expression throughout thymopoiesis diminishes the DN4 cell pool due to DP cell overconsumption and reduces thymic cellularity and peripheral T cell numbers. 12 Moreover, IL7RA gene amplification and human gain-of-function mutations that increase IL-7 sensitivity associate with T-ALL, 13,14 whereas IL7R overexpression may also protect preleukemic T cells from apoptosis, allowing time for additional mutations to accumulate. 15 Ultimately, it has been shown that IL7R overexpression predisposes mice to thymoma 16 and inflammatory bowel disease. 17 Taken together, these observations highlight the need of tight IL7RA transcriptional regulation for therapeutic correction of IL7R-SCID.
While the introduction of IL7RA-specific regulatory regions in the viral vector design could serve this purpose, no studies have succeeded so far in correcting this disease through viral gene therapy, mostly due to: (1) difficulties in defining the minimum regulatory regions required to recapitulate endogenous IL7R expression; (2) the complex regulatory networks needed to control IL7RA transcription and translation are prohibitively large for incorporation into a viral vector to achieve tight physiological regulation; (3) semi-random vector integration pattern, which may impact on transgene expression.
An alternative to using virus-based gene therapy is to utilize genome editing, to correct the endogenous IL7RA locus while avoiding issues of unregulated transgene expression. Gene editing uses programmable nuclease to generate site-specific genomic double-strand breaks (DSBs) in which desired alterations can be introduced during DNA repair. The major repair pathway is nonhomologous end joining (NHEJ), in which the DSB site gains a random assortment of small insertions and/or deletions (indels) and point mutations. The alternative pathway is homology-directed repair (HDR), in which a donor template with flanking arms homologous to the DSB-surrounding region accurately integrates into the DSB site. This allows targeted therapeutic transgene integration as a platform for correcting multiple mutations. 18
Robust protocols have been developed for CRISPR/Cas9 gene editing in HSPCs, 19 –21 and several gene editing platforms to treat primary immunodeficiency diseases relying on high-fidelity HDR to integrate a therapeutic transgene in its own locus have been developed preclinically (reviewed in Naseem et al. 22 ), reaching up to 70% of HSPC's correction and complete hematopoietic reconstitution in mice, establishing the safety and efficacy of this approach.
In this study, we aim to develop a safe and effective CRISPR/Cas9-mediated genome editing platform to treat IL7R-SCID by directly knocking-in a IL7RA cDNA in frame with the endogenous IL7RA translational start codon, allowing regulated transcription from the promoter and enhancers naturally present in the locus, as well as the functional correction of all the mutations in the IL7RA gene responsible for the onset of the disease. Given the rarity of the disease and the difficulty in accessing patient blood samples, we devised a disease modeling strategy based on a multiplexed HDR platform to mimic both IL7R deficiency in T cells and HSPCs from healthy donors, as well as its restoration by monoallelic or biallelic knockin (KI) of the corrective IL7RA cDNA through gene editing.
By taking advantage of this system, we showed almost complete restoration of IL7R expression and function in IL7RA cDNA knocked-in T cells, as well as rescue of T cell development when the corrective transgene is incorporated in IL7RA knockout HSPCs. Overall, this study demonstrates the feasibility and safety of a CRISPR/Cas9-based platform as a viable therapeutic approach to treat IL7R-SCID and paves the way for its potential clinical translation.
MATERIALS AND METHODS
Culture of human CD34+ HSPCs and T cells
Under written informed consent, CD34+ HSPCs were isolated from GCSF mobilized healthy donor apheresis using the CD34+ Microbead Kit (Miltenyi Biotec). The percentage of purified CD34+ cell was analyzed by flow cytometry staining with anti-human CD34 PE antibody (BioLegend). The cells were cultured in StemSpan SFEM II medium (StemCell Technologies) supplemented with Stem Cell Factor (100 ng/mL; PeproTech), Thrombopoietin (50 ng/mL; PeproTech), Fms-like Tyrosin kinase 3 Ligand (100 ng/mL; PeproTech), Interleukin-3 (30 ng/mL; PeproTech), Interleukin-6 (50 ng/mL; PeproTech), StemReginin-1 (1 μM; Sigma), and UM171 (35 nM; StemCell Technologies). The cells were incubated at 37°C/5% CO2 for 2 days before gene editing.
To isolate T cells, human peripheral blood (PB) from healthy donors were first collected under written informed consent. After Ficoll gradient, the isolated PB mononuclear cells were cultured in X-VIVO 15 medium (Lonza) supplemented with 5% human AB serum (Lonza), Interleukin-2 (50 ng/mL; PeproTech), Interleukin-7 (10 ng/mL; PeproTech) and activated by CD3-CD28 Dynabeads (Thermo Fisher Scientific). After incubating at 37°C/5% CO2 for 3 days, the Dynabeads were removed by DynaMag Magnet (Thermo Fisher Scientific) before performing any gene editing experiment.
Methylcellulose CFU assay
The colony-forming unit (CFU) assay was performed by seeding 500 cells in six-well plates containing MethoCult Enriched (StemCell Technologies) after 4 days of editing. After 14 days of incubation at 37°C/5% CO2, different types of colonies, including CFU-Erythroid (E), CFU-Macrophage (M), CFU-Granulocyte (G), CFU-GM, and CFU-GEM were counted based on their morphological appearance.
Selection of gRNAs
All the gRNAs used in this study, each with 20 nucleotide length sequences, were identified using the online design tool created by the Zhang laboratory (
Cloning of donor templates and AAV6 production
Both KO and KI AAV6 donor vectors carrying 400 bp left and right homology arms on each side were cloned into the pAAV-MCS plasmid containing AAV2-specific ITRs. Each of the two KO constructs comprised either EFS-GFP (AAV6_EFS-GFP) or EFS-mCHERRY cassette (AAV6_EFS-mCHERRY), whereas the two IL7R corrective AAV6 contained 1,380 bp codon-optimized IL7R cDNA followed by either an EFS-GFP (AAV6_coIL7RA_EFS-GFP) or EFS-mCHERRY cassette (AAV6_coIL7RA_EFS-mCHERRY_KI). HEK293T cells were transfected with the AAV6 donor plasmid and pDGM6 helper plasmid, and after 72 h the viral particles were collected by iodixanol gradient and purified using 10K MWCO Slide-A-Lyzer G2 Dialysis Cassette (Thermo Fisher Scientific). AAV6 titer was determined by the Quick Titer AAV Quantification Kit (Cell BioLabs).
Electroporation and transduction of cells
CD34+ HSPCs, human primary T cells, and DG-75 cells were electroporated using the Neon Transfection Kit (Thermo Fisher Scientific) at 1,600 volts/10 ms/3 pulses. The ribonucleoprotein (RNP) complex was made by combining High-Fidelity Cas9 protein (IDT) with IL7RA targeting gRNAs at a molar ratio of 1:2 (Cas9:gRNA). To generate biallelic knockout (KO−/−), the electroporated cells were transduced with AAV6_EFS-GFP and AAV6_EFS-mCHERRY. For biallelic correction (KO+/+), the cells were transduced with AAV6_coIL7RA_EFS-GFP and AAV6_IL7R_EFS-mCHERRY, whereas for monoallelic correction (KO+/−), cells were transduced with the AAV6_EFS-GFP and AAV6_coIL7RA_EFS-mCHERRY. The multiplicity of infection (MOI) used for each AAV6 was 25,000 vector genomes/cell. After 48 h in culture, the gene-edited cells were sorted by flow cytometry for the collection of highly enriched GFP+, mCherry+, and GFP+/mCherry+ populations.
Artificial Thymic Organoid system
To generate Artificial Thymic Organoids (ATOs), 0.15 × 106 of murine stromal cells expressing human delta-like ligand 1 (Sigma) were combined with 7.5 × 103 of sorted and enriched KO−/−, KI+/−, or KI+/+ CD34+ HSPCs. ATOs seeded with healthy donor HSPCs (wild-type, WT) were used as a positive control. Each ATO pellet was resuspended in 5 μL of RB-27 medium comprising RPMI 1640 (Thermo Fisher Scientific), 4% B-27 supplement (Thermo Fisher Scientific), 30 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma), 1% penicillin–streptomycin (Thermo Fisher Scientific), 5 ng/mL human FLT3 ligand, and 5 ng/mL human IL-7. Five microliters of ATO suspension was seeded on 0.4 μm Millicell Transwell insert (Sigma) and placed in a six-well plate containing 1 mL of RB-27 medium. The medium was changed and replaced with fresh RB-27 medium every 3–4 days. Each ATO was harvested for flow cytometry analysis at week 4, 6, and 8 postseeding.
Flow cytometry analyses
BD FACSAria II (BD Bioscience) instrument was used for cell sorting of CD34+ HSPCs and T cells. For surface and intracellular staining, CytoFLEX (Beckman Coulter) instrument was used while FlowJo v10 software (FlowJo LLC) was used to analyze subsequent data.
For IL7R protein detection, bulk unsorted KO−/−, KI+/−, and KI+/+ T cells were fixed with 1 × Fixation buffer (Thermo Fisher Scientific) for 30 min at room temperature. After washing, the fixed cells were permeabilized in 1 × Permeabilization buffer (Thermo Fisher Scientific) and stained with IL7R PE antibody (BioLegend) for 30 min at room temperature and analyzed on flow cytometry. The percentage of total IL7R expression on KO−/−, KI+/−, and KI+/+ were determined by gating on the GFP+/mCherry+ double-positive population.
For pSTAT5 detection, bulk unsorted KO−/−, KI+/−, and KI+/+ T cells were cultured in X-VIVO 15 medium (Lonza) in the absence of serum and cytokines for 12 h. After stimulation with 10 ng/mL of either IL-2 or IL-7 cytokines for 10 min, the cells were fixed in 4% PFA for 15 min at room temperature and permeabilized in Perm III buffer (BD Bioscience) for 30 min on ice. The cells were stained with pSTAT5 BV421 (BioLegend) antibody and analyzed by flow cytometry. The precise impact of KO−/−, KI+/−, and KI+/+ on activated STAT5p was determined by gating on GFP+/mCherry+ double-positive population. Stimulated and unstimulated healthy donor T cells were used as positive and negative control, respectively.
Digital droplet PCR analysis
The frequency of integrated GFP and mCherry cassettes per IL7RA allele was quantified by Digital droplet PCR. For each reaction in a total volume of 27 μL, 60 ng of genomic DNA was combined with 1 × PerfeCT Mulitplex qPCR ToughMix (Quantabio), 0.05 μM Fluorescein (Thermo Fisher Scientific), 1 μM each of GFP forward and reverse primer, 1 μM each of mCherry forward and reverse primers, 0.25 μM FAM-labeled probe specific for GFP target amplicon, 0.25 μM HEX-labeled probe specific for mCherry target amplicon, 1 μM each of reference SPDR forward and reverse primer, 0.25 μM Cy5-labeled probe specific for reference SPDR amplicon, and nuclease-free water. All the primers and probes were synthesized by Eurofins Genomics. Each reaction was inserted into Sapphire chips and run on Naica 3 Color system (Stilla Technologies). The PCR conditions used were: 1 cycle of initial denaturation at 95°C for 10 min and 50 cycles comprising of denaturation (95°C for 30 s), annealing (54°C for 30 s), and extension (60°C for 6 min). After PCR, the chips were scanned and analyzed by CrystalReader and CrystalMiner software (Stilla Technologies), respectively.
The percentage of GFP or mCherry cassette integrated per haploid genome is calculated initially by the number of target amplicon-positive droplets from either FAM or HEX channel divided by the number of reference amplicon-positive droplets acquired from Cy5 channel and multiplied by 100%. The corresponding value is then divided by 2 to account for the diploid IL7RA locus.
Validation of predicted off-target sites by deep sequencing
Potential off-target sites for IL7RA gRNA detected by COSMID webtool 23 and GUIDE-seq were validated by high-throughput next-generation sequencing. CD34+ HSPCs from three healthy donors were edited with IL7R RNP complex as described above, and genomic DNA was harvested after 72 h. PCR-purified amplicons of 200 bp were generated from the list of off-target primers. End repair, adaptor ligation, and PCR indexing were performed on the denatured amplicons using the NEB Next Ultra II DNA Library Prep Kit for Illumina (New England BioLabs). The resulting FASTQ files from RNP-treated samples for each of the off-target amplicons were analyzed for indels through CRISPResso2 webtool 24 by comparing them with untreated samples.
GUIDE-seq
Identification of potential off-target sites by GUIDE-seq 25 was performed by Creative Biogene. One million HEK293T cells were transfected with 12 μg HIFI Cas9, 4 μg IL7RA gRNA4, and 5 pmol of dsODN using Lonza Nucleofector 4-D (program CM-137). At 48 h posttransfection, genomic DNA was extracted and sheared using a Covaris S220 Focussed-ultrasonicator to an average length of 500 bp. After end-repaired, A-tailed, and ligation with adaptors containing 8-nt random molecular index, the DNA library was sequenced using Illumina Miseq. The subsequent datasets were analyzed using the guideseq Python package software. 26
CAST-seq
Chromosomal aberration analysis by single targeted LM-PCR (CAST-seq) was performed and analyzed as described in Turchiano et al. 27 Briefly, 1 × 106 HSPCs were nucleofected with spCas9;gRNA4 RNP targeting IL7RA or mock nucleofected in biological duplicates. At day 4 postnucleofection, genomic DNA was extracted and 500 ng of gDNA from each sample were randomly digested with the NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina (NEB #E6177) to obtain fragments of ca. 350 bp, and linkers were ligated. The first PCR was performed with IL7RA-specific primer and decoy oligonucleotide together with the linker-specific primer. A nested PCR was performed with the IL7RA and linker-specific primers carrying Truseq adaptor sequences. Samples of high-throughput sequencing were performed with the MiSeq V2 500 cycle Kit (MS-102-2003; Illumina). Data were analyzed by filtering out nonspecific reads utilizing the mock nucleofected sample as a control and by comparing duplicate samples.
Finally, the retrieved hits were classified as chromosomal aberrations coming from the on-target specific activity (ON), spCas9 OFF-target activity (OT), homology-mediated recombination events (HR), and from natural breaking sites. Frequency of detection is calculated as the number of hits retrieved divided by alleles at testing (150,000 in 500 ng) and multiplied by 100.
Ethics and animal approval statement
For usage of human CD34+ HSPC from healthy donors, informed written consent was obtained in accordance with the Declaration of Helsinki and ethics approval from the Great Ormond Street Hospital for Children NHS Foundation Trust and the Institute of Child Health Research Ethics (08/H0713/87).
RESULTS
Design and testing of a CRISPR/Cas9 platform to edit the IL7RA locus
To mediate the site-specific integration of a corrective IL7RA cDNA in the IL7RA genomic locus (Fig. 1A), we designed different gRNAs targeting the first exon of the IL7RA gene and tested their activity in Jurkat cells. Allelic disruption (indels' formation) rates of up to 84% were obtained with gRNA4 (Supplementary Fig. S1), which were utilized for all further experiments. Delivery of the gRNA precomplexed to a Cas9 protein as RNP to PB-derived CD34+ HSPCs from healthy donors yielded up to 81% of indel formation (Fig. 1B). To deliver the donor DNA molecule, which serves as a template for HDR-mediated repair, we created an AAV6 vector that contains a GFP reporter cassette flanked by sequences homologous to the IL7RA genomic regions surrounding the gRNA cut site (Fig. 1C). By RNP electroporation followed by transduction with the AAV6 donor vector, we observed targeted integration of the PGK-GFP reporter cassette in up to 52% of HSPCs, with no significant decrease in cell viability compared with mock-targeted HSPCs (Fig. 1B–D).

Development of a stem cell gene editing platform for IL7RA-SCID.
To evaluate the capacity of edited HSPCs to differentiate into multiple lineages, cells were subjected to in vitro CFU assays. Edited cells retained their clonogenic potential and produced similar frequencies of erythroid and myeloid cells without lineage skewing compared with controls, while yielding similar number of colonies of mock-treated cells (Fig. 1E).
We next assessed the ability of our gene editing protocol to restore functional IL7R expression when a corrective cDNA is knocked-in at the selected site. The PGK-GFP reporter cassette in the AAV6 donor vector was therefore replaced with a codon divergent and promoterless IL7RA cDNA (coIL7RA) followed by a synthetic Bovine Growth Hormone polyadenylation (pA) signal (Fig. 1F). Codon optimization introduced changes in the Cas9 target sequence of the coIL7RA cDNA to prevent Cas9 from recutting the integrated cassette. To quantify the level of IL7R protein expressed from the coIL7RA cDNA knocked-in in frame with IL7RA promoter and enhancer, we developed an in vitro model of IL7R deficiency using a human B-lymphocyte DG-75 cell line that expresses IL7R in normal conditions. To generate IL7RA knockout (KO) cell clones, we designed five different gRNAs targeting IL7RA exons 2–5 to introduce mutations that would abrogate the IL7RA open reading frame (Fig. 1G).
The RNP complexes containing gRNA5–9 were nucleofected in DG-75 cells and the efficacy of gene editing assessed by a T7 Endonuclease 1 (T7E1) assay and TIDE analysis, showing up to 37% of cutting frequency when gRNA6 was employed, which in turn led to a significant reduction of IL7R expression as detected by flow cytometry (Fig. 1H–J). Clonal populations of IL7RA KO cells were manually selected from the edited cell bulk and assessed for IL7R deficiency by immunoblotting; clone #65 showed complete abrogation of IL7R expression (Fig. 1K), further confirmed by IL7RA open reading frame disruption downstream of the gRNA6 cut site (Supplementary Fig. 1D) and was thus utilized as an IL7R deficiency cell model. Electroporation of clone #65 with the Cas9:gRNA4 RNP complex followed by transduction at increasing doses of the AAV6 donor template carrying the coIL7RA-pA cassette (Fig. 1F) led to the successful insertion of the therapeutic cDNA at the IL7RA locus by HDR at a frequency plateauing 40% of the cells, when a MOI of 50,000 was used (Fig. 1L).
This corresponded to a proportional restoration of IL7R protein expression (38% of the protein level detected in wild-type DG-75 cells; Fig. 1M), suggesting that our strategy has the potential to fully rescue physiological IL7R expression in the target cell upon KI of the corrective cDNA.
Modeling and correcting IL7R-SCID in T-lymphocytes by multiplexed gene editing
Expression of appropriate levels of IL7R in target cells is critical to ensure therapeutic effectiveness. However, to evaluate this acquiring a considerable number of T cells or HSPCs from the PB or the bone marrow of IL7RA-SCID patients would be required, which poses challenges due to the very rare nature of the disease, the early age of IL7R-SCID infants, and the invasiveness of the procedure. To overcome this challenge, we devised a HDR multiplexing platform to mimic both IL7RA KO and coIL7RA cDNA monoallelic and biallelic KI in primary hematopoietic cells derived from healthy donors. The IL7RA KO multiplexing strategy entails the use of two distinct AAV6 donor vectors carrying either an elongation factor 1α short (EFS) promoter-driven GFP or mCherry reporter cassette. The integration of these cassettes into the IL7RA exon 1 at the gRNA4 cut site through HDR simultaneously abrogates endogenous IL7RA transcription (and thus protein expression) and expresses reporter genes permitting the selection of GFP+/mCherry+ double-positive cells that underwent biallelic gene KO (Fig. 2A, top panel).
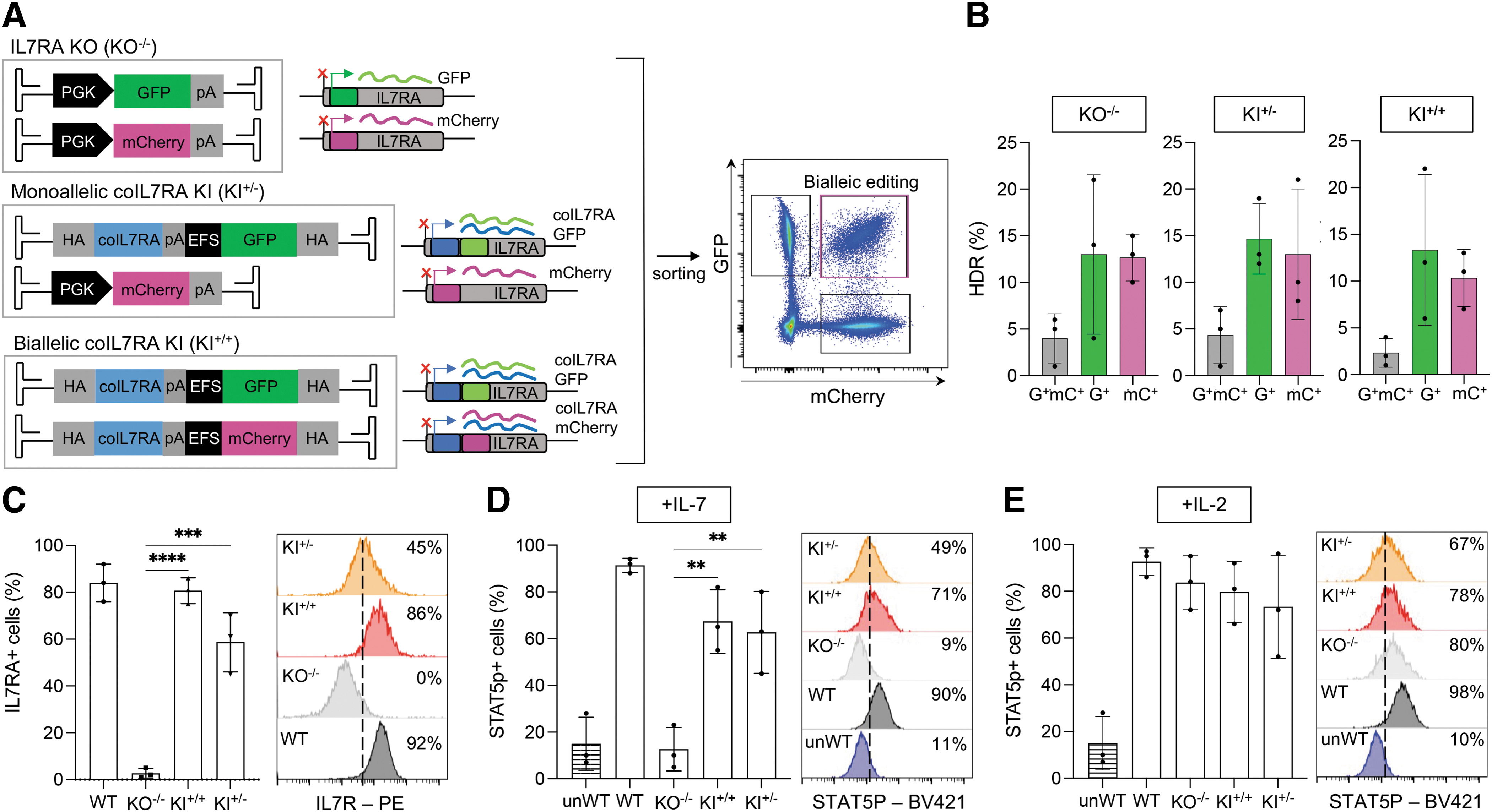
IL7R-SCID disease modeling and correction in primary T cells.
A similar strategy allows selection of cells that bear biallelic KI of the corrective transgene, by utilizing AAV6 donor vectors that carry the GFP or mCherry reporter gene preceded by the coIL7RA cDNA; the simultaneous integration of these cassettes in the IL7RA locus will again abrogate endogenous IL7RA transcription while expressing the therapeutic cDNA together with the respective reporter gene (Fig. 2A, bottom panel). As monoallelic correction of the IL7RA locus is in principle sufficient for the disease treatment due to the functional immune systems observed in heterozygous parents of IL7R-SCID patients, 8 we also paired one KO with one KI AAV6 donor vector carrying two different reporter genes, to mimic KO of the endogenous IL7RA gene in both alleles and KI of the corrective cassette in one allele (Fig. 2A, middle panel).
T cells isolated from the PB of three different healthy donors were electroporated with the Cas9:gRNA4 RNP complex and transduced with the combination of AAV donor vectors required to achieve either complete IL7RA KO (KO−/−), monoallelic coIL7RA cDNA KI coupled with monoallelic IL7RA KO (KI+/−), or biallelic coIL7RA cDNA KI (KI+/+). HDR-mediated transgene integration was successfully achieved as measured by flow cytometry, with an average of 2.5–4.5% of biallelic and 10–15% of monoallelic integration per each reporter gene, respectively, totaling an average of 25–34% of editing frequency of the IL7RA locus across conditions (Fig. 2B). GFP+/mCherry+ double-positive cells were FACS sorted to obtain pure populations of cells with the desired genotype; flow cytometry gating showed sufficient separation between populations for effective purification, which was further confirmed by ddPCR analysis on individually sorted populations showing correct integration of each reporter cassette in ∼50% of the alleles in GFP+/mCherry+ double-positive cells (Supplementary Fig. S2).
To evaluate the restoration of IL7R expression in T cells in which the coIL7RA cDNA was precisely inserted in frame with the translational start site of the endogenous IL7RA gene, cells were fixed, permeabilized, and stained for IL7R intra- and extracellular expression. A proportional analysis of IL7R expression compared with WT cells is necessary as IL7R is usually stochastically expressed in only a minority of cells even in healthy donor samples, reflecting its status as an altruistically regulated receptor. 12 Flow cytometry analysis showed a significantly increased proportion of IL7R-positive cells in WT and edited cells relative to KO−/− cells, with biallelic coIL7RA cDNA KI (KI+/+) cells recapitulating WT expression and monoallelic KI+/− cells exhibiting an intermediate proportion of IL7R-positive cells (Fig. 2C).
Comparison of IL7R mean fluorescence intensity (MFI) in edited cells showed that biallelic KI of the coIL7RA cassette led to the expression of the protein at levels comparable to those observed in WT cells, while monoallelic KI+/− cells expressed it at reduced levels, likely reflecting the presence of only one functioning copy of the corrective cDNA per cell (Fig. 2C). Data were confirmed by IL7R immunoblotting (Supplementary Fig. 2G, H).
To assess whether integration of the coIL7RA cDNA restores IL7R functionality, we evaluated the levels of phosphorylated STAT5 protein (pSTAT5) in KI+/−, KI+/+, KO, and WT T cells upon IL-7 stimulation. Indeed, binding of IL-7 to its receptor activates a downstream signaling cascade, which results in the phosphorylation of STAT5 and activation of an IL7R-dependent transcriptional profile. 28 Intracellular pSTAT5 staining in IL-7-stimulated cells indicated a trend toward restoration of the proportion of pSTAT5+ T cells in both biallelic and monoallelic KI cells, whereas KO cells showed no IL7R functionality, mimicking pSTAT5 levels detected in unstimulated IL-7 WT cells (Fig. 2D). While pSTAT5 is a common signaling effect of many cytokines acting through the common gamma chain receptor in T-lymphocytes, 29 we confirmed that signaling disruption in IL7RA KO and its restoration in KI cells is strictly dependent on the presence of IL7R, as no significant difference in the frequency of pSTAT5+ cells was detected in all experimental groups when cells were stimulated with IL-2 (Fig. 2E).
KI of a corrective IL7RA cDNA in HSPCs restores T cell development
Having demonstrated the successful development of a gene editing strategy to model IL7R deficiency and its correction by HDR-mediated gene KI in primary human T cells, we next assessed if the same approach would be effective when applied to HSPCs, the target cell type for the definitive treatment of IL7R-SCID. Because IL7R expression is neither detectable nor required in these cells, therapeutic success of the gene editing platform applied to HSPCs would be demonstrated by differentiation into mature TCRαβ+CD3+ cells, which is abrogated in the SCID phenotype. Furthermore, during the differentiation process from HSPCs to T cells, tight regulation of IL7R expression is necessary to faithfully recapitulate its physiological role during T cell development and homeostasis. To evaluate these aspects, we took advantage of the 3D ATO system, which models the in vitro differentiation and maturation of T cells from HSPCs, allowing us to elucidate the precise stages in which T cell developmental blocks occur in SCIDs and if gene editing is able to ameliorate such developmental obstructions. 30,31
To this aim, HSPCs isolated from the PB of three different healthy donors were electroporated with the Cas9:gRNA4 RNP complex and transduced with the combination of AAV donor vectors required to obtain IL7RA KO−/−, KI+/− and KI+/+. HDR-mediated transgene integration was successfully achieved as measured by flow cytometry, with a frequency of 0.4–4% of biallelic and 6–21% of monoallelic integration per each reporter gene, respectively, totaling an average of 28.4% editing frequency of the IL7RA locus across conditions (Fig. 3A). As performed for T cells, GFP+/mCherry+ HSPCs were FACS sorted and the purity of the populations were confirmed by ddPCR, showing correct integration of each reporter cassette in ∼50% of the alleles (Supplementary Fig. S2F). HSPCs retained their clonogenic potential and produced similar frequencies of erythroid and myeloid colonies without lineage skewing compared to controls, although we could observe a slight reduction in colony number formed by KO and KI HSPCs transduced with two AAV6 donor vectors (Supplementary Fig. S3B).
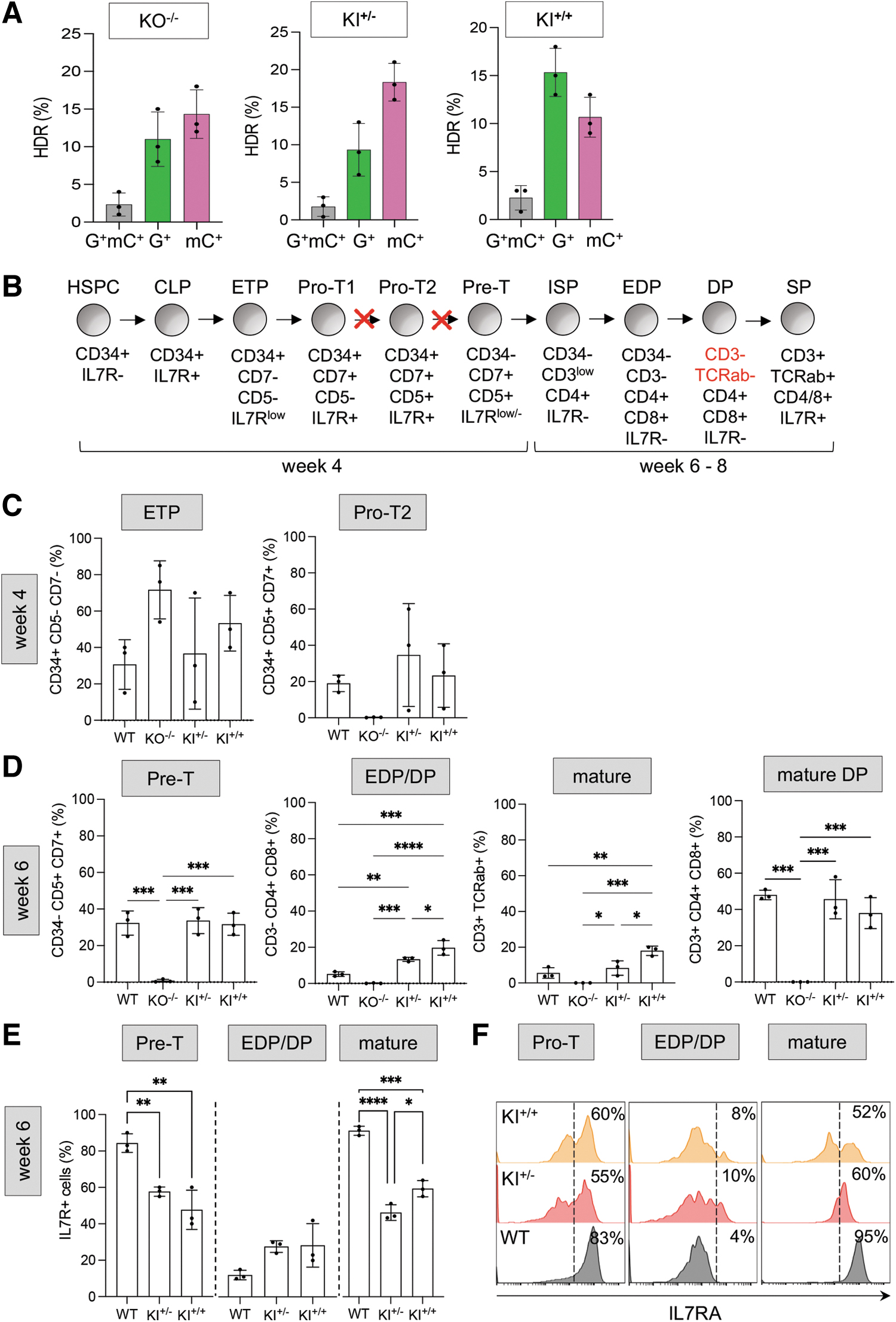
Modeling IL7R-SCID correction in HSPCs.
ATOs seeded with edited and FACS sorted cell populations, alongside unmanipulated wild-type HSPCs, were cultured for 6–8 weeks and characterized by flow cytometric analysis of thymopoietic surface markers at different time points to track T cell development (Fig. 3B and Supplementary Fig. S3A, C). By week 4 postseeding, ATOs were able to recapitulate the two early phenotypic stages of thymic T cell progenitors: multipotent CD34+CD7−CD5− early thymic progenitors (ETP) and developmentally downstream CD34+CD7+CD5+ pro-T2 progenitors (Fig. 3C), identified using a CD5 and CD7-based classification scheme. 32 At this stage, WT, KI+/−, KI+/+, and KO-/- groups showed similar proportions of ETPs; however, pro-T2 progenitors were already absent from the KO−/− sample, reflecting IL7R dependence of pro-T2 cell survival during thymopoiesis. 32 Contrary to this, the frequency of both precursor types in KI+/− and KI+/+ resembled those observed in the WT samples, suggesting that KI of the corrective coIL7RA cDNA does rescue pro-T2 cell survival and the block in T cell development observed in patients.
ATOs were further assessed for complete T cell maturation at week 6 and week 8 of culture. Similar trends as observed at earlier time points were borne out in the pre-T (CD34– CD7+ CD5+), early double-positive (EDP; CD34–CD3–CD4+CD8+), and mature T cell (CD34– CD3+ TCRαβ+) populations at 6 and 8 weeks of culture, when the frequencies of these populations in the KI samples again recapitulated the behavior of WT samples, whereas KO−/− ones kept exhibiting curtailed thymocyte proportions (Fig. 3D and Supplementary Fig. S3B). These results indicate that IL7RA KO recapitulates the expected pro-T2 block in thymopoiesis and that coIL7R KI rescues all thymocyte populations downstream of the initial IL7R dependence at the pro-T2 stage. As previously discussed, IL7R expression derived from a gene addition approach must be tightly controlled throughout T cell development to avoid the occurrence of adverse events and dysregulated immunity.
When looking at IL7R protein abundance in the samples seeded on ATOs, we observed correct restoration of protein expression mediated by the coIL7RA cDNA knocked-in in the IL7RA locus in both KI experimental groups, with biallelic KI achieving an average of 65% of WT IL7R expression in both pre-T and mature T cells. Most importantly, IL7R was not constitutively expressed throughout cell differentiation, but correctly shut down at the immature EDP stage and subsequently reactivated at the mature DP cell stage (Fig. 3E, F). Overall, these data show that HDR-mediated KI of a coIL7RA cDNA in frame with its endogenous regulatory regions mediates sufficient protein expression to relieve the block in T cell development observed in IL7R-deficient patients and does so in a physiologically regulated fashion.
Off-target analysis confirms the safety of the gene editing platform
One of the potential concerns associated with gene editing is the introduction of unwanted genetic modifications at off-target sites and the occurrence of gross chromosomal rearrangements, posing a huge risk for clinical therapeutic applications involving engineered nucleases. 33 To determine the specificity of our gRNA targeting IL7RA, we delivered the CRISPR/gRNA4 RNP to HEK293T cells and assessed the presence of indels at nonspecific sites using GUIDE-seq (genome-wide, unbiased identification of DSBs enabled by sequencing) an unbiased genome-wide analysis tool, 25 as well as the bioinformatic prediction tool COSMID. 23 Analysis of the GUIDE-seq sequencing data retrieved 10 different off-target sites at extremely low read numbers; 8 sites were mapping to gene introns of which only 1 gene (ZFHX3) has been linked to oncogenesis (Fig. 4A and Supplementary Table S1). However, targeted deep sequencing of these sites in CRISPR-treated and mock-treated HSPCs from three different healthy donors demonstrated no significant gene disruption at the genomic locations tested (Fig. 4B).
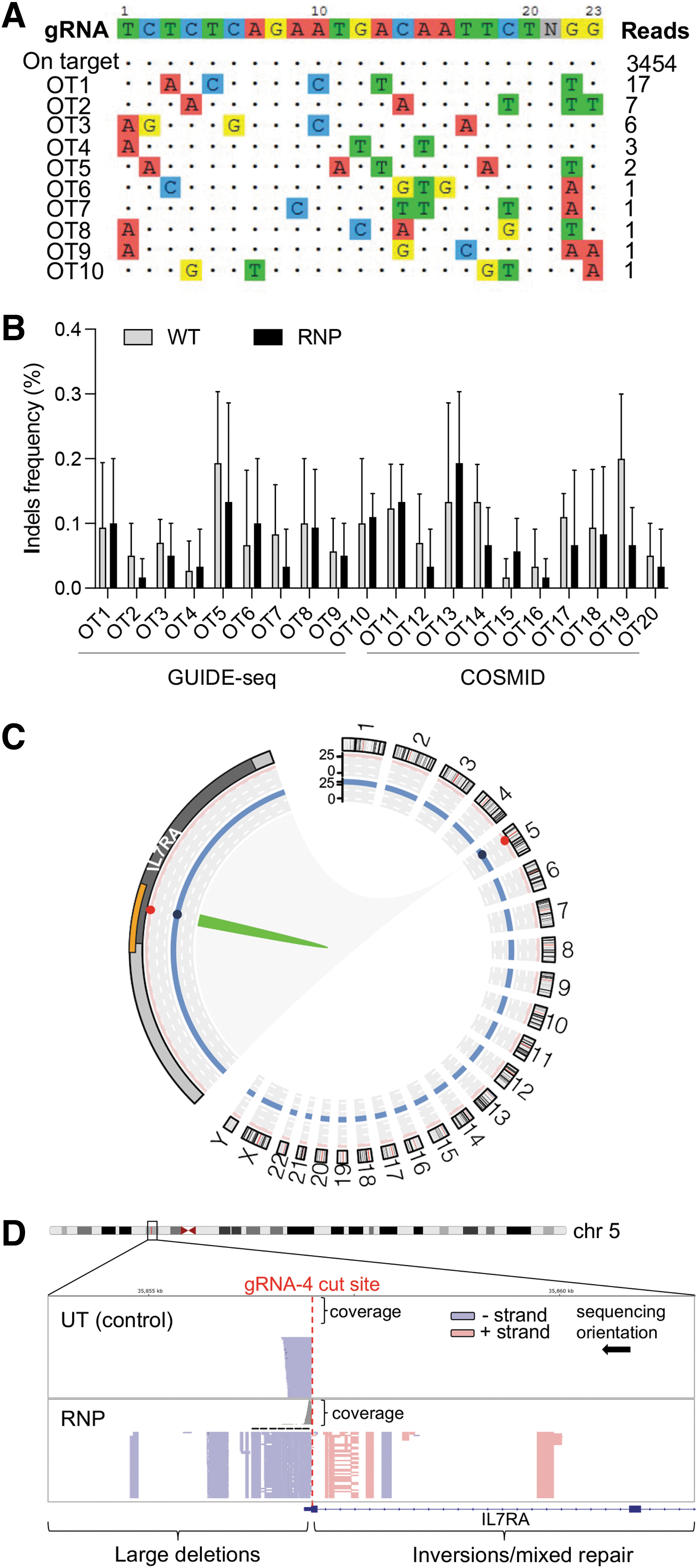
Genotoxicity analysis in edited HSPCs.
In parallel, off-target activity at the top 10 genomic sites showing high homology with IL7RA gRNA target sequence (up to five mismatches) by COSMID was measured by targeted deep sequencing in treated and mock-treated HSPCs. Of these sites, 7 were in gene introns, with no oncogenes or protooncogenes involved (Supplementary Table S1). At a read depth of 50,000 × we could not detect any genetic disruption at a statistically significant frequency compared with mock-treated controls in all genomic sites tested (Fig. 4B). Lastly, we sought to identify the potential occurrence of large chromosomal aberrations, such as translocations, insertions, or deletions, at off- and on-target gRNA cutting sites by using the CAST-seq technology. 27 For this analysis, we edited HSPCs with a Cas9:gRNA4 RNP complex containing a wild-type recombinant Cas9 protein, to be able to evaluate the presence of remote and rare rearrangement events that would not be detectable when using a high-fidelity Cas9.
Despite this, the CAST-seq analysis confirmed the safety of this gene editing platform, with only rearrangements at the IL7RA on-target locus being visible because of non-HDR-mediated DNA repair at the CRISPR/Cas9 cut site (Fig. 4C, D), as previously shown for other loci. 27,34 These rearrangements (large deletions and inversion) were clustered in a 8.1 kb region spanning the IL7RA gene transcriptional start site and did not affect other genes apart from the on-target site; moreover, they were detected at very low frequency in the analyzed HSPC populations (0.07% in HSPC donor 1 and 0.32% in HSPC donor 2).
DISCUSSION
In this study, we report the successful application of a CRISPR/Cas9-based gene editing platform to model and correct a rare and devastating primary immunodeficiency, IL7R-SCID, through editing of the IL7RA locus in primary human T cells and HSPCs. There has been a limited number of studies showing treatment options for this disease and this work represents the first evidence of therapeutically relevant genetic correction in primary cells using a gene editing approach.
IL7R-SCID represents an ideal candidate for gene editing applications for many reasons: (1) previous studies demonstrated the efficacy of genetically corrected HSPCs in curing different forms of SCID 35 –37 ; (2) safety issues raised by preclinical studies using viral gene therapy approaches for IL7R SCID suggest the need of a physiologically regulated and restricted IL7R expression during T cell development, 11,16 which can be achieved through in situ gene editing; (3) the tremendous selective advantage that functionally corrected cells have compared with mutated cells in a SCID setting 35,38 can compensate for the relative low HDR correction rates that can be obtained in more primitive, long-term repopulating HSCs 21 and could ideally allow infusion of autologous gene-edited cells without pharmacological conditioning; (4) the absence of a definitive treatment available to all the patients urges the need for new, innovative treatments to be established.
Our strategy restores IL7R expression by integrating a codon-optimized IL7RA cDNA next to IL7RA endogenous translational start site on chromosome 5, utilizing a highly specific gRNA cutting at the beginning of the IL7RA first exon and an AAV6 donor vector carrying a promoterless therapeutic cassette. This approach mediates the targeted integration of one/two correct copy/ies of the coIL7RA cDNA per cell, maintaining a normal gene copy number and reducing the risks of genotoxicity caused by the semi-random integration of the cDNA in the genome of patient's cells as in viral gene therapy approaches. 39 Moreover, controlled integration of the corrective cDNA in the IL7RA locus allows the transcriptional regulation by endogenous regulatory elements, ensuring physiological IL7RA expression upon integration of the cassette. Lastly, our platform can treat all disease-causing mutations with one set of reagents thus representing a universal platform that could be applied to all IL7R-SCID patients.
The gene editing platform devised by us performed efficiently when tested in an IL7RA KO B cell line, in T cells and in healthy human HSPCs, with high levels of targeted integration of the cassette in the desired locus and no major disruption in cell viability and HSPC's colony-forming and differentiation capacity in vitro. However, the limited access to precious SCID patient cells limits our ability to evaluate whether expected thresholds for clinical efficacy are met in terms of editing efficiency, protein expression and function, as well as toxicity. To overcome this barrier, we have developed a multiplexing gene editing system that can efficiently mimic gene knockout and KI by taking advantage of the simultaneous HDR-mediated integration of two expression cassettes with distinct reporter genes that allow FACS sorting of pure cell populations with the desired genotype. While modeling strategies similar to ours have been already devised, 40,41 in this study, we show its versatility in modeling both monoallelic and biallelic gene correction in two human primary cell types, T cells and HSPCs.
By applying our multiplexed targeted integration approach to T cells, we were able to completely abrogate IL7R expression and downstream signaling, thus obtaining a pure population of IL7R KO cells that recapitulate the IL7R-SCID defect. On the other hand, efficient monoallelic and biallelic KI of the corrective coIL7RA cDNA restored near physiological levels of protein expression, especially in the biallelic configuration, validating the use of our gene editing platform for therapeutic purposes. Assaying IL7R expression and signaling pathways is important not only for evaluating whether cDNA integration restores physiological IL7R expression and functionality, but also whether these lie below thresholds associated with oncogenic transformation risk. 11,38 We indeed confirmed that KI+/+ and KI+/− cells showed significantly greater IL7R expression and pSTAT5 levels than KO T cells, while no IL7R overexpression or pSTAT5 overphosphorylation were observed in these samples relative to WT T cells.
Moreover, correction of the pSTAT5 signaling cascade by gene KI was IL7R specific, as no changes in the signaling pathways were observed across experimental conditions when T cells were stimulated with IL-2 instead of IL-7.
Because a potential gene editing application for IL7R-SCID would require the manipulation of HSPCs, we implemented the same disease modeling strategy to those cells, to assess (1) achievable rates of gene correction; (2) restoration of HSPCs' capability to give rise to mature T cells once corrected; and (3) restoration of physiological protein expression in mature T cells. Evidence from bone marrow transplantation in IL7R-SCID patients and preclinical gene therapy studies have shown that as little as 10% of corrected cells engrafting the host bone marrow is sufficient to functionally cure T-B+ SCIDs due to the selective advantage of corrected cells over SCID cells. 8,35 While the efficiency of biallelic KI of the corrective cassette in HSPCs failed to meet the 10% threshold, total editing efficiencies, including monoallelic KI cells frequently exceeded 25%. As such, total KI efficiency achieved is a clinically relevant figure for IL7R-SCID as, being an autosomal recessive disorder, even monoallelic correction alone is sufficient to enable normal hematopoiesis in carrier parents. 8
Previous work by our group 43 and others 20 with similar gene editing strategies applied to HSPCs reached KI rates sensibly higher than those achieved in our modeling system. We believe that this discrepancy could be due to the increased viral burden of transduction with two AAV viruses required for multiplexed HDR, which likely artificially suppressed edited cell survival as a result of cell toxicity. 44 The AAV-mediated toxicity is reflected by the decrease in the CFU ability in edited versus nonedited cells, particularly observed in KI and KO samples transduced with two AAV vectors at the same time. Moreover, the increased length of the HDR donor molecules due to the inclusion of an EFS-GFP/mCherry cassette also likely reduced KI efficiency compared with smaller clinical constructs lacking reporter genes. 19,45 This is backed up by results obtained in previous works using multiplexed HDR platforms, 40,41 and additionally by the increased KI rates (up to 58%) achieved in this study when only one AAV donor vector carrying the shorter coIL7RA cDNA cassette was employed to edit HSPCs.
Overall, the evidence suggests that by further counteracting gene editing- and AAV-related toxicities (e.g., by reducing the MOI used for AAV transduction, by inhibiting DNA Damage Repair pathways 45 or by optimizing HSPC culture conditions 21 ), we could further improve our overall KI rates in vitro to ensure engraftment of corrected IL7R-SCID HSPCs at therapeutically beneficial levels in vivo.
As the hallmark of IL7R-SCID is the immunodeficiency caused by an IL7R-dependent T-lymphocyte developmental block, we sought to understand if our gene editing platform could restore HSPCs' ability to give rise to mature T cells in vitro, by taking advantage of the ATO system. The IL7RA-KO ATO model successfully recapitulated the block in thymopoiesis and absence of mature T cells seen in IL7R-SCID patients. The observation of diminished cell populations arising from differentiating KO−/− HSPCs from the pro-T stage onward agrees with previous works showing that abrogation of IL7R function causes blocks at the pro-T2/DN2 stage of thymopoiesis. 6 On the other hand, coIL7RA-KI ATOs showed the successful overcoming of the abovementioned developmental block when IL7R expression is reinstated, with cell frequencies detected at every developmental stage tested being comparable to WT ATOs in both mono- and biallelic KI conditions.
Importantly, we demonstrated that IL7R expression is tightly regulated during lymphopoiesis in the ATO system, with correct downregulation of the protein expression at the intermediate EDP/DP stages followed by its reintegration in mature TCR+ TCRαβ+ cells, highlighting the indisputable advantage of our gene editing strategy that relies on endogenous regulatory regions for gene expression. Although there appears to be a small fraction of EDP/DP cells still expressing IL7R in mono- and biallelic KI samples, we do not expect any unintended consequences as it has been shown that the presence of signaling inhibitor SOCS1 in EDP/DP cells effectively blocks downstream signaling from IL7R introduced exogenously. 46 When checking the frequency of cells expressing IL7R upon gene editing and T cell differentiation, we observed an average of 65% IL7R+ cells (as a fraction of WT IL7R+ cells) in both monoallelic and biallelic KI samples at both the Pre-T and mature T cell stages analyzed, exhibiting an MFI equivalent to that detected in T cells derived from WT HSPCs.
The fact that we do not see an increase in IL7R expression going from monoallelic to biallelic cDNA KI equaling WT levels suggests that: (1) there may be a maximum threshold of protein expression that can be achieved with our system; (2) the codon-optimized IL7RA cDNA used in our system may lead to suboptimal IL7R protein expression; or (3) the populations analyzed by FACS using established panels of markers may contain heterogeneous population of T cells at different developmental stages and thus with different IL7R-expressing levels when pushed to differentiate in the ATO system. The final, intriguing possibility is that intronless coIL7RA cDNA used in our KI strategy lacks important regulatory elements that limit transgene expression. 47 Intronic IL7R-SCID-associated mutations have been reported as causing splicing aberrations, 13 but they may also have unexplored regulatory significance. Future work could include bioinformatic assessment of putative regulatory regions in IL7RA introns, generation of cDNA constructs incorporating critical sequences, and reassessment of KI cell function and HDR rates.
Nevertheless, as heterozygous parents of SCID patients have functional immune systems, an IL7R protein expression of at least 50% of WT levels is thought be sufficient to rescue the disease phenotype, thus validating the efficacy of our gene editing platform in its current design. Moreover, the strong selective advantage of IL7R-corrected cells over deficient ones in a SCID setting may lead to eradication of the disease even when infusing HSPCs with much lower correction rates and protein expression levels than those achieved here. 35 Lastly, the lack of an increase in protein expression in biallelic versus monoallelic KI cells and relative to WT cells does imply that coIL7RA integration does not predispose T cells to overexpression of IL7R, an important safety aspect given that IL7R overexpression is associated with clonal dominance and thymocyte lymphoma. 16
While a limitation of this study is that it mostly relies on an in vitro assessment of the platform, it suggests that our gene editing strategy successfully rescues thymopoiesis through IL7R expression reconstitution when applied to primary human HSPCs and has the potential to perform similarly when assessed in clinical trials using patient-derived cells. This could be tested at the preclinical level in vivo by performing xenotransplantation experiments of edited IL7R-SCID HSPCs in immunodeficient NSG-SGM3 mice, which allow robust human hematopoietic reconstitution with a superior T cell output compared with other NSG strains. 49
CRISPR/Cas9-mediated targeted integration through HDR provides a potentially safer strategy of gene correction than viral gene therapy, as it mitigates the risk of oncogenic transformation and genotoxicity associated with the use of viral vectors. 39 While the safety of engineered nucleases for therapeutic purposes is still under investigation, with indels' generation at off-target sites being a potential threat to their safe clinical application, 33 we demonstrated the absence of nonspecific targeting of our gRNA and of major chromosomal rearrangements upon DNA cutting, using biased and unbiased detection tools. Indeed, deep-sequencing of 20 putative off-target sites in HSPCs detected through either GUIDE-seq or a sequence similarity prediction algorithm returned no significant modifications at those sites when our optimized CRISPR/Cas9 reagents were used. This finding was further confirmed by a CAST-seq analysis in HSPCs, which showed no evident large chromosomal rearrangements between on- and putative off-target sites.
While we could detect a cluster of chromosomal rearrangements at the IL7RA locus caused by CRISPR/Cas9 editing, 27 these aberrations were found to be quite rare in the analyzed HSPC population and affected no other genes, except for IL7RA proximal promoter region. In this scenario, we expect gene editing-related deletions happening at this locus in patient-derived IL7R-SCID HSPCs to have no genotoxic outcome, as the gene is already not functional in these patients, and loss of IL7R has not been linked to tumorigenesis.
CONCLUSIONS
Our study provides proof of concept of the efficacy and potential safety of a CRISPR-based gene editing approach to treat IL7R-SCID in primary human T cells and HSPCs using an in vitro disease modeling system. This strategy could provide a valuable therapeutic alternative for all patients affected by this disease and could enable the translation of such technology to a much wider range of HSC blood disorders.
Footnotes
ACKNOWLEDGMENTS
The authors thank Dr. Ayad Eddaoudi (Flow Cytometry Core Facility, University College London) for assistance with flow cytometry and cell sorting.
AUTHORs' CONTRIBUTIONS
R.R., Z.S., M.R., F.Z., Y.H., N.W., A.N., and G.T. performed experiments and analyzed data; A.C., G.T., and A.J.T. contributed to the study design; A.C. initiated the study, designed experiments, illustrated data, and wrote the article, with inputs from all the authors.
AUTHOR DISCLOSURE
A.J.T. is on the Scientific Advisory Board of Orchard Therapeutics and Rocket Pharmaceuticals. The other authors declare no competing interests.
FUNDING INFORMATION
R.R., Y.H., G.T., N.W., and A.J.T. were supported by the Wellcome Trust (104807/Z/14/Z). A.C., and M.R. were supported by the Sparks-GOSH Children's Charity Grant (V4318); A.C. and A.N. were supported by the Sparks-GOSH Children's Charity Grant (V4522); A.C., Z.S., and F.Z. were supported by the UKRI MRC Molecular and Cellular Biology Board Grant (MR/W001314/1); A.C., and G.T. were also supported by the University College London Therapeutic Acceleration Support fund (MRCCIC7554230 and ICH/GOSH BRC Award 175540) and the NIHR Biomedical Research Center at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. For the purpose of Open Access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.