
Abstract
Fabry disease (FD) is an inherited lysosomal storage disease caused by deficiency of α-galactosidase A (α-Gal A), an enzyme that hydrolyzes glycosphingolipids in lysosome. Accumulation of glycosphingolipids, mainly globotriaosylceramide (Gb3) in tissues, induces cellular dysfunction leading to multi-organ disorder. Gene therapy is a promising strategy that can overcome these problems, and virus vectors such as adeno-associated virus (AAV) have been used for study on gene therapy. We used human Gb3 synthetase-transgenic (TgG3S)/α-Gal A knockout (GLAko) mice. TgG3S/GLAko mice have elevated Gb3 accumulation in the major organs compared with GLAko mice, which have been widely used as a model for FD. At the age of 6 weeks, male TgG3S/GLAko were injected with 2 × 1012 vector genome AAV9 vectors containing human α-Gal A cDNA. Eight weeks after intravenous injection of AAV, α-Gal A enzymatic activity was elevated in the plasma, heart, and liver of TgG3S/GLAko mice to levels corresponding to 224%, 293%, and 105% of wild-type, respectively. Gb3 amount 8 weeks after AAV injection in the heart and liver of this group was successfully reduced to levels corresponding to 16% and 3% of untreated TgG3S/GLAko mice. Although the brain and kidney of AAV9-treated TgG3S/GLAko mice showed no significant increases in α-Gal A activity, Gb3 amount was smaller than untreated littermates (48% and 44%, respectively). In this study, systemic AAV administration did not show significant extension of the lifespan of TgG3S/GLAko mice compared with the untreated littermates. The timing of AAV injection, capsid choice, administration route, and injection volume may be important to achieve sufficient expression of α-Gal A in the whole body for the amelioration of lifespan.
Introduction
Fabry disease (FD) is an X-linked lysosomal storage disease caused by mutations in GLA (GenBank: NM_000169.3), the gene encoding α-galactosidase A (α-Gal A). 1 –3 α-Gal A hydrolyzes glycosphingolipids, such as globotriaosylceramide (Gb3), in lysosomes. Deficiency of α-Gal A induces the progressive accumulation of incompletely catabolized glycosphingolipids within lysosomes, mainly in vascular endothelial cells, smooth muscle cells, and renal cells (podocytes, tubular cells, glomerular endothelial, mesangial, and interstitial cells). 3,4
Lysosomal accumulation of Gb3 causes cellular dysfunction, which contributes to progressive multiorgan disorder, including cerebrovascular disease, cardiac disease, and chronic renal dysfunction. 5 The standard treatment for FD is enzyme replacement therapy (ERT) with biweekly intravenous injection of human recombinant α-Gal A, which is taken up in the tissues via mannose-6-phosphate (M6P) receptors in the cell membrane. 6
Although ERT is effective to reduce the progression of organ damage, especially is started early in the course of the disease, 7 this approach has a number of limitations, including short half-lives, production of antibodies to the recombinant enzyme, and inability to cross the blood-brain barrier (BBB). Recently, an oral pharmacological chaperone therapy has been approved for FD. 8 However, this treatment is only available for patients with amenable GLA gene mutations. Therefore, gene therapy will be a favorable approach for FD in terms of systemic, long-standing, therapeutic efficacy with a single application.
Adeno-associated viral (AAV) vectors have been shown to be safe and effective to deliver genes for experimental and clinical purposes. 9 –12 AAV is a non-pathogenic single-stranded DNA virus that can infect both non-dividing and dividing cells. To date, AAV1, AAV2, AAV6, AAV8, and AAV9 serotypes have been used as vectors to transduce human GLA gene into GLAko mice, where they were shown to restore α-Gal A activity and decrease Gb3 accumulation in the organs (Supplementary Table S1). 13 –17
However, GLAko mice do not show symptomatic phenotypes with normal lifespan, which have been widely used in the earlier mentioned studies of FD. 18 Taguchi et al. demonstrated that Gb3 accumulation in GLAko mice was not sufficient to have phenotypic manifestations, and generated a symptomatic model mouse line (called TgG3S/GLAko mice) by cross-breeding GLAko mice and human Gb3 synthetase (G3S)-transgenic (TgG3S) mice with higher levels of Gb3 accumulation in major organs than GLAko mice, resulting in albuminemia followed by polyuria and neurological symptoms, including tremor, slow movements, and gait disturbance. 19,20 This TgG3S/GLAko mice was reported to die by 36 weeks of age, with a median lifespan 27 weeks in male and female. 19
Most AAV serotypes transduce genes predominantly into neurons, although the mechanisms of gene transduction have not yet been elucidated. 21,22 AAVrh8, 9, and rh10 have been reported to transduce genes throughout the central nervous system when administered intravenously. 23,24 We previously reported that AAV9 carrying human GLA expressed in major organs including brain, heart, liver, and kidney using GLAko mice. In this study, we further investigated the effect of AAV-based gene therapy using the clinically relevant TgG3S/GLAko mice.
We administered 2 × 1012 viral genomes (vg) of AAV9 carrying human GLA into adult TgG3S/GLAko mice at the age of 6 weeks, and killed the animals at the age of 14 weeks for quantification of α-Gal A activity, viral genome copy numbers (VGCNs), Gb3 content, and histological analysis for Gb3. In heart and liver, α-Gal A activity of the AAV-treated group was significantly higher than the phosphate-buffed saline (PBS)-treated group, and the Gb3 content of the AAV-treated group was significantly lower than the PBS-treated group, although the α-Gal A activity and Gb3 content showed the same tendency in the brain and kidney that were not significant.
Histological analysis showed reduction of Gb3 accumulation of brain, heart, liver, and kidney after AAV treatment. In this study, AAV treatment did not show extension of the lifespan of TgG3S/GLAko mice. We need further research for the best gene therapy to cure FD effectively.
Materials and Methods
Preparation of AAV-hGLA viral vectors
Human GLA was subcloned in the pUC57 plasmid (Azenta Life Sciences, Chelmsford, MA), and then subcloned into the pAAV-MCS plasmid (Agilent Technologies, Santa Clara, CA) by digestion with NotI. The expression cassette contained human GLA driven by the cytomegalovirus (CMV) immediate–early promoter, followed by the simian virus 40 (SV40) polyadenylation (pA) signal sequence.
Recombinant AAV9 vectors carrying human α-Gal A cDNA (AAV9-hGLA) were produced by triple plasmid transfection of human embryonic kidney (HEK) 293 cells, as described previously. 11 Briefly, 60% HEK293 cells (Agilent Technologies) were incubated in large culture vessels and cotransfected with the pAAV-CMV-GLA plasmid, adenoviral helper plasmid pHelper (Agilent Technologies), and helper plasmid containing AAV2 Rep and AAV9 Cap genes. Crude viral lysates were purified using an AAV purification kit (Takara Bio, Shiga, Japan). Viral titers were determined by quantitative PCR of the SV40 pA sequence (forward, 5′-CAAATAAAGCAATAGCATCACAAA-3′; reverse, 5′-ATGAGTTTGGACAAACCACAAC-3′).
Animals
All experiments were performed in full compliance with the guidelines of the Institutional Animal Care and Use Committee of Jichi Medical University. TgG3S/GLAko mice were generated by cross-breeding homozygous female B6;129-Glatm1Kul/J (GLAko) mice (#3535; Jackson Laboratory, Bar Harbor, ME) with male TgG3S mice (nbio232; Laboratory Animal Resource Bank, National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, Japan) and maintained in the Center for Experimental Medicine at Jichi Medical University.
In GLAko mice, a neomycin cassette replaced exon 3 and intron 3 of the galactosidase α (Gla) gene. TgG3S mice overexpressing human Gb3 in major organs with the cytomegalovirus immediate early enhancer-chicken β-actin hybrid (CAG) promoter were generated by Shiozuka et al. 25 The G3S transgene was kept in a single allele in the TgG3S and TgG3S/GLAko mice. Groups of 6-week-old male TgG3S/GLAko mice were injected with 2 × 1012 viral genomes (vg) of AAV9-hGLA, or PBS via the retroorbital vein (in a volume of 200 μL).
These AAV- or PBS-treated groups were maintained for 8 weeks (n = 8 in each group). Age- and sex-matched C57BL/6J (wild-type [WT] mice), TgG3S mice, and GLAko mice were used as controls (n = 7 in each group). Blood samples were collected in heparinized capillary tubes from the mandibular vein just before and at 3 and 8 weeks after injection. Plasma was isolated by centrifugation at 1,000 g for 15 min. At the endpoint, organs (brain, heart, liver, and kidney) were harvested and snap-frozen in liquid nitrogen for preservation at −80°C. Experimental timeline is summarized in Fig. 1. In addition, Kaplan–Meier survival curves were constructed to measure lifespans of the mice in each group (n = 5 in each group). Observations were censored at the age of 18 months, except for animals that died at an earlier time point.
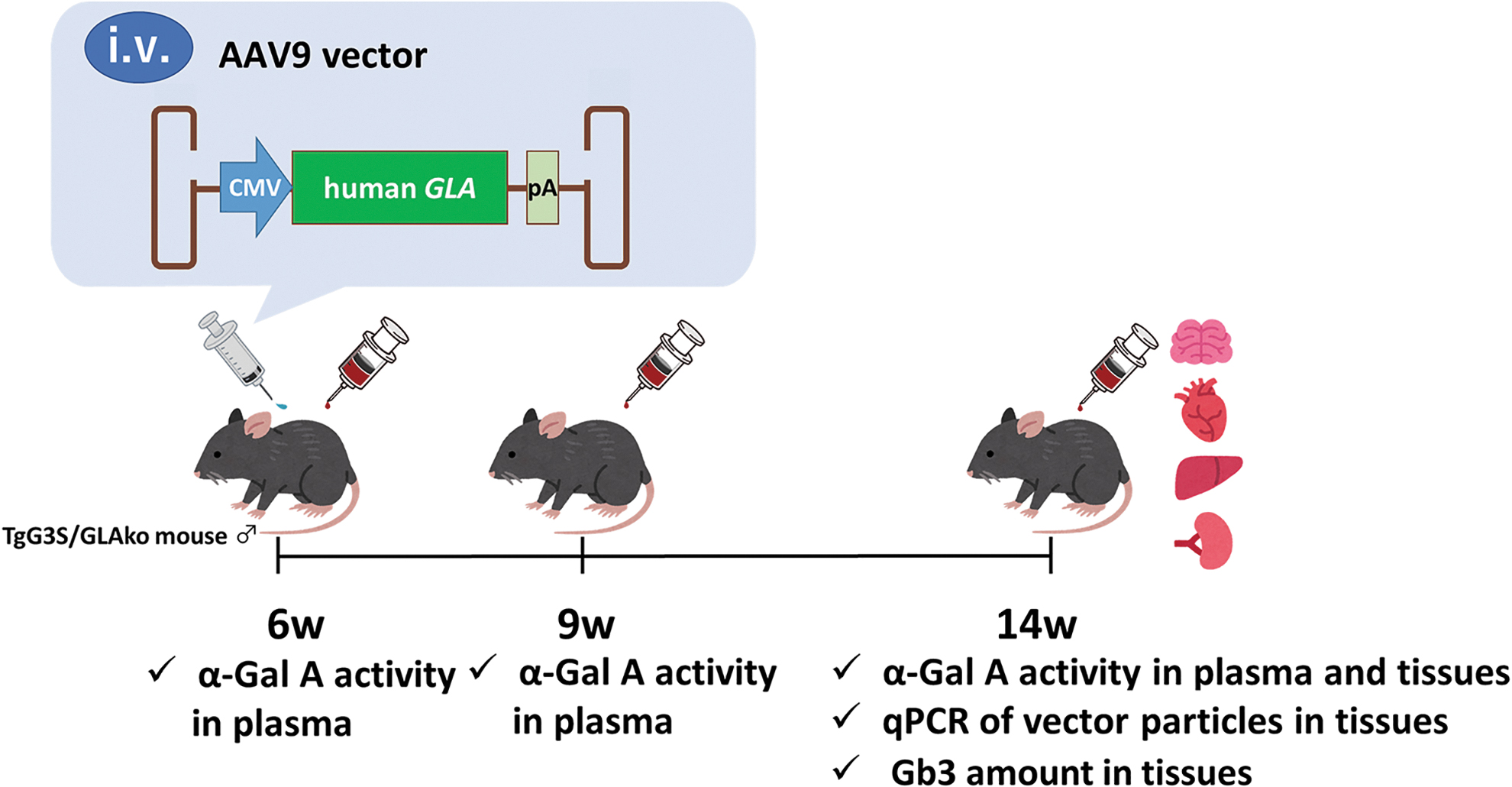
Experimental timeline of AAV-mediated treatment of human globotriaocylseramide (Gb3) synthetase-transgenic (TgG3S)/α-Gal A knockout (GLAko) mice. Male TgG3S/GLAko mice were intravenously injected with AAV9 vectors carrying human GLA expressing cassette or PBS at the age of 6 weeks. Age- and sex-matched WT controls, TgG3S mice, GLAko mice, and TgG3S/GLAko mice were also analyzed (n = 7–8 in each group). The plasma in each group was collected just before and at 3 and 8 weeks after injection for α-Gal A activity assay. At 8 weeks after injection, brain, heart, liver, and kidney were harvested for α-Gal A activity assay, quantitative PCR, and Gb3 measurement. AAV, adeno-associated virus; CMV, cytomegalovirus immediate-early promoter; Gb3, globotriaosylceramide; α-Gal A, α-galactosidase A; GLA, gene encoding α-galactosidase A; GLAko, α-galactosidase A knockout; i.v., intravenous injection; pA, simian virus 40 polyadenylation; PBS, phosphate-buffed saline; qPCR, quantitative PCR; TgG3S, human globotriaocylseramide synthetase-transgenic; WT, wild-type.
α-Gal A enzymatic activity assay
α-Gal A enzymatic activity was examined with an Alpha Galactosidase Activity Assay Kit (Biovision, Waltham, MA) according to the manufacturer's protocol. Tissue samples were homogenized with the buffer and centrifuged at 12,000 g at 4°C for 10 min. The supernatant was incubated at 37°C for 2 hours with α-Gal A substrate, which releases a fluorophore by enzymatic reaction. The fluorescence was quantified at 360 nm excitation and 445 nm estimation wavelengths using Fluoroskan Ascent FL (Thermo Fisher Scientific, Waltham, MA).
Specific sample α-Gal A activity was expressed as nanomoles of hydrolyzed substrate per hour and milligram of total protein, which were estimated from a standard curve (fluorescence vs. known concentrations of 4-methylumbelliferone). Total protein concentration was determined using a BCA Protein Assay Kit (Takara Bio). The plasma samples were incubated as well, and α-Gal A activity was expressed as nanomoles of hydrolyzed substrate per hour and milliliter of plasma (n = 7–8 in each group).
Biochemical examination of plasma
Plasma was collected from each group 8 weeks after injection (n = 2–4). The plasma levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and lactate dehydrogenase (LDH) were measured by Oriental Yeast Co., Ltd. (Tokyo, Japan).
Genomic DNA purification and viral genome quantification
Genomic DNA was extracted from brain, heart, liver, and kidney using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). The DNA samples were analyzed by quantitative real-time PCR, using SYBR green master mix and a StepOne real-time PCR system (Thermo Fisher Scientific), according to the manufacturer's protocol. Primers specific for the SV 40 pA signal sequence were used to detect AAV viral genomes (described above). Genomic DNA was quantified in parallel samples using hypoxanthine phosphoribosyltransferase 1 (HPRT1)-specific primers: forward, 5′-AGGCCCAGCTCTACAAGTTT-3′; reverse, 5′-TGATTGCGCATTCCCACAAA-3′. Negative control (no template control) was included in each assessment. Results are presented as viral genomes per diploid genome (vg/dg) (n = 7–8 in each group).
Gb3 extraction and quantification
Gb3 (mixture of various Gb3 isoforms) and Gb3 (C17:0) were purchased from Matreya, LLC (Pleasant Gap, PA), and used as standards in the experiments. Gb3 were extracted from brain, heart, liver, and kidney according to the protocol described by Sueoka et al. 26 The organ tissues were homogenized in methanol and diluted with chloroform/methanol (1/2, v/v).
Methanol containing Gb3 (C17:0) as internal standard was added. After centrifugal separation, the supernatant was applied to a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay system. Gb3 isoforms were separated by high-performance LC and detected by LCMS-8060 triple quadrupole mass spectrometer (Shimadzu, Kyoto, Japan). The settings of LC/MS-MS system were the same as that described by Kodama et al. 27 The calculations for quantification of Gb3 isoforms were performed using LabSolutions (Shimadzu). The total Gb3 contents were calculated from the sums of those of Gb3 isoforms (n = 3 in each group).
Gb3 staining with anti-Gb3 monoclonal antibody
Ten-micrometer-thick frozen tissue sections of the brain, heart, liver, and kidney that were taken from the PBS- or AAV9-treated TgG3S/GLAko mice were incubated with primary antibody against Gb3 (diluted at 1:1 for brain, heart, and liver; diluted at 1:4 for kidney, #A2506; Tokyo Chemical Industry, Tokyo, Japan) overnight at 4°C followed by incubation with a biotinylated secondary antibody at room temperature for 10 min (#414321; Nichirei Biosciences, Tokyo, Japan). Then, the sections were incubated with 3,3′-diaminobenzidine for 20 min.
Measurement of neutralizing antibody against AAV9 vectors
Plasma was collected from the AAV9-treated TgG3S/GLAko mice just before and 8 weeks after injection and tested for neutralizing antibody against AAV9 (n = 3 in each group). Assays for detection of neutralizing antibodies against AAV9 were performed as previously reported. 28 Briefly, the day before transduction, 5 × 104 of HEK293-derived 2V6.11 cells in 100 μL of DMEM/HAM F12 supplemented with 10% fetal bovine serum (FBS) were seeded into the wells of 96-well culture plates.
Ponasterone A was added to induce the E4 gene expression of 2V6.11 cells. On the day of transduction, 10 μL of test serum and 10 μL of AAV9-CMV-LacZ prepared to 1,000 vector genome copies per cell were mixed, incubated at 37°C for 1 h, and added to the wells of 96-well culture plates. Sucrose solution was added to the culture medium at a final concentration of 125 mM. After 48 h of incubation, the culture medium was removed, β-galactosidase activity was quantified using ortho-nitrophenyl-β-D-galactopyranoside (Invitrogen, Carlsbad, CA) as substrate, and color change at 405 nm was quantified using a microplate reader SpectraMax 190 (Molecular Devices, San Jose, CA).
If the β-galactosidase activity of the test serum inhibited more than 50% of the control fetal bovine serum, it was considered positive for neutralizing capacity. The inhibitory titer of the test serum was expressed as the highest final dilution of the serum in the culture medium that showed inhibitory activity.
Statistical analysis
The results are presented as the mean ± standard error of the mean (SEM). Statistical comparisons between groups were performed by one-way analysis of variance with Tukey's post hoc test for multiple comparisons. p < 0.05 was taken to indicate statistical significance. Comparison of survival distributions in Kaplan–Meier survival curves was performed using the log-rank test. p < 0.05 was taken to indicate statistical significance. All statistical analyses were performed using GraphPad Prism software version 9.1.1 (GraphPad Software, San Diego, CA).
Results
Systemic injection of AAV9-hGLA increased plasma α-Gal A activity in AAV9-treated TgG3S/GLAko mice
To evaluate the chronological change of plasma α-Gal A activity, the plasma α-Gal A activity in each group was assessed just before and at 3 and 8 weeks after injection (Fig. 2). Just before injection, the α-Gal A activities in TgG3S mice, GLAko mice, PBS- or AAV9-injected TgG3S/GLAko mice were 139.5%, 27.8%, 49.0%, and 12.8% of WT controls, respectively. Three weeks after injection of AAV9, TgG3S/GLAko mice showed an increase in α-Gal A activity comparable to that of WT mice (TgG3S: 98.6%, GLAko: 18.7%, PBS-injected TgG3S/GLAko: 34.5%, AAV9-injected TgG3S/GLAko: 91.1% to WT controls; PBS-TgG3S/GLAko vs. WT, p < 0.05; AAV9-TgG3S/GLAko vs. WT, not significant).

Plasma α-Gal A activity in TgG3S/GLAko mice, WT controls, TgG3S mice, and GLAko mice following injection of AAV9 or PBS. The plasma α-Gal A activity was analyzed by fluorescence-enzymatic activity assay in the TgG3S/GLAko mice receiving 2 × 1012 viral genomes (vg) of AAV9 or PBS alone just before and at 3 and 8 weeks after injection (n = 8 in each group). Age- and sex-matched WT controls, TgG3S mice, and GLAko mice were also analyzed (n = 7 in each group).
At 8 weeks after injection of AAV9, the α-Gal A activity in TgG3S/GLAko group was 223.7% of that in WT mice (TgG3S: 103.6%, GLAko: 33.6%, PBS-injected TgG3S/GLAko: 40.4%, AAV9-injected TgG3S/GLAko: 223.7% to WT controls; PBS-TgG3S/GLAko vs. WT, not significant; AAV9-TgG3S/GLAko vs. WT, not significant). α-Gal A activities of the other groups did not change at each time point. AAV9-hGLA administration increased plasma α-Gal A activities to the same level as the WT controls at 3 weeks after injection and to about twice as high as WT controls at 8 weeks after injection.
Systemic injection of AAV9-hGLA increased α-Gal A activity, and decreased Gb3 content and its isoforms significantly in the heart and liver but not significantly in brain and kidney of TgG3S/GLAko mice
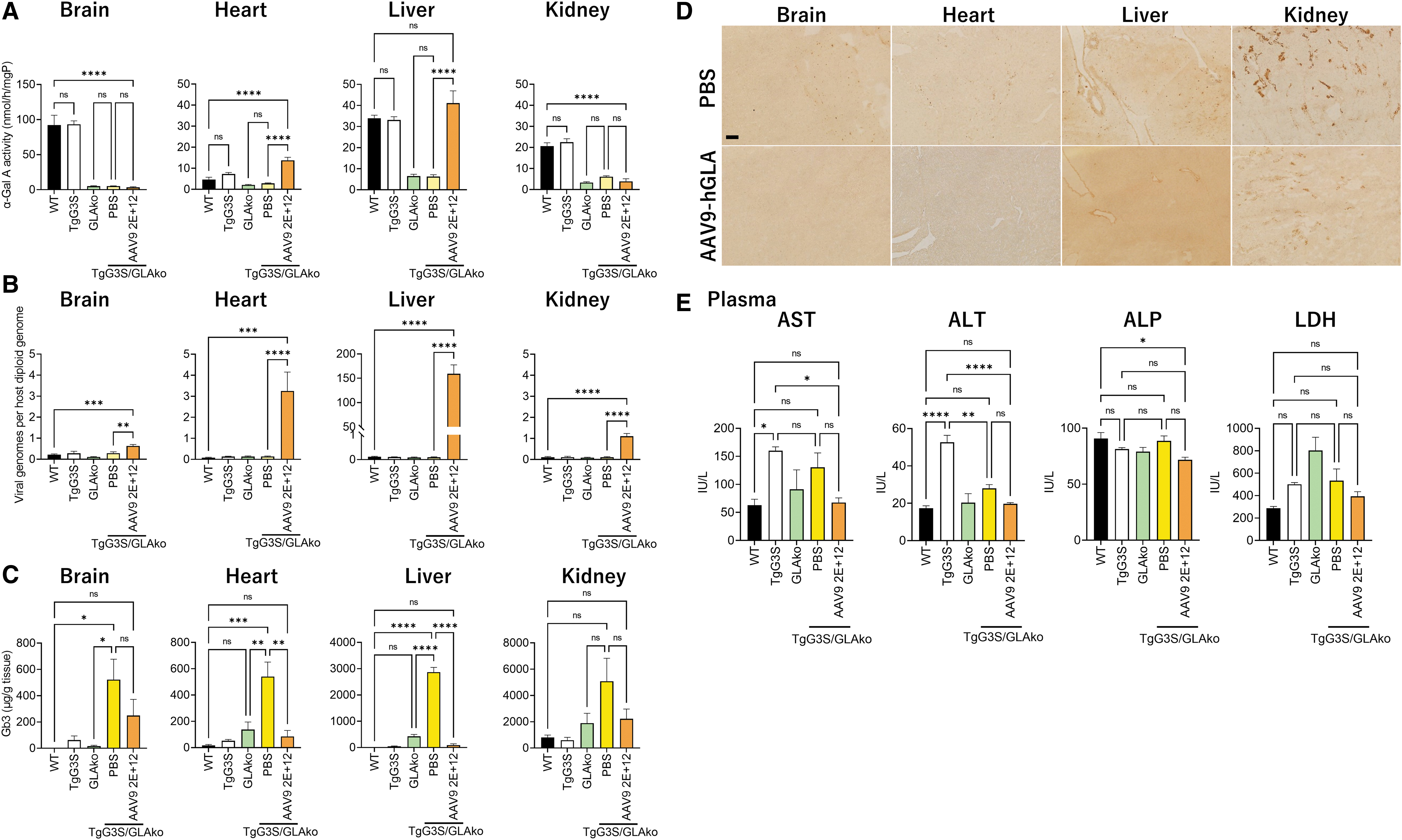
To evaluate the gene expression after AAV injection, α-Gal A activity, VGCNs per host diploid genome, and Gb3 amount in the brain, heart, liver, and kidney of each group were measured at 8 weeks after AAV9 injection (Tables 1 and 2 and Fig. 3). In the heart and liver of AAV9-treated TgG3S/GLAko mice, the VGCNs were significantly increased, and α-Gal A activities were the highest among the all groups with levels of 2.9-fold and 1.1-fold higher than in the WT controls, respectively. Gb3 amount in these two organs was successfully reduced in the AAV9-treated TgG3S/GLAko mice with the levels corresponding to 16% and 3% of PBS-treated littermates, respectively.
Quantification of α-Gal A activity
α-Gal A activity in the brain, heart, liver, and kidney of TgG3S/GLAko mice, WT mice, TgG3S mice, and GLAko mice after AAV9 injection
The α-Gal A activity is expressed means ± SEM. The unit: nmol/h/mg protein.
AAV, adeno-associated virus; GLAko, α-galactosidase A knockout; ND, not detected; PBS, phosphate-buffed saline; SEM, standard error of the mean; TgG3S, human globotriaocylseramide synthetase-transgenic; WT, wild-type
α-Gal A activity in the brain, heart, liver, and kidney of TgG3S/GLAko mice, WT mice, TgG3S mice, and GLAko mice after AAV9 injection
The α-Gal A activity is expressed means ± SEM. The unit: nmol/h/mg protein.
In the brain and kidney, there was no significant difference in the α-Gal A activity between PBS-treated TgG3S/GLAko mice and AAV9-treated TgG3S/GLAko mice, although the VGCNs of AAV9-treated TgG3S/GLAko mice were significantly increased. Notably, Gb3 content in the brain and kidney of AAV9-treated TgG3S/GLAko mice showed smaller tendency than PBS-treated littermates (48% and 44%, respectively; not significant compared with the WT controls).
There was no significant difference in α-Gal A activity in each organ between WT controls and TgG3S mice, or between GLAko mice and PBS-treated TgG3S/GLAko mice. Plasma levels of AST, ALT, ALP, and LDH at 8 weeks after injection showed lower tendency in AAV9-treated TgG3S/GLAko mice than in PBS-treated mice (reduction of 48% in AST, 29% in ALT, 19% in ALP, and 26% in LDH; not significant compared with the PBS-treated group), which means that the tissue damage of whole bodies including liver can be ameliorated by AAV9 administration.
Although the VGCNs of AAV9-treated TgG3S/GLAko mice were significantly increased in all the major organs, including brain, heart, liver, and kidney, significant increase of α-Gal A activity and significant decrease of Gb3 contents in the AAV9-treated TgG3S/GLAko mice were found in heart and liver, compared with the PBS-treated mice.
Neutralizing antibody did not preexist and increased after AAV9-hGLA injection
To evaluate the preexisting neutralizing antibody and generated antibody after injection, we measured the titers of neutralizing antibody against AAV9 before and at 8 weeks after injection. We did not detect neutralizing antibody before injection, which increased to 1:44,800 or 1:89,600 at 8 weeks after injection (Supplementary Table S2).
AAV9-hGLA did not extend lifespan of TgG3S/GLAko mice
To evaluate the lifespan extension effect of AAV-mediated gene therapy, mice were kept raised until 18 months. There was a significant difference in the lifespan among the PBS-treated and AAV-treated TgG3S/GLAko mice, WT mice, TgG3S mice, and GLAko mice (Fig. 4). TgG3S/GLAko mice showed shorter lifespan than WT controls. There was no change in lifespan between PBS-treated and AAV-treated TgG3S/GLAko mice; both of them showed a median survival time of 11 months, and they all died by 16 months. WT controls and GLAko mice were alive at 18 months.

Kaplan–Meier survival curves of TgG3S/GLAko mice, WT mice, and TgG3S mice and GLAko mice. The median survival time was 11 months in both AAV-treated TgG3S/GLAko mice and PBS-treated TgG3S/GLAko mice, which had shorter lifespan compared with WT controls, TgG3S mice, and GLAko mice (n = 5 in each group). ND, not detected.
Discussion
ERT and molecular chaperone therapy are effective and reasonable treatments for FD. However, there remain various problems with these treatments, including short half-lives, inability to cross the BBB, and production of neutralizing antibodies. Gene therapy is a potentially useful strategy to achieve a long-standing therapeutic effect in the whole body with a single systemic application.
Although the GLAko mouse has been widely used as a mouse model of FD, it lacks the FD phenotype and has a normal lifespan. The TgG3S/GLAko mouse with higher accumulation of Gb3 in the major organs would be a more appropriate model. In this study, we expressed human α-Gal A in TgG3S/GLAko mice by an intravenous injection of AAV9-hGLA vectors. After injection of AAV9-hGLA, we found the significant increase of transduced genes in brain, heart, liver, and kidney, although the significant increase of α-Gal A activity and reduction of Gb3 was seen in the heart and liver.
Taguchi et al. 19 showed that the amount of Gb3 accumulation in the brain of 25-week-old TgG3S/GLAko mice in which lipid inclusions were observed in neurons and cells around blood vessels, was 19-fold higher than that of GLAko mice. The low transduction efficiency of AAV9-hGLA in the TgG3S/GLAko mouse brain may have been due to the cell damage that had already occurred. Early administration of AAV, such as neonatal injection, may be effective related to higher permeability of BBB as Biferi et al. reported using an AAV9 vector in GLAko mice. 17
We chose AAV9 as it has tropism for nervous system and the ability to cross the BBB of nonhuman primates. 29 –31 The recently developed artificial AAV, AAV-PHP.B, which is BBB-permeable, may be a powerful tool to deliver transgenes to systemic organs including brain, although the AAV-PHP.B was proved to be BBB-impermeable in nonhuman primates.
Because the mechanisms by which AAV9 cross the BBB remain unelucidated, the randomization of outermost layer protein, capsid, has now been undergoing. 32 In 2022, artificially developed AAV, AAV-CAP.B, was reported to be BBB-permeable in marmosets. 33 This kind of capsids can be a choice for future studies in FD. Also, to increase the transduction efficiency of AAV into the central nervous system, administration route of AAVs can be reconsidered, including intrathecal, and the combination of systemic and intrathecal. 28,29
The efficacy of systemic AAV injection for target gene delivery to the kidney is controversial. Several reports showed sufficient α-Gal A expression in the kidney of GLAko mice following systemic injection of AAV-hGLA. 15 –17,34,35 However, other reports failed to achieve delivery of AAV vectors to the kidney by systemic injection 13,14,36,37 and suggested that direct injection into kidney was a favorable route. 36,37
The lower transduction efficacy in kidney would be due to the structure of the glomeruli, which are unable to filtrate molecules with molecular weights above 50 kDa, while most gene delivery vectors, including AAV, have masses in the MDa range. In addition, slit diaphragms formed by podocytes in the glomeruli are only 10 nm in diameter, which is smaller than the diameter of AAV particles. 37 Circulating α-Gal A in the plasma, which represents as a homodimer with 49 kDa subunits, might have helped to reduce Gb3 accumulation in the kidney. Taguchi et al. 19 reported that TgG3S/GLAko mice showed albuminuria and polyuria as a result of dysfunction of proximal tubules, distal convoluted tubules, and collecting ducts in which lipid inclusions with lamellar structures were detected by electron microscopy.
On the other hand, no structural abnormalities were observed in the Bowman's capsule and the level of creatine excreted into the urine was constant over time, indicating that glomerular filtration function in TgG3S/GLAko mice was normal throughout their lifespan.
In this study, we used only male mice. Because GLA gene is located on the X chromosome,
TgG3S/GLAko mice showed significantly shorter lifespan than GLAko mice, as reported previously. 19 Although the cause of early death in these transgenic mice is still unknown, the deficiency of α-Gal A in the kidney and brain may play a critical role similar to that in human FD patients, because there was no significant difference in lifespan between PBS-treated TgG3S/GLAko mice and AAV-treated TgG3S/GLAko mice in this study, which showed increased α-Gal A activity in the heart and liver.
Although AAV9-hGLA showed limited effects in this study, it was meaningful to assess the efficacy of AAV vector in a symptomatic FD model. Further investigations are required for the timing of AAV injection, capsid modification, administration route, and injection volume to achieve sufficient AAV-hGLA transduction in all organs.
Conclusions
We demonstrated α-Gal A expression in the plasma and organs of a TgG3S/GLAko mouse model, which is more similar to FD patients compared with the conventional mouse model, following a single intravenous injection of AAV9-hGLA. The transduction efficacy in the brain and kidney was low, whereas the heart, liver and plasma showed increased α-Gal A activity with higher levels than seen in WT mice. Reconsideration of the timing of AAV injection, capsid choice, administration route, and injection volume may be the key to achieve sufficient α-Gal A expression in all organs.
Footnotes
Acknowledgments
The authors thank Akane Hirasawa, Momoko Aoyama, and Miyuki Watanabe for their technical assistance. They also thank Dr. Atsumi Taguchi for her helpful advice. The image of ![]() was created by Takashi Mifune. The TgG3S mice (Nbio232) used in this study were provided by the Japanese Collection of Research Bioresources (JCRB) Laboratory Animal Resource Bank of the National Institutes of Biomedical Innovation, Health and Nutrition (NIBIOHN).
was created by Takashi Mifune. The TgG3S mice (Nbio232) used in this study were provided by the Japanese Collection of Research Bioresources (JCRB) Laboratory Animal Resource Bank of the National Institutes of Biomedical Innovation, Health and Nutrition (NIBIOHN).
Authors' Contributions
Y.H. and Y.Se. conceived and designed the study. Y.H.,
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by JMU Graduate Student Start-Up Award (to Y.H.), JSPS KAKENHI (grant number 22H03441), AMED (grant number JP21lm0203003 and JP22ym0126805), The Ichiro Kanehara Foundation, Taiju Life Social Welfare Foundation, and Japan Brain Foundation (to Y.S.).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.