
Abstract
The clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated (Cas) system is a powerful genomic DNA editing tool. The increased applications of gene editing tools, including the CRISPR-Cas system, have contributed to recent advances in biological fields, such as genetic disease therapy, disease-associated gene screening and detection, and cancer therapy. However, the major limiting factor for the wide application of gene editing tools is gene editing efficiency. This review summarizes the recent advances in factors affecting the gene editing efficiency of the CRISPR-Cas9 system and the CRISPR-Cas9 system optimization strategies. The homology-directed repair efficiency-related signal pathways and the form and delivery method of the CRISPR-Cas9 system are the major factors that influence the repair efficiency of gene editing tools. Based on these influencing factors, several strategies have been developed to improve the repair efficiency of gene editing tools. This review provides novel insights for improving the repair efficiency of the CRISPR-Cas9 gene editing system, which may enable the development and improvement of gene editing tools.
INTRODUCTION
In 2020, C
In brief, two NHEJ repair factors (XLF and XRCC4) capture the damaged terminals of broken double-stranded DNA (dsDNA) and combine them in a highly stable, but mobile complex. 4 The terminal linkages of DSB were then directly linked and repaired by DNA ligases in the NHEJ repair pathway. 5 Although the homology-directed repair (HDR) is a homology-mediated dsDNA repair without nucleotide deletions or additions at the broken DNA terminals, it can accurately repair DSBs. 6,7 In general, the terminals of DSB will be encapsulated by various HDR factors when DSB occurs in the S or G2 phase of the cell cycle, and the homologous sequences will be presented in the vicinity of the DSB, which will then invade the homologous dsDNA to form an exchange intermediate, such as the displacement loop (D-loop) structure. 8
Of interest is that the key step in the HDR is the BRCA2-mediated ligation of the RAD51 enzyme to the excised broken DNA. 9 A difference between NHEJ and HDR-mediated DNA repairs is that the HDR-mediated DNA repairs occur during the meiotic prophase or the S and G2 phases of the cell cycle, whereas the NHEJ produces DSBs in the absence of sister chromatid monomers. 10
The ability of CRISPR-Cas9 to induce and subsequently repair DSBs has been utilized to knock out or silence target genes. 11 Thus, the CRISPR-Cas system can also be used to knockout or repair mutated human genes, indicating its potential application in the treatment of genetic diseases. At present, the CRISPR-Cas9 gene editing system has been extensively studied and applied in different fields.
CRISPR-Cas 9 GENE EDITING TOOL
Bacteria and archaea can recognize and resist foreign invading viruses and plasmids through the CRISPR system. 2 In brief, the CRISPR array is transcribed into a long precursor CRISPR RNA (pre-crRNA). The transactivating crRNA (tracrRNA) base pairs with the repeat sequences in pre-crRNA, promoting RNase III-mediated cleavage and Cas9 binding. The processed crRNA:tracrRNA duplex then directs the bound Cas9 to DNA sequences matching the crRNA guide sequence (protospacer) and flanked by a PAM. 12 Subsequently, double-strand DNA begins unwinding and induces RNA-DNA heteroduplex formation. Finally, Cas9 cleaves the dsDNA and induces the formation of a blunt-ended DSB near the specific PAM sequences 3 (Fig. 1a).

Mechanisms of the CRISPR-CRISPR-associated protein 9 (Cas9), ZFN, and TALEN gene editing tools.
The CRISPR-Cas9 gene editing system, which has both advantages and drawbacks, can be applied to target and edit different targets with specifically designed guide RNAs (gRNAs). 13 These gRNAs can be easily generated in vitro via genetic transcription and modification. Cas9 can break the methylated DNA and introduce DSBs at multiple loci in human cells. 3 Compared with other gene editing systems, such as the classical zinc-finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) gene editing tools (Fig. 1b, c), the CRISPR-Cas9 system does not require complex protein engineering. In addition to multisite gene editing, the CRISPR-Cas9 system can activate or repress the expression of genes. 14 Only specific gRNAs for different target genes must be designed for the application of the CRISPR-Cas9 system. 15 However, the CRISPR-Cas9 gene editing system can exert off-target effects in different cells of the same species. 16
Recently, some researchers mitigated the off-target effects of the CRISPR-Cas system by modifying the gRNA structure and developing various Cas protein mutants. 17,18 Huang et al. used phage-assisted noncontinuous evolution and eVOLVER-supported phage-assisted continuous evolution to obtain a Cas9 variant, which targets most pyrimidine-rich PAM sequences in the human genome and exerts decreased off-target effects in human cell types with various N4CN PAM sequences. 19 Compared with unmodified Cas9, Chen et al. fused the Cas9 construct with PRDM9 (a chromatin remodeling factor), which increased the efficiency of HDR by threefold but did not increase the incidence of off-target gene editing. 20 The key components of Cas9 protein were redesigned according to the resolved CRISPR-Cas9 structural features described by the research group of Bravo to generate Cas9 mutants with low off-target effects and high cleavage efficiency. The off-target efficiency of these Cas9 mutants decreased by 500-fold compared with that of the wild-type Streptococcus pyogenes Cas9. 21
OTHER GENE EDITING TOOLS BASED ON THE CRISPR-Cas SYSTEM
In addition to the CRISPR-Cas9 gene editing system, several gene editing tools based on the CRISPR-Cas system, such as the CRISPR-Cas12 and CRISPR-Cas13 systems have been developed for a wide range of applications. In the CRISPR-Cas12 gene editing system, crRNA guides the Cas12 nuclease to the target DNA. Cas12 nuclease cleaves the target DNA near the distal PAM end of the interspace sequence to generate a cohesive terminus. The CRISPR-Cas12 gene editing system induces DSBs through a single RuvC-like nuclease functional domain but cleaves multiple crRNAs containing expression arrays through its RNase domain, enabling the simultaneous editing of multiple genes. 22 After the recognition of target sites and the activation of the nuclease activity, several Cas12 homologs indiscriminately cleave single-stranded DNA (ssDNA) and single-stranded RNA (ssRNA). This characteristic is widely used for nucleic acid detection. 23 The CRISPR-Cas13 system can target RNA 24 and does not permanently alter the genome.
Furthermore, the RNA editing effect of the CRISPR-Cas13 system can be adjusted in a dose-dependent manner and is reversible, which is a major advantage for the treatment of acquired metabolic diseases. 25 However, the CRISPR-Cas13 RNA gene editing system exhibits a general collateral activity in mammalian cells. 26 The CRISPR-Cas13 RNA gene editing system may randomly cleave nontarget ssRNAs in the presence of both crRNA and target RNA, which can lead to severe off-target effects 27 and major toxic effects in animal cells or individuals. 28
More than half of human genetic diseases are caused by point mutations. 29 Thus, the correction of mutated genes with gene editing tools is a good therapeutic strategy for genetic diseases. Various CRISPR-Cas9–based single-base gene editing systems have been developed, such as the adenine base editors (ABEs) and cytosine base editors (CBEs) (Fig. 2). The ABEs and CBEs can directly interact with genomic DNA or cellular RNA to produce precise point mutations without inducing DNA DSBs. 30,31 In addition, ABEs and CBEs do not require DNA donors as templates for cell-dependent HDR. 32 Thus, the target sequences of ABEs or CBEs are edited without the introduction of any small indels.
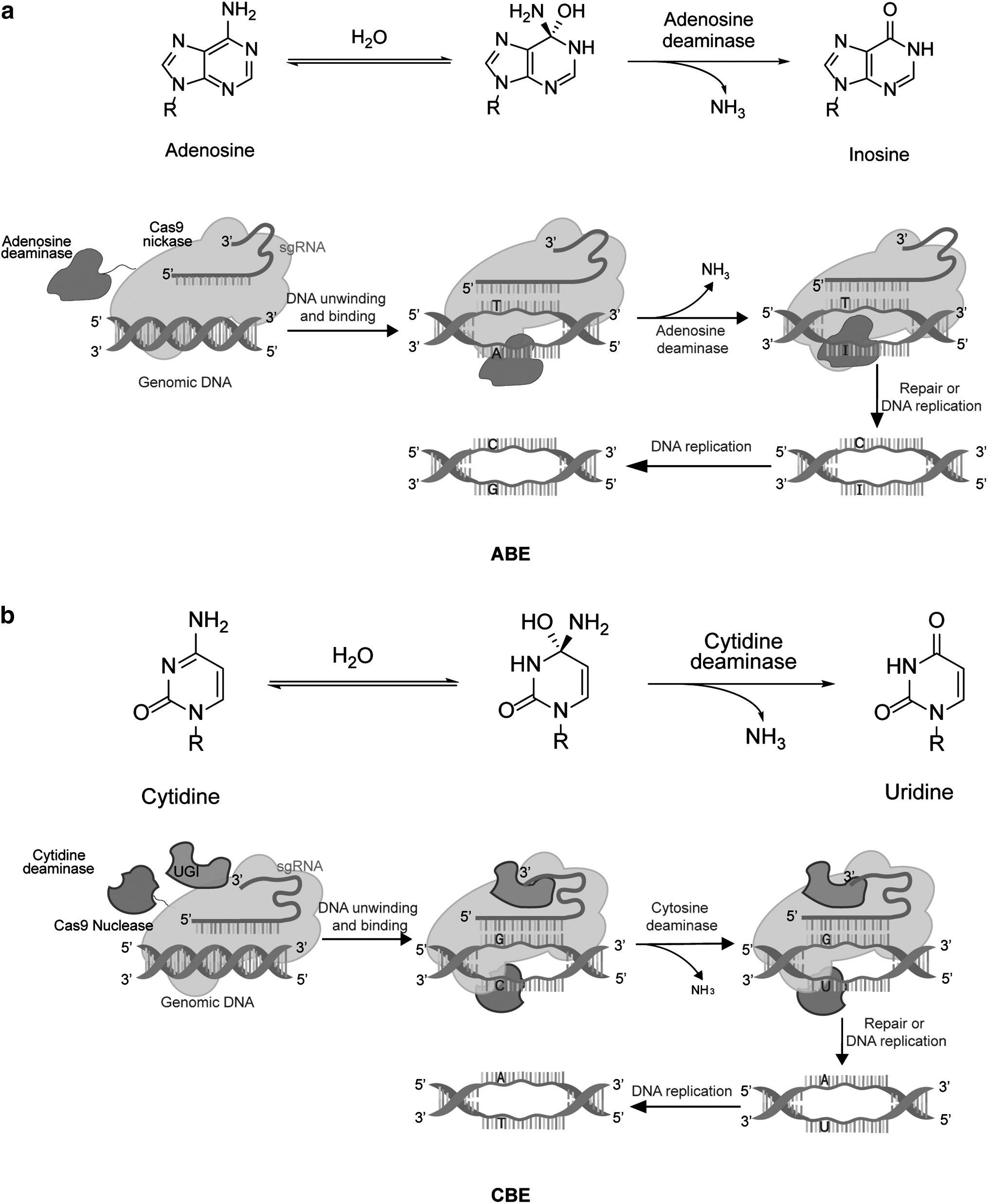
Schematic of the single-base editors (ABEs and CBEs).
The CRISPR-Cas gene editing technology has potential therapeutic applications in genetic diseases. The application of gene editing tools in genetic disease therapy is limited by their low repair efficiency. Precise targeting of target genes and mitigating off-target effects are critical for gene editing. 33 The ABEs and CBEs based on the CRISPR-Cas gene editing system were mainly used to correct single-base mutations in genetic diseases. 34 CRISPR-Cas9, Cas12, and Cas13 were widely used in genetic correction and detection. Significantly, Cas9 and Cas12 can target targeting DNA and cleave its 5′ PAM side sites, Cas12 also cleaves the 3′ PAM side sites of target DNA. 35 Both Cas9 and Cas12 can cleave dsDNA or ssDNA. Target sequences matched by Cas12 is usually smaller than 17 base pair (bp), it usually forms to an R-loop, which makes base-pair hybridization between the crRNA and the target DNA strand. 36
The Cas9 matches sgRNA sequences ranging in size between 20 and 24 nt. 36 The Cas13 can target ssRNA, and the size of matched sgRNA sequence to the Cas13 ranges from 22 to 28 nt. 37,38 The CRISPR-Cas13 system is mainly used for RNA gene editing. 39 The CRISPR-Cas12 gene editing system nonspecifically cleaves ssDNA and is usually applied for DNA detection. 40 The CRISPR-Cas9 gene editing tool is adapted to edit GC-rich PAMs, which are abundant in the mammalian genome 41 and can correct both single-base and multibase mutations. Thus, the CRISPR-Cas9 gene editing system is widely used in mammalian systems for genetic disease therapy. The gene repair efficiency and targeting accuracy are the major limiting factors of the CRISPR-Cas9 gene editing system. For example, the CRISPR-Cas9–mediated HDR efficiency is only ∼15% upon transfection of sgRNA, DNA templates, and CRISPR-Cas9 into HEK293T cells via electroporation in vitro. 42
This gene editing efficiency is not optimal for clinical genetic disease therapy. The research group of Liang et al. suggested that the CRISPR-Cas9 gene editing system exerts off-target effects in human trinuclear zygotes. 43 Unexpected mutations resulting from the off-target effects of the CRISPR-Cas9 system limit its applications in genetic disease treatment. 44 In the following sections, recent advances in factors affecting the gene editing efficiency of the CRISPR-Cas9 system and CRISPR-Cas9 system optimization strategies have been reviewed.
CORRELATION BETWEEN HDR EFFICIENCY AND GENE REPAIR EFFICIENCY OF THE CRISPR-Cas 9 SYSTEM
Cas9-generated DSB can be repaired through the NHEJ and HDR pathways. HDR is the most common pathway used by the CRISPR-Cas9 gene editing system to introduce specific changes to target DNA sequences. In cases where the endogenous or exogenous DNA repair template is homologous to a certain region of target DNA around the DSB sites (also known as the donor DNA), the DNA DSB is repaired by related repair factors through the HDR pathway. This enables the precise insertion, deletion, and alteration of bases during the exchange of new gene fragments with target DNA (Fig. 3). Previous studies have suggested that inefficient HDR during gene editing may be a limiting factor for CRISPR-Cas9 gene editing efficiency. 45 The enhancement of HDR efficiency can improve the gene editing efficiency of the CRISPR-Cas9 system. 46
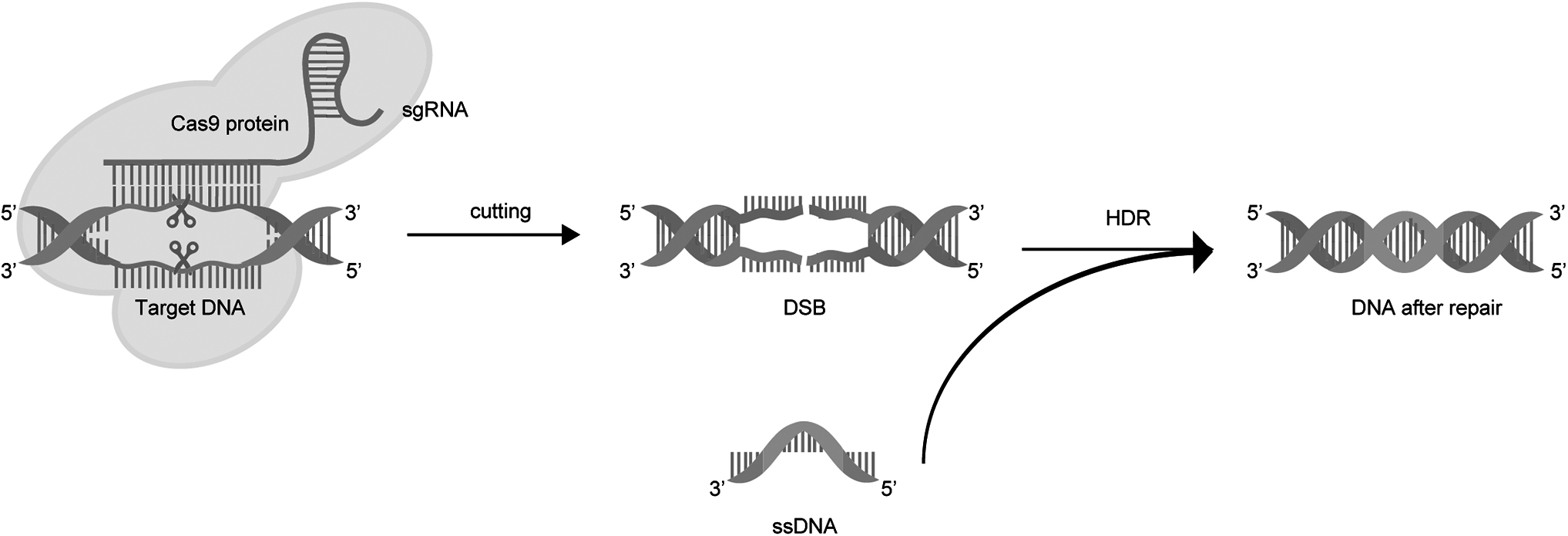
Schematic of HDR mechanism. The target DNA is cleaved by the CRISPR-CRISPR-associated (Cas) system to induce a DSB. After DSB formation at the target DNA, an exogenous ssDNA with a homologous arm is integrated into the target DNA via DNA repairing factors through the HDR pathway. HDR, homology-directed repair; ssDNA, single-stranded DNA.
HDR efficiency is limited by the NHEJ pathway in some cell types. The NHEJ pathway predominantly repairs DSBs and can suppress the HDR pathway through various mechanisms. 47 The cell cycle phase markedly affects the HDR pathway. The NHEJ repair pathway is highly active throughout the cell cycle, whereas the HDR pathway is restricted only to the S/G2 phases. DSB repair via the NHEJ repair pathway is faster than that via the HDR pathway. 48 The knockdown of Ku70/80 heterodimer or DNA ligase IV, which are two key factors involved in the NHEJ pathways, can improve HDR efficiency. 49,50 The small molecule compounds M3814 and trichostatin A (TSA), which are both NHEJ inhibitors, affect HDR efficiency to varying degrees. The application of M3814 markedly improved HDR efficiency after Cas9-mediated gene editing in different cell types.
Moreover, the combination of M3814 and TSA increased HDR efficiency by three times compared with the DMSO control. 45 Similarly, treatment with the DNA ligase IV inhibitor Scr7 increased HDR efficiency in cells. 49 In addition, the overexpression of RAD51/52/54, BRCA1/2, or HDR-related genes improved HDR efficiency. 51 The synchronization of cells at the G2/M cycle using CCND1 and nocodazole compounds increases HDR efficiency in induced pluripotent stem cells. 52 Therefore, the overexpression of HDR pathway-related genes, the knockout or suppression of NHEJ pathway-related genes, or the synchronization of cell cycle can improve HDR efficiency and the gene editing efficiency of the CRISPR-Cas9 system. 53
EFFECTS OF DIFFERENT DONOR FORMS ON GENE EDITING EFFICIENCY OF THE CRISPR-Cas 9 SYSTEM
Different DNA donor forms significantly affect the HDR and gene editing efficiencies. Common sources of donor DNA include plasmids, dsDNA, ssDNA, and chemically modified DNA. For example, one study reported that transfecting mammalian cells with asymmetric homology arm donor DNA, Cas9, and nickase mutants for gene editing improved HDR efficiency to ∼60%. 54 Compared with that of vectors harboring nonlinear donors, the HDR efficiency of ZFN-containing multiple tandem donor DNAs was ∼30-fold higher in 293T cells. 55 After the cleavage of the target gene by the CRISPR-Cas9 system, a double cleavage donor with two flank sgRNA and PAM sequences is released. The HDR efficiency of this double cleavage donor was two- to fivefold higher than that of the cyclic plasmid donor. 52 Linear donors or changing donor sources can improve HDR efficiency during cellular gene editing events.
In 2018, one study reported that the 5′ modification of the dsDNA donor preserved its monomeric conformation, maximally enhanced HDR efficiency, and facilitated target gene replacement and gene labeling. 56 The fusion of Cas9 to a single avidin domain to bind with a biotinylated donor DNA increased the HDR efficiency to up to 90% during gene editing. 57 The chemically modified donor DNA exhibits increased intracellular stability, HDR efficiency, and DSB repair ability. 58
The impact of the proximity of CRISPR components to donor DNA also influences HDR efficiency. Such as, it has been demonstrated that when Cas9 proteins genetically edit the target fragment to produce DSBs, the higher concentration of ssDNA was presented in the vicinity of the DNA incision, the higher the probability of HDR repair favors. 58,59 Besides, a Cas9–avidin–biotin ssDNA (CAB) system that consists of Cas9 fused with avidin, sgRNA and biotin-modified single-strand donor DNA (biotin-ssDNA) was developed by Ma et al. and applied to gene editing in mouse zygotes. By utilizing the high affinity between avidin and biotin, biotin-ssDNA would be enriched near the cleavage sites, which also improved the precision of HDR insertion by threefold compared with the traditional CRISPR-Cas9 system. 60
In addition, Lee et al. have demonstrated that the gRNA and donor DNA can be directly conjugated together into one molecule, and they also showed that the HDR efficiency of this gRNA–donor DNA conjugate improved three times than that of transfecting cells with cationic polymers, unconjugated gRNA and donor DNA. 58 Moreover, according to evidences from Farbiak et al., they showed that when the CRISPR-Cas9 system was used to edit the same gene site by liposomal encapsulation of three nucleic acids (Cas9 mRNA, sgRNA, ssDNA) in different weight ratios, with the percentage of sgRNA increasing, the HDR efficiency also improved. 61
The length of the homologous arm at both ends of donors and the donor insert segment sizes affect the efficiency of gene integration and editing. Yang et al. reported that HDR donors with a length of 90 nt exhibit excellent gene editing efficiency. 62 Jian-Ping Zhang et al. constructed donors with different homologous arm lengths for CRISPR-Cas9 gene editing events and reported that a linear donor is more efficient than a circular donor for HDR-mediated gene integration. The HDR efficiency was ∼6–10% for a linear donor whose left and right homologous arms had a length of 50 bp and increased by twofold when the length of the left and right homologous arms was increased from 50 to 100–150 bp. Additionally, the HDR efficiency increased to 26% when the length of the double-cut donor homologous arms was increased to 900 bp. 52 Thus, different donor DNA forms or the lengths of donor homologous arms markedly affect HDR efficiency, as well as the repair efficiency of the CRISPR-Cas9 gene editing system. 63
EFFECT OF THE CELLULAR DELIVERY MODE OF CRISPR-Cas 9 GENE EDITING SYSTEM ON GENE EDITING EFFICIENCY
The CRISPR-Cas9 gene editing system can be delivered into cells in the form of plasmids, mRNAs, or proteins. The delivery form of the CRISPR-Cas9 gene editing system into cells also affects its gene editing efficiency. For example, the transfection efficiency of linear DNA (ssDNA and dsDNA) is higher than that of plasmid donor DNA at the same transfection dosage. 52 The preparation of plasmid DNA of the CRISPR-Cas 9 gene editing system is affordable, and this plasmid DNA can be easily delivered into cells. However, the plasmid DNA can overexpress Cas9 protein in cells for a prolonged duration, resulting in off-target effects. 64 The cellular delivery of the CRISPR-Cas9 gene editing system in the mRNA form enables the direct translation of Cas9. Subsequently, the translated Cas9 can rapidly edit genes without the need for DNA transcription. Therefore, the CRISPR-Cas9 gene editing system delivered in the mRNA form can efficiently and rapidly edit the target genes. 65
However, mRNA is susceptible to degradation and is difficult to prepare and preserve in practical applications. The chemical modification of mRNA enhances its stability. Thus, the delivery of the CRISPR-Cas9 gene editing system in the chemically modified mRNA form, which has high efficiency and decreased off-target effects, will have wide applications in gene editing. 66 The target genes can be immediately edited after the cellular delivery of the CRISPR-Cas9 gene editing system in the ribonucleoprotein (RNP) form. 67 The gene editing duration of the CRISPR-Cas9 delivered in the RNP form is markedly shorter than that delivered in the DNA and mRNA forms. 68 In addition, the off-target effect of the CRISPR-Cas9 delivered in the RNP form is lower than that delivered in the plasmid DNA and mRNA forms. 69
EFFECTS OF DIFFERENT TRANSFECTION METHODS FOR CELLULAR CRISPR-Cas 9 SYSTEM DELIVERY ON GENE EDITING EFFICIENCY
The CRISPR-Cas9 gene editing system can be introduced into eukaryotic cells via physical, chemical, or biological transfection. 70 Different transfection methods for cellular delivery of CRISPR-Cas9 and their gene editing efficiencies are given in Table 1.
Different transfection methods for cellular delivery of clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated (Cas) protein 9 system
DOPE, 1, 2-dioleyl-sn-glycero-3-phosphoethanlamine; NA, not applicable; PEG, polyethylene glycol; RNP, ribonucleoprotein; sgRNA, single-guide RNA.
Microinjection and electroporation are the major physical transfection methods for introducing the CRISPR-Cas9 gene editing system into cells. In the microinjection method, the cells are injected with genetic materials via a microinjection needle. The microinjection method is efficient to load the CRISPR-Cas9 gene editing system into cells. However, highly trained personnel and sophisticated equipment are needed to perform microinjection, which has limited its wide application. At present, microinjection technology is widely used for the transfection of fertilized eggs in animals to generate transgenic animal models. 99 The microinjection of a base editor comprising CRISPR-Cas9–fused cytidine deaminase into mouse fertilized eggs resulted in a nonsense mutation. The gene editing efficiency was 44–57%, whereas the allele frequency was up to 100% in F0 mice. This indicated that the microinjection technology was an efficient method to generate mice with target point mutations. 31
The electroporation transfection method is commonly used for gene editing. In the electroporation transfection method, the cell membrane is stimulated by applying pulses of electric current, which promotes the formation of temporary pores and allows the delivery of materials into cells. Electroporation-mediated transfection is not limited by the cell type and has good transfection efficiency in both walled and suspended cells. Several studies have applied electroporation transfection–mediated ex vivo gene editing, which has been widely used in the treatment of hematological malignancies. 100 This method has contributed to the development of stem cell therapies. However, the strong current generated during electroporation can induce cell death. Hence, this method is not suitable for electroporation-sensitive cell types.
Chemical transfection methods have been commonly used. Chemical transfection methods that mediate the delivery of the CRISPR-Cas9 gene editing system into cells include cationic liposome transfection, calcium phosphate coprecipitation, and diethylaminoethyl (DEAE)-dextran. The transfection efficiency of cationic liposome transfection and calcium phosphate coprecipitation methods for adherent cells is higher than that for suspended cells. Although these methods are widely used, they exert cytotoxic effects on cells. 101 Therefore, the amount of chemical transfection reagent must be regulated and the new culture medium must be replaced at an optimal time when these two methods are used. DEAE-dextran, which was one of the first reagents used for mammalian cell transfection, is a cationic polymorph that binds to negatively charged nucleic acids close to the cell membrane and is easily taken up by transfected cells. 102
DEAE-dextran–mediated transfection, which requires a small amount of DNA, has been successfully used in transient expression studies but is less reliable for stable transfection. Similar to cationic liposome-mediated transfection and calcium phosphate–mediated transfection, DEAE-dextran–mediated transfection can exert toxic effects on cells. 103 In addition, the serum must be removed when DEAE-dextran–mediated transfection is used. The lipid nanoparticle (LNP) delivery method can help to realize the instantaneous expression of the Cas9 effector. The immunogenicity of LNP is lower than that of virus expression vectors. In addition, LNP can be incorporated into the CRISPR-Cas9 system and improve the in vivo delivery safety of the system. 104 Several chemical transfection methods have been developed for delivering the CRISPR-Cas9 gene editing system into cells and are used in different cell types. However, the chemical transfection method–mediated gene editing is associated with decreased transfection efficiency compared with other transfection methods (Table 1).
Biological methods for the cellular delivery of the CRISPR-Cas9 gene editing system include adenovirus (Ad)-mediated, lentivirus (LV)-mediated, and adeno-associated virus (AAV)–mediated transfection. These viral transfection methods are associated with high transfection efficiency. In addition, the viral transfection of gene editing tools increases gene editing efficiency. For example, the lentiviral CRISPR-Cas9 system successfully knocked out melanoma cell adhesion molecule (MUC18) in human primary nasal airway epithelial cells with high efficiency (>90%). 105 One study demonstrated that AAV6 donor can effectively edit hematopoietic stem and progenitor cells upon electroporation of Cas9/sgRNA RNP, achieving up to 94% monoallelic editing frequency at the gene encoding the hemoglobin subunit β (HBB) locus. 90 The AAV-mediated transfection method is characterized by low immunogenicity, serotype dependence, and stability of transgene expression 106 and can be used for the cellular delivery of Cas9, sgRNA, or donor templates.
The size of the cargo that AAVs can carry is <4.7 kb, which limits their application in carrying gene editing–related proteins. AAVs can be used to transfect cells with donor DNA. 107 In contrast to AAV-mediated transfection, the Ad-mediated transfection method can transfect large genetic material (8–36 kb). 108 However, Ads are highly immunogenic. 109 LV is regarded as a quality delivery tool for carrying the CRISPR-Cas9 gene editing system. 110 Lenti viruses can package up to 10 kb, and the LV-mediated transfection has the advantage of low cell cycle propensity. 94,111 For example, Cas9 protein was persistently expressed in cells by packaging the sgRNA and Cas9 protein expression genes into the same LV expression vector. Moreover, this LV expression vector has a good ability to infect both dividing and nondividing cells. 112
However, LV-mediated CRISPR-Cas9 gene editing is a safety hazard because the LV genome can randomly integrate into the genomes of somatic cells, limiting its clinical applications. 113 Recently, a peptide-assisted genome editing system that enables powerful and rapid editing of primary cells with minimal cytotoxicity was developed by Zhang et al. 97 Kreitz et al. developed a novel protein delivery system that can precisely deliver any protein to any human cell. 114 Thus, peptide-mediated or protein-mediated delivery is a new research trend in gene editing tool delivery.
In summary, the CRISPR-Cas9 gene editing system can be delivered to cells through different methods, which are associated with differential gene editing efficiency. The optimal mode of CRISPR-Cas9 system delivery is determined based on the advantages of different transfection methods and the application.
DISCUSSION
This review demonstrated that the components of the CRISPR-Cas9 system, HDR efficiency–related factors, and the delivery forms and modes of the CRISPR-Cas9 gene editing system into cells affect the gene editing efficiency of the CRISPR-Cas9 system. Optimization strategies to improve the gene editing efficiency of the CRISPR-Cas9 system were also discussed. This may guide in improving the gene editing efficiency and developing new gene editing tools.
For example, DSBs of target DNA are generally repaired through the NHEJ pathway, which can generate indels, especially in nondividing cells. Unrepaired DSBs may cause unpredictable gene safety risks, resulting in unnecessary genomic changes, such as chromosomal translocations and large fragment deletions, and activation of the p53 pathway. Thus, improving the HDR efficiency of the CRISPR-Cas9 gene editing system has potential applications in the precise treatment of genetic diseases and precise DNA repair. This review discussed the factors affecting HDR efficiency and CRISPR-Cas optimization strategies, which may enable the development of precise gene editing tools and genetic disease therapy.
Viral vector-mediated transfer of the CRISPR-Cas gene editing system has markedly improved gene editing efficiency. The commonly used viral vectors include AAVs, LVs, and Ads. More interesting, the integration-deficient lentiviral vector (IDLV) was obtained by mutation of the D64V gene of integrase. 115 The development of IDLV is highly anticipated. It can overcome the genotoxicity associated with LV when used for CRISPR-Cas9 delivery. 116 At present, a research has reported that IDLV has significant lower levels of indel formation and other off-target effects compared with that of integrase-competent lentiviral vectors, both in vitro and in vivo. 116 Moreover, Michael Chavez et al. utilized an IDLV to encode the homology arm and the payload (large segments of DNA) on both sides of the “cleavage site” of DSBs, which has achieved stable expression of a large transgene by integrase-deficient LV knock-in, leading to HDR at an endogenous essential gene locus. 117
Thus, the advantages of IDLV-mediated CRISPR-Cas9 delivery would make it to be a more and more popular tool in the future study of gene therapy. However, a single optimal viral vector that can deliver different components of the CRISPR-Cas gene editing system is not available. The advantages and disadvantages of different viral vectors vary. Hence, the immunogenicity, integrability, and extra-target effects must be considered when viruses are used as vectors to deliver gene editing components. Delivery of the CRISPR-Cas via the nonviral vectors LNPs and polymer nanoparticles is associated with some advantages for gene therapy. The transfection of target-editing plasmids and donor DNA using multiple transfection methods can improve the integration efficiency after gene editing. 118 Therefore, an appropriate delivery mode must be selected to ensure that the CRISPR-Cas gene editing components are efficiently delivered to the most relevant cell populations, which will improve the gene editing efficiency of the CRISPR-Cas9 system under different application conditions and in different cell types.
The gene editing efficiency can be improved through several other methods. For example, base editing proteins can be optimized for specific therapeutic targets. The optimized protein can maximize the recruitment of gene editing products to the target site and minimize off-target effects, improving gene editing efficiency. 119 In addition, reducing the size of the effector protein will enable the incorporation of other regulatory elements in the delivery system, which can potentially increase gene editing efficiency. A small Cas9 system (named Nme2-Cas9) that was optimized and modified through the protein-directed evolution system by Huang et al. successfully overcame the limitations of traditional PAM (N4CC). This system could effectively recognize the PAM of N4YN and achieved efficient gene editing, which markedly expanded the application scope of accurate editing tools. 19
However, some changes in gRNA may lead to the loss of gene editing activity and limit the application of highly specific Cas9 mutants. 120 Therefore, gene editing efficiency can be improved by reducing off-target effects and maintaining effective target site editing activity of the CRISPR-Cas system, as well as by ensuring the compatibility between gene editing systems and various gRNA variants.
Several recently developed novel gene editing technologies have markedly improved gene editing efficiency, such as, a eukaryotic transposon-encoded first RNA-guided DNA-cutting enzyme (Fanzor) system has been recently verified that it can derive the gene editing in eukaryotes but not undergo collateral cleavage. 121,122 It enables precise gene editing. The novel gene editing technologies have several advantages, which must be investigated in the future. A recently developed CRISPR SpRYgests, which is a PAM-less in vitro system, can cleave DNA at any sequence, including sites refractory to wild-type SpCas9-mediated cleavage. 13 Thus, CRISPR SpRYgests has potential applications in the molecular cloning field. 13 Moreover, Durrant et al. discovered a series of novel large serine recombinases (LSRs) that can precisely insert a large DNA fragment (>7 kb) into the human genome without the involvement of host proteins and guide RNA.
As the average molecular weight of LSRs is two times lesser than that of Cas9, they can be easily delivered into cells. 123 Lampe et al. successfully performed a DSB-free DNA knock-in in mammals using the type I–F CAST system. This system, which was derived from Vibrio cholerae and comprises CRISPR-associated transposases, can mediate large DNA segment integration into the genome without the induction of DSBs. 124 Some transposons can also edit specific genes, such as Tol, ProtoRAG, IscB, Tn7, and IS200/IS605 transposons. These transposons are associated with high gene editing efficiencies and activities. 125 –129
Some transposons have been developed as novel gene editing tools, such as Tol2 transposon, Sleeping Beauty, and piggyBac. 130 –132 The ProtoRAG transposon has two effectors (RAG1 and RAG2) for DSB formation, which is the main difference compared with other transposon gene edit tools. 126,133,134 This may be used for new gene editing tool developing. The development of transposon gene editing tool may be a following worth noted direction in the future.
In conclusion, several factors affect the gene editing efficiency of the CRISPR-Cas system. Appropriate methods to improve the gene editing efficiency of the CRISPR-Cas system must be considered based on the application, cell types, DNA damage and repair pathways, and the forms and delivery modes of gene editing tools into cells.
Footnotes
AUTHORs' CONTRIBUTIONS
S.L., H.S. and J.L. conceived and designed the structure of this review, J.L., C.T., G.L., H.T., Y.W., S.L., and G.L. drafted the article. H.S., S.L., and W.Z. reviewed and edited the article, and supervised the study. All authors reviewed and approved the final version of this article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by National Natural Science Foundation of China (No. 32100705), Science and Technology Program of Guangzhou, China (Nos. 202201010036, 202201010154), Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515012324), the Medical Scientific Research Foundation of Guangdong Province of China (No. A2021236).
The work is also granted by the National Key Clinical Specialty Construction Project (Clinical Pharmacy), and High Level Clinical Key Specialty (Clinical Pharmacy) in Guangdong Province, College Students' Innovation and Entrepreneurship Training Project of Guangdong Province (No. 202210573054), the Special Fund for the Cultivation of Scientific and Technological Innovation of College Students in Guangdong Province of China (No. pdjh2022b0270), the Special Fund for the Cultivation of National Natural Science Foundation of China in School of Clinical Pharmacy, Guangdong Pharmaceutical University (No. SCP2022-03), 2021 Open Project Fund of Guangdong Provincial Key Laboratory of Medicinal Functional Gene Research.