
Abstract
As the clinical experience in adeno-associated viral (AAV) vector-based gene therapies is expanding, the necessity to better understand and control the host immune responses is also increasing. Immunogenicity of AAV vectors in humans has been linked to several limitations of the platform, including lack of efficacy due to antibody-mediated neutralization, tissue inflammation, loss of transgene expression, and in some cases, complement activation and acute toxicities. Nevertheless, significant knowledge gaps remain in our understanding of the mechanisms of immune responses to AAV gene therapies, further hampered by the failure of preclinical animal models to recapitulate clinical findings. In this review, we focus on the current knowledge regarding immune responses, spanning from innate immunity to humoral and adaptive responses, triggered by AAV vectors and how they can be mitigated for safer, durable, and more effective gene therapies.
INTRODUCTION
From bacteria to vertebrates, life has established sophisticated mechanisms to detect and eliminate foreign molecules or to restrict its function and replication. In mammalian cells, pattern recognition receptors (PRR) have a central role as they recognize evolutionarily conserved structures on pathogens, termed pathogen-associated molecular patterns within the specific compartments that they patrol. Engagement of toll-like receptors (TLRs) or of the cytosolic nucleic acid-detecting immune receptors such as the RIG-I family of helicases (RIG-I, MDA5, LGP2), cGAS, IFI16, or AIM2 will ultimately elicit type-I interferon (IFN)-mediated antiviral responses. 1 –3 Because all current and emerging gene transfer and editing technologies are bound to expose cells to exogenous nucleic acids and, most often, also to viral vectors, host antiviral factors and nucleic acid sensors may play a pivotal role in the efficacy and safety of gene therapy. 4
These cell-intrinsic innate immune responses may also promote both humoral and cellular adaptive immune responses against components of the viral vector and, potentially, its cargo. The relevance of immune responses in the context of other delivery platforms such as integrating Lentiviral Vectors or nonviral delivery systems such as Lipid Nanoparticles and their nucleic acid cargos have been extensively reviewed elsewhere. 4 –8 For adeno-associated viral (AAV) vectors, studies indicate that the establishment of adaptive immune responses starts by the recognition of the AAV vector capsid and genome by the innate immune system. 9
In particular, the stimulation of different TLRs present on antigen-presenting cells (APCs) has been shown to be involved in initial innate sensing of the vector and subsequent establishment of adaptive responses, but other PRRs and nonimmune cell types may also contribute to this initial priming. As wild-type AAV naturally infects humans, cross-reactive pre-existing immunity to AAV vectors, both humoral and cell-mediated, is highly prevalent and can also interfere with gene transfer.
Immunogenicity of AAV vectors in humans has been associated with several observations, including lack of efficacy due to antibody-mediated neutralization, tissue inflammation, and loss of transgene expression. In some cases, acute toxicities have also been documented, triggered by complement activation. Importantly, the lack of preclinical animal models that robustly recapitulate findings in patients further adds to the complexity of the understanding of the interaction between the immune system and AAV vectors. This review will focus on the current knowledge regarding immune responses, spanning from innate immunity to humoral and cellular adaptive responses, triggered by AAV vectors, and how they can be mitigated for safer, durable, and more effective gene therapies.
INNATE IMMUNITY TO AAV
Capsid sensing
In primary human nonparenchymal liver cell cultures, including Kupffer cells (KCs) and liver sinusoidal endothelial cells, the viral capsid has been seen to contribute to innate immunity mainly through binding to TLR2 expressed on the cell surface 10 (Fig. 1). TLR2 stimulation by capsid proteins has been shown to signal through MyD88/nuclear factor kappa B (NFκB) and transiently upregulate proinflammatory cytokines such as interleukin (IL)-8, IL-1β, tumor necrosis factor-α, or IL-6. 10 However, there is no clear evidence so far of the role of TLR2 sensing in the induction of adaptive immune responses. 11,12 Work by Kuranda et al., explored innate responses stimulated by the AAV2 capsid in the context of human peripheral blood mononuclear cells (PBMCs), reporting that peripheral monocyte-derived dendritic cells (moDCs) play a key role in the establishment of humoral and cellular responses through IL-1β and IL-6 secretion. 13
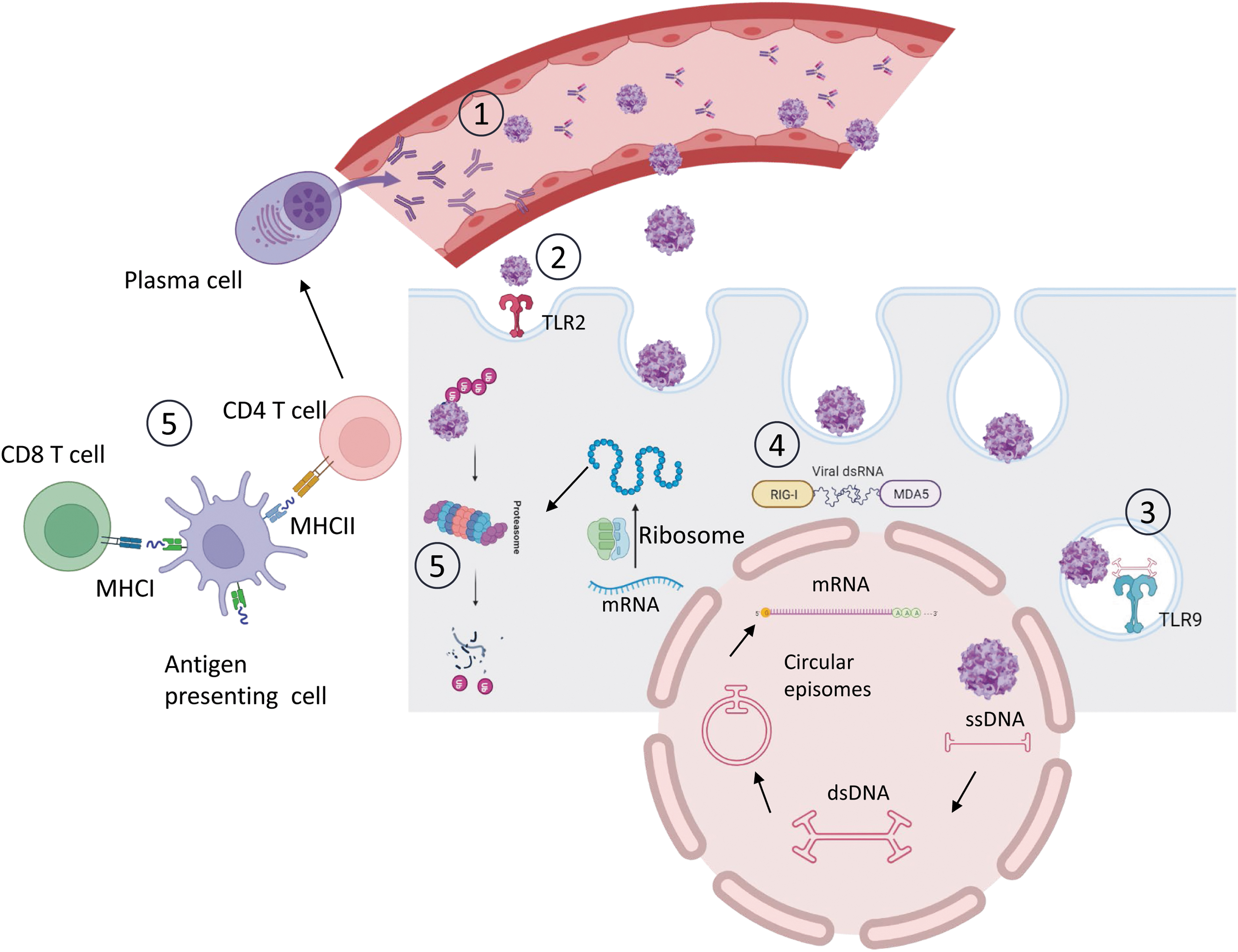
Immunological barriers to AAV-mediated gene therapy. (1) Pre-existing NAbs bind to AAV capsids hampering successful liver transduction. (2 and 3) TLRs recognize vital capsid (TLR2) and viral genomes (TLR9) and trigger innate immune pathways. (4) dsRNA can induce RLRs and MDA5 sensors that will trigger an IFN type I immune response. (5) Antigenic capsid or recombinant protein peptides generated by proteasome degradation are presented by APCs via MHC class I to CD8+ T cells and via MHC class II to CD4+ T cells. After presentation, antigen-specific cytotoxic CD8+ T cells can eliminate transduced cells and CD4+ T cells stimulate the activation of antibody producing plasma B cells. AAV, adeno-associated viral; APCs, antigen-presenting cells; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; IFN, interferon; MHC, major histocompatibility complex; NAbs, neutralizing antibodies; RLRs, Rig-I like receptors; ssDNA, single-stranded DNA; TLRs, toll-like receptors.
Exposure of human PBMCs to either full AAV2, AAV2 empty particles, or capsid-derived peptide pools resulted in increased IL-1β and IL-6 levels in supernatants independently of the serological status of the donors, with the main source being moDCs compared to plasmacytoid (pDCs) or conventional dendritic cells (cDCs). In subjects previously exposed to wild-type AAV, these cytokines triggered the differentiation of capsid-specific memory B cells into antibody-secreting cells resulting in anti-capsid antibody production, and this process could be inhibited in vitro and in vivo by IL-6 and IL-1β blockade. On the contrary, the AAV-seronegative individuals responded to the AAV capsid by transient NK activation. The innate receptors involved in the response were not directly assessed in this study, and whether moDC activation results from direct interaction with AAV peptides or via interaction with other APCs remains to be elucidated. 13
Vector genome sensing
As opposed to the TLR2-dependent sensing of the viral capsid, sensing of the AAV vector genome through endosomal TLR9 has been clearly linked with the subsequent activation of innate and adaptive responses against the AAV capsid and the transgene product 11,14 –16 (Fig. 1). TLR9 recognizes unmethylated CpG sequences present in viral or bacterial but not mammalian DNA. TLR9 signals through the adapter molecule MyD88, which induces the expression of proinflammatory cytokines through NFkB activation, as well as type I IFNs via IRF3 and IRF7.
In 2009, Zhu et al. reported that exposure of murine bone marrow-derived pDCs to single-stranded AAV (ssAAV) led to the induction of type I IFN responses, whereas no evidence of proinflammatory signaling was detected in cDCs, macrophages, or KCs cultured in vitro. 14 Type I IFN signaling in pDCs was shown to be dependent on the TLR9-MyD88 pathway and independent of the nature of the transgene. Later studies have shown that TLR9 stimulation and type I IFN responses mediated by pDCs are important to promote cross-presentation of AAV capsid antigens by cDCs to CD8+ T cells, being pivotal for anti-capsid cellular responses. 12 In vivo, Tlr9− /−, and Myd88− /− mice injected intramuscularly with a ssAAV2 encoding the influenza virus hemagglutinin showed diminished T cell responses against both capsid and transgene compared to wild-type controls. 14
Moreover, the authors reported that the anti-HA and anti-capsid humoral responses were also significantly diminished in both models. Cytotoxicity was also absent in Ifnr−/− mice together with reduced antibody responses to HA and AAV, leading to long-term transgene expression, highlighting a role for type I IFN in induction of adaptive immunity in mice. Moreover, stimulation of PBMC-derived human pDCs ex vivo with AAV2 encoding for lacZ led to the upregulation of human IFN-α (hIFN-α) and hIFN-β mRNA. The response was blocked by the TLR9 antagonist H154 ODN, suggesting a similar mechanism as observed in mice.
A different study addressing innate immune responses in the liver showed that proinflammatory responses to ssAAV were mild in this tissue, whereas they were enhanced when using self-complementary AAV (scAAV) vectors, correlating with stronger cellular and humoral responses to the AAV capsid, but not to the transgene product. 17 Innate responses to scAAV in the liver were also dependent on TLR9 signaling and could be prevented by transient inhibition of TLR9. Some inconsistent results have been obtained regarding the role of TLR9 signaling on humoral responses to the transgene product upon intramuscular AAV injection. While Wu et al., reported stronger transgene-specific cellular and humoral responses induced by scAAV compared to ssAAV, 18 Rogers and colleagues observed enhanced cellular responses but unchanged antibody responses. 19
Regarding the different impact of modulating TLR9 signaling on the regulation of cellular and humoral responses, different roles have been attributed to TLR9 and MyD88 in the establishment of adaptive responses to the capsid and transgene product. 11 In intramuscular setting, cellular but not humoral responses to transgene product were reported to depend both on TLR9 and MyD88. As for anti-AAV capsid responses, only humoral immunity seemed to partially depend on MyD88, but not on TLR9, indicating the involvement of additional sensing mechanisms in shaping adaptive responses. 11 Altogether, different works evidence that TLR9 is not required for antibody formation, yet, it may have a modifying effect on anti-AAV IgG titers and subclasses.
Another factor shown to affect the magnitude of TLR9-mediated signaling in addition to the vector DNA structure is the content of CpG motifs in regions such as the inverted terminal repeats (ITRs), the enhancer and promoter regions, intronic sequences, and polyA tail. 16,20 –22 Importantly, it has been suggested that the unexpected loss of FIX expression observed in patients administered the investigational product BAX335 was due to an increase in CpG content on the expression cassette resulting from codon-optimization. 21,23
In the setting of ex vivo gene editing, while AAV efficiently avoids activation of type I IFN responses in human hematopoietic stem and progenitor cells (HSPC), it triggers p53-mediated DNA damage responses (DDRs) in this cell type. 24 –26 AAV vector DNA genome is sensed in the nucleus, where it triggers p53-mediated DDR upon recruiting the MRE11-RAD50-NBS1 (MRN) complex on the AAV ITRs. 24,25 AAV-mediated DDR induced apoptotic responses in vitro and reduced engraftment of short-term (ST)-HSC in vivo. 24,27
Complement activation
Recent clinical studies have shown that complement responses constitute another toxicity risk associated with high-dose AAV gene transfer, causing variable clinical manifestations, including thrombotic microangiopathy (TMA), kidney injury, thrombocytopenia, and cardiopulmonary insufficiency. 28 –32 Complement-related acute toxicities have been observed only at vector doses above 1 × 1013 vector genomes (vg)/kg, and not consistently across trials.
Although complement factors have been shown to directly bind the AAV capsid ex vivo in the absence of anti-capsid antibodies, 33,34 leading to increased AAV uptake and innate responses in APCs, 33,35 the correlation between pre-existing antibodies and the risk for complement activation is unclear. Additional studies have shown increased complement factor binding and activation in the presence of anti-AAV IgG, 34,35 in particular IgG1, 34 constituting a risk in particular when dosing patients with pre-existing humoral immunity. Nevertheless, complement responses have been reported in clinical studies of gene therapy for diseases, including Fabry disease, spinal muscular atrophy (SMA), Methylmalonic acidemia, 28,31,36 and Duchenne muscular dystrophy (DMD), 32 in which patients had been prescreened for pre-existing anti-AAV antibody titers, whereas no complement responses were reported in an hemophilia B clinical trial, in which seropositive patients were treated with AAV vectors. 37,38
Therefore, the risk for complement activation could be influenced by the total vector dose administered and the subclass of pre-existing or de novo formed anti-AAV antibodies, the AAV serotype, or the underlying genetic background of the host. Furthermore, while similar level of complement activation has been observed to full and empty AAV particles in the presence of immunoglobulins in vitro, 35 a recent study presented at the 26th annual meeting of American Society of Gene and Cell Therapy (ASGCT) by Buchlis and colleagues suggests that the vector genome could also be involved in the mechanism of complement activation upon liver transduction in vivo. 39
Other mechanisms potentially contributing to AAV innate immune activation
Among the potential alternative mechanisms contributing to AAV-induced innate immunity, one study reported the induction of type I IFN responses by AAV through the MDA5/MAVS axis, due to the detection of double-stranded RNA generated from the intrinsic promoter activity of the ITR regions (Fig. 1). However, the impact of MAVS signaling on adaptive immune responses has not been demonstrated. 40
Importantly, AAV gene transfer was also shown to induce dose-dependent toxicity in sensory neurons of the dorsal root ganglia (DRG) upon systemic and local AAV administration in large animal models. 28,41,42 The mechanism responsible for this toxicity remains still unclear since evaluation of T cell responses and studies in the presence of immunosuppressive drugs do not support a clear role of T cell-mediated immunity, 41,43 but studies suggest a correlation between overabundance of the transgene product and neuron loss, 42 as reported also by Henry et al., at the 2023 ASGT meeting. 39 Ongoing works recently presented at the ASGCT annual meeting have detected the presence of immune cell infiltrates in spinal cord as well as different cytokines in cerebrospinal fluid of injected nonhuman primates (NHPs), suggesting the involvement of innate immune mechanisms which require further investigations. 39
Finally, one, although rare but lethal innate immune response, was recently described as cytokine-mediated capillary leak syndrome in a N-of-1 clinical trial for DMD. 44 The patient experienced cardiac and pulmonary toxicities associated to AAV9-mediated innate signaling, which were complicated by the advanced DMD disease in the patient, resulting in cardiorespiratory failure. Postinjection studies in serum and pericardial fluid had revealed elevation of proinflammatory cytokines and complement factors before the cardiorespiratory arrest.
Restriction factors
The high AAV doses still required for therapeutic efficacy in some applications of gene transfer are a potential risk factor contributing to the adverse toxicity observed in recent clinical trials. The efficiency of viral vector-based genetic manipulation may be limited by recognition of exogenous components by host cell restriction factors (RF). 4 RF are intrinsically present in different cell types and usually part of the interferon stimulated genes, thus further inducible upon type I IFN responses. 45
The antiviral mechanisms that interfere with AAV transduction are not completely understood. Thus far, the factors identified to block AAV transduction act at key steps of the viral life cycle such as vector entry or DNA genome conversion into dsDNA or target the AAV capsid for proteasomal degradation (Fig. 2). A whole-genome siRNA screen led to the identification of members of the small ubiquitin-like modifier pathway as critical RF for AAV transduction that acts at the level of vector entry within the cells and are shared across different AAV serotypes. 46 More recently, the VP2 capsid protein was proposed as a direct target of SUMOylation. 47 Restriction of AAV gene transduction can be mediated by direct SUMOylation of the AAV capsid or by AAV-mediated SUMOylation of cellular proteins, which then indirectly influence vector transduction.
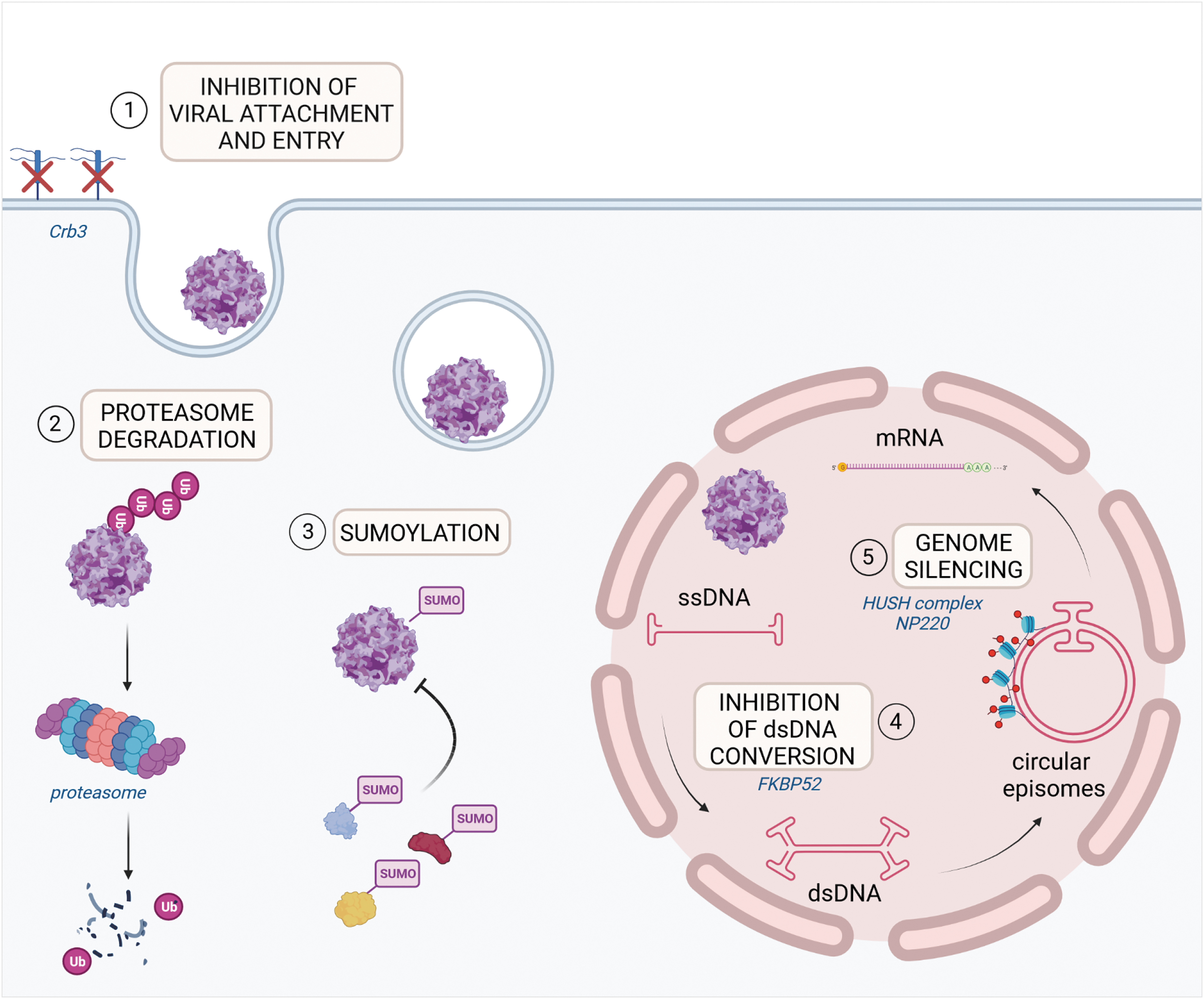
Innate immune restriction mechanisms to efficient AAV transduction. Host RF have been shown to limit AAV transduction efficiency acting at different steps of the AAV life cycle. The main mechanisms are reported in the figure: (1) inhibition of viral attachment and entry (Crb3); (2) capsid ubiquitination and proteasome degradation; (3) capsid SUMOylation and/or AAV-mediated SUMOylation of putative host AAV RF; (4) viral ITR binding and inhibition of dsDNA conversion (FKBP52); (5) AAV genome silencing by epigenetic modification of associated histones (NP220; HUSH complex). HUSH, human silencing hub; ITR, inverted terminal repeat; RF, restriction factors.
More recently, the apical polarity determinant Crumbs 3 (Crb3) protein emerged as a key RF playing a role in viral attachment and entry from a CRISPR screen in a human hepatic cell line. 48 FKBP52, a cellular chaperone protein, is among the factors reported to act at the level of dsDNA conversion. Phosphorylated FKBP52 binds AAV2 ITR 49,50 inhibiting viral second-strand DNA synthesis and affecting transduction efficiency. The dephosphorylated protein loses the ability to bind AAV genome thus allowing efficient transgene expression. PHF5A, a U2 snRNP-associated protein, is an additional critical host factor identified through a siRNA screening as able to limit AAV transgene expression in a serotype and cell type-independent manner, influencing a step after second-strand synthesis. 51
Disruption of PHF5, a subunit of the splicing factor 3b protein complex that forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP) together with other proteins, is sufficient to increase AAV transcript levels, suggesting a critical role for the U2 snRNP spliceosome complex in host-mediated restriction of AAV. 51
AAV proteasome degradation can also potentially affect transduction efficiency. Different AAV capsid surface residues are targets of cellular protein kinases. 52,53 Phosphorylated capsids become a substrate for ubiquitin conjugation and proteasome-dependent degradation 53,54 with overall decrease in transduction efficiency. 55
AAV genome silencing is another barrier to efficient long-term transgene expression. It has been shown that the dsDNA binding protein NP220 and the human silencing hub (HUSH) complex mediate transcriptional silencing of single-stranded and self-complementary rAAV genomes by epigenetic modification of associated host histones in a serotype-dependent manner. 56 Lack of NP220 or HUSH complex favors higher AAV transcript levels with reduced H3K9 histone methylation marks. 56
Many other proteins involved in different cellular processes such as cell cycle regulation, DDR, chromatin remodeling, and transcriptional regulation, have also been identified through cellular screenings as potential RF limiting AAV transduction efficiency. 57,58 Silencing or knock-out of these proteins results in improved transgene expression from different AAV serotypes. The potential benefits of inhibiting proteins such as the Fanconi anemia protein FANCA, the HUSH-associated methyltransferase SETDB1, or the nuclear matrix protein MORC3 have been demonstrated in human primary cells, highlighting their potential relevance in therapeutic settings. 58 CRISPR screenings in cell lines were also used to identify factors critical in facilitating AAV transduction. Among these factors, knock-out of the GPR108 and TM9SF2 proteins decreased AAV transduction using different serotypes. 59 Since GPR108 localizes to the Golgi, it has been suggested that it may interact with AAV, playing a critical role in viral escape or trafficking. 59
Strategies to overcome innate immune barriers to AAV transduction
Although most strategies currently tested to prevent innate immune responses to AAV are still under preclinical research, several investigations have shown the influence of the AAV vector genome composition on the induction of proinflammatory signals through TLR9 and the magnitude of cellular and humoral responses to both capsid and transgene product, in particular, when using single-stranded or self-complementary vectors. 11,17,18 In agreement, reducing the CpG content 16,20 –22 or including TLR9 inhibitory sequences in cis 60 can dampen this cascade of immune responses. CpG-depleted AAV vectors have shown reduced CD8+ T cell responses to both capsid and the transgene in liver and muscle gene transfer settings. 16,20,22
Yet, despite the amount of evidence regarding the link between TLR9 stimulation and cytotoxic responses to the capsid and transgene product, it seems that the beneficial effect of avoiding TLR9 signaling is vector-dose dependent, and responses are still induced above a certain threshold 61,62 as reported also by Kumar et al., at the 26th annual meeting of the ASGCT. 39 Although removing CpG dimers from the transgene cDNA is beneficial in reducing AAV immunogenicity, it is inherently impossible to completely eliminate TLR9-mediated immune sensing. As a result, even CpG depleted vectors stimulate TLR9 above a certain dose. Moreover, additional pathways have been seen to contribute to induction of such responses, such as the IL-1 receptor pathway reported by Kumar and colleagues, 39,63,64 suggesting that multiple pathways may need to be blocked at high vector doses for effective immune modulation.
To prevent cell toxicity derived from DNA-damage responses, inhibition of Ataxia-telangiectasia mutated kinase, upstream of p53 activation, can be exploited to prevent vector-induced DDRs in HSPC, rescuing delayed engraftment. 24,27 Similarly, transient overexpression of GSE56, a p53 dominant negative mRNA, is exploited in AAV/Cas9 gene-editing procedure to rescue engraftment defects and clonality of manipulated HSPC when coelectroporated with the genome-editing components. 25 –27
The C5 inhibitor Eculizumab has been used to treat complement-associated symptoms such as TMA in clinical trials of SMA 36 and DMD, 32,65,66 however, further studies are needed to determine the efficacy of this drug in limiting complement responses. Other strategies have attempted detargeting transgene expression from unwanted tissues through the introduction of microRNA (miR) target sites on the transgene sequence, such as the introduction of targets for miR12267 or miR18342 to prevent transgene overexpression and associated toxicity in liver and DRG, respectively, as well as the use of miR142 targets to detarget transgene expression in APCs and thus decrease immune sensing of the transgene product. 67 –69 The use of miR targets to improve the tissue-specificity of transgene expression is already being used in an ex vivo gene transfer clinical trial for glioblastoma for the delivery of IFNα specifically into the tumor microenvironment. 70
Strategies aimed at preventing AAV vector restriction are also important to improve transduction efficiency and decrease the vector doses needed to achieve therapeutic benefit. For instance, targeting SUMOylation has been reported to increase AAV transduction, constituting a potential strategy to enhance AAV-based transgene delivery. 47 Proteasome inhibitors have also shown potential to increase AAV transduction efficiency in different cell types, both in vitro and in vivo, 71 as well as epigenetic modulation of target cells to prevent vector silencing. 56
As the molecular understanding of the early innate immune activation associated with AAV transduction increases, it will be possible to design and test improved vector configurations that avoid cellular sensors such as TLRs or DNA recognizing proteins. Transduction protocols adjusted to transiently prevent activation of these pathways through targeted inhibitor delivery behold potential to dampen innate immune activation and thereby potentially diminish also subsequent humoral and adaptive responses in vivo. While it is still unclear whether infectious AAV encodes for antagonists of some of the identified RF, the field of virology offers plenty of examples of how viruses effectively counteract these innate immune barriers. 72,73 Further studies will help elucidate if AAV-encoded proteins could be harnessed for such purposes or if other viral factors or synthetic agonists could be exploited to improve target cell permissivity to transduction, allowing the lowering of vector doses with consequent lowering of the risks of adverse immune toxicity.
ADAPTIVE IMMUNITY TO AAV
Humoral responses
Humoral immunity directed against the AAV capsid represents one of the most important barriers to successful gene transfer. Depending on the route of vector administration, the impact of anti-capsids neutralizing antibodies (NAbs) can be major (Fig. 3A), with systemic vector administration being the setting, in which exposure to antibodies and vector neutralization is the highest. The antibody titer and the total capsid dose administered 74 may also contribute to the outcome of gene transfer in the presence of NAbs. Studies in animal models and in humans indicate that even low titer NAb can majorly affect the outcome of gene transfer. 37,75,76 For example, NAb titers of just 1:5 were shown to block AAV transduction of the liver at vector doses of 5 × 1012 vg/kg in NHPs. 77
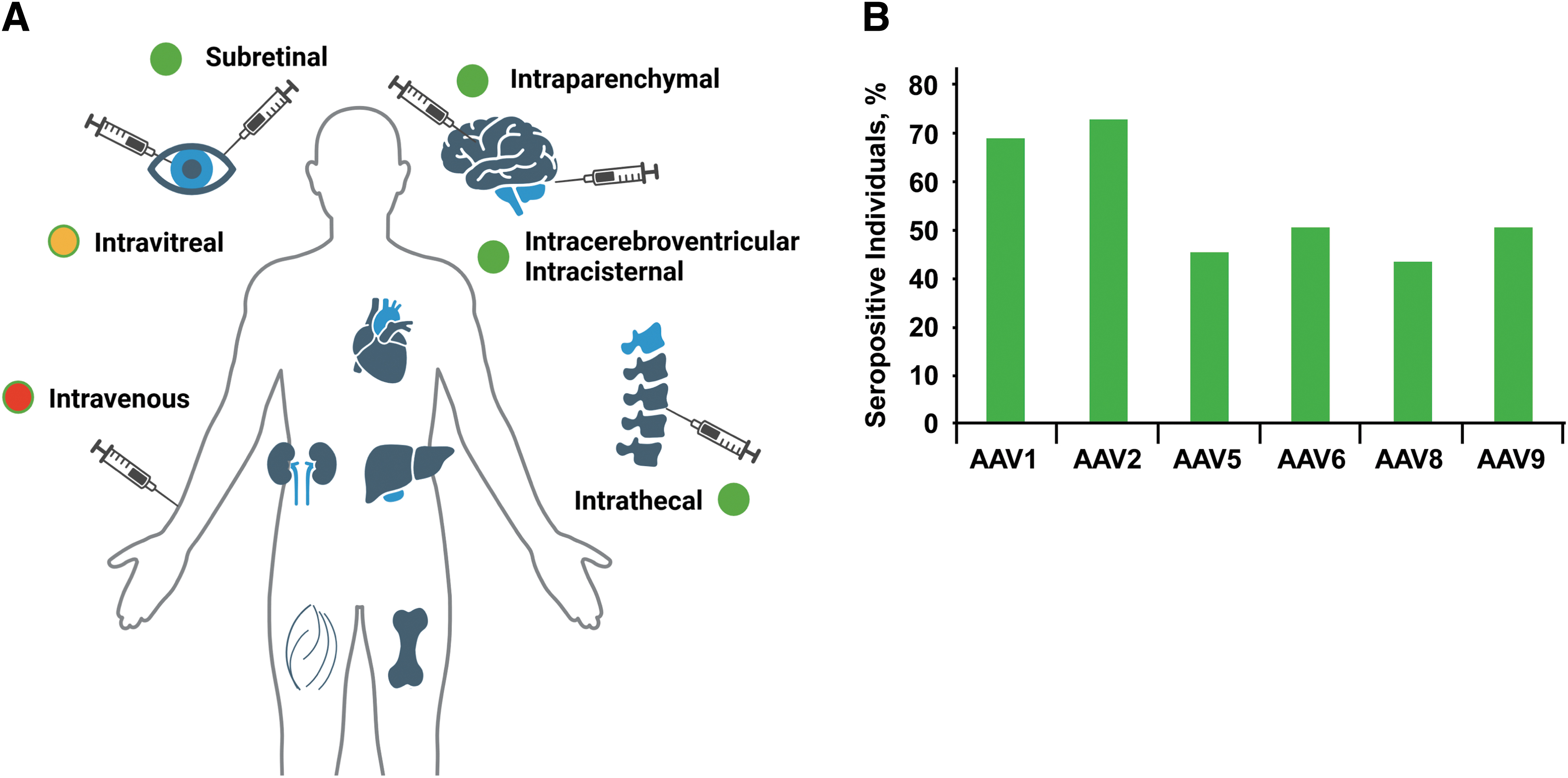
Influence of pre-existing immunity on AAV delivery efficacy.
Similarly, in humans, in the context of hemophilia B trials, antibody titers of 1:17 were shown to prevent liver transduction at a dose of 2 × 1012 vg/kg, 37 whereas titers of just 1:1 can reduce transgene expression by about 50%. 75 Nevertheless, an important aspect to be considered is that NAb titers may vary from laboratory to laboratory depending on the specific design of the NAb assay, due to the lack of a standardized protocol.
Humoral immunity to AAV can be categorized in pre-existing and postdosing. Pre-existing antibodies to AAV originate from the exposure to the wild-type virus, which usually occurs early in life. 78 Depending on the AAV serotype, up to ∼70% of humans can be seropositive, with an average of 30–40% positivity for anti-AAV NAbs across serotypes in adults (Fig. 3B), and lower seroprevalence in young children. 78,79 IgGs binding to AAV are the antibody isotype mostly found in humans, with positivity for IgM detected in a minority of individuals. 80 IgG-binding titers correlate with NAb titers, 81 and in the context of natural immunity to AAV tend to be low to moderate. Upon AAV vector administration, early production of IgM is commonly seen, which is rapidly followed by production of high-titer IgG binding to AAV 82 and, consequently, NAbs.
Post-treatment anti-AAV antibodies tend to persist at high titers for several years and can cross-react with different AAV serotypes. 83 While pre-existing antibodies to AAV affect eligibility to receive gene transfer, postdosing humoral immunity prevents vector readministration, if needed.
Anti-AAV antibodies are mainly measured with two assays, a neutralization assay which measures inhibition of transduction of a cell line by a reporter AAV vector, and a binding assay measuring antibodies binding to the AAV capsid. 84,85 Various protocols have been developed for both the binding and neutralizing assays, and both have been used for the prescreening of subjects ahead of AAV vector administration. While binding and neutralizing antibody titers correlate, some discrepancies can be found, particularly at lower titers, with subjects scoring seronegative in one assay and positive in the other. The choice of a method to screen for antibodies to AAV has important implications, including ease of assay setup, validation, and throughput; ultimately the selection of the assay platform needs to be defined in the context of both preclinical and clinical studies and efforts to develop a companion diagnostic for an investigational gene therapy need to start early in the development.
Several solutions have been proposed to address the issue of anti-AAV antibodies. Broadly, they can be categorized as (1) methods to prevent antibody formation; (2) methods to protect the vector from neutralization; and (3) methods to remove or inactivate circulating NAbs. Antibody prevention is a potential effective strategy to allow for vector readministration, unlikely to be useful in the context of pre-existing immunity. In general, most strategies work best in the context of low to moderate NAb titers, like those found in the context of natural immunity to the virus, while are less effective in the context to high titer antibodies. Thus, redosing of AAV vectors will possibly require a combination of NAb reduction strategies.
Methods to prevent antibody formation
Prevention of antibody formation via immunomodulatory regimens has been proposed to allow for vector readministration. Various combinations of T and B cell targeting drugs have been tested to block antibody formation postvector dosing. Some of these combinations have been tested in the clinic. 86 Overall, the task has been challenging, with preclinical data not easily scalable to humans, possibly reflecting the higher immunogenicity of AAV vectors in humans and the long-term persistence of the AAV capsid antigen postvector dosing. While immunomodulation is almost universally used in AAV gene transfer to block detrimental inflammatory responses, the risk benefit of the approach in the context of vector readministration requires careful evaluation.
The idea of tolerizing the host immune system against the AAV capsid has been explored using various strategies. For example, the coadministration of tolerogenic nanoparticles containing sirolimus has shown promising results in preclinical studies. 87 In a recent healthy volunteer clinical trial, empty AAV capsids were administered together with tolerogenic nanoparticles. 88,89 The approach was effective in reducing NAb titers in the ST, while antibodies appeared to rebound at later times to levels comparable to the control group, suggesting that repeated antigen-nanoparticle (i.e., AAV capsid-nanoparticle) or sirolimus-nanoparticle exposures may be required to allow for a more persistent suppression of antibody formation.
Vector engineering and serotype switching. Several methodologies can be used to engineer AAV capsids with better tissue tropism or the ability to cross physical barriers. 61 Similarly, the development of non-natural variants and their selection against pooled human sera can help identify novel AAV capsids with low seroprevalence. 90 In the context of vector readministration, serotype switch has been shown to be effective in preclinical models. 91 While the effectiveness of the approach needs clinical validation, it should be kept in mind that the use of a different capsid for redosing purposes is highly resource intensive as in many ways it is equivalent to developing a new investigational gene therapy product.
Chemical modification of AAV vectors and the copurification of AAV with exosomes have been shown to provide some resistance to Nabs. 92 Both approaches are less effective in evading high titer antibodies, thus being potentially useful only in the context of pre-existing humoral immunity to AAV. Also physical isolation of a target organ, like the liver, with saline flushing has been shown to be effective against low to moderate NAb titers. 93 Ultimately the clinical feasibility of the approach needs to be established.
As a broadly available and safe, clinically established technology, the use of plasmapheresis/plasma exchange has been shown to be effective in lowering antibody titers to AAV in preclinical studies and in human samples in the context of natural immunity to AAV. 94 While no data are available in the context of gene therapy trials, based on the data available, the strategy is likely to be effective in the context of low anti-AAV NAb titers. Lowering of high-titer NAbs would likely require several cycles of plasma absorption/exchange, due to the rapid rebound postprocedure linked to antibodies residing in the extravascular space.
More recently, it has been shown that the application of immune-adsorption procedure enables successful readministration of AAV5 in NHPs 95 and AAV-specific columns have been developed to remove AAV-specific Nabs from circulation. 96 In addition, empty AAV capsid decoys have been explored as a potential way to absorb pre-existing antibodies during vector administration. 74 Nevertheless, as empty AAV capsids alone have been suggested to trigger innate immune responses, 10 the impact of such strategies on overall immune activation will need to be carefully assessed.
IgG-cleaving endopeptidases are both in development and approved drugs for kidney transplant and other antibody-mediated diseases. 97 Following a single enzyme infusion in human trials, a rapid cleavage of IgG was observed. In the context of gene transfer, the approach has been shown to be effective in allowing for the rapid inactivation of NAbs, allowing for vector administration in the context of pre-existing immunity to AAV and, potentially, readministration. 98 –100 While only preclinical data are available to date, gene therapy trials set to start soon will have to confirm the safety and efficacy of the approach.
A key question is the efficacy of the approach at high NAb titers, as F(ab’)2 fragments released upon IgG cleavage retain some neutralization activity that can potentially affect vector transduction. Related to that, the performance of current antibody detection methods needs to be assessed carefully in the context of this technology. Several endopeptidase variants are in development, including a novel IgG and IgM cleaving enzyme. 101
Inhibitors of the neonatal Fc receptor are approved drugs for antibody-mediated diseases such as myasthenia gravis. 102 The chronic use of this class of drugs is safe, and human studies showed lowering of antibody titers by up to 70% of baseline. Early preclinical studies show that the approach can be useful alone or in combination with IgG-cleaving endopeptidases to address Nabs. 103 More studies are likely needed to define the efficacy of the approach, along or in combination with other strategies, at different NAb titers.
As efforts to tackle the issue of anti-AAV NAbs continue, more strategies are being developed to address the issue. With clinical trials set to start, the issue of pre-existing immunity to AAV, mostly characterized by low NAb titers, is likely to be addressed effectively with some of the approaches outlined above. Vector readministration will likely require the combination of approaches, for example immunomodulation at the time of the first administration to lower antibody production followed by IgG removal, or reduction of IgG half-life followed by IgG removal. Of note, successful AAV administration in the presence of humoral immunity to the capsid requires only a temporally limited window of time to allow for the vector to reach the target tissue. 104 Nevertheless, risk benefit evaluation and development of suitable biomarkers to monitor residual neutralization activity will be key to gather much needed clinical learnings on this critical challenge of in vivo gene transfer.
T cell immunity
The human immune system has evolved powerful cell-mediated adaptive mechanisms to fight against and prevent viral recurrent infections, which hampers the efficacy of viral vector-based gene therapies. As nonpathogenic viruses, and originally not associated with T cell activation in mouse studies, AAVs were put forward as ideal viral vectors invisible to the adaptive T cell surveillance. However, the host cellular immune responses to AAV have been challenging to elucidate due to several factors: (1) AAV vector immunogenicity depends on serotype, expression cassette, route of administration, target tissue, and dose 105 ; (2) differences in T cell responses between animal models and humans 106 ; and (3) low sensitivity of AAV-reactive lymphocyte detection assays in peripheral blood as well as lack of correlation with humoral response. 81,107
In fact, most of what is known about the T cell responses to AAV come from lessons learned from gene therapy clinical trials. As a general mechanism, upon AAV systemic administration, transduced APC can recognize and present capsid-derived antigens to lymphocytes. Antigen presentation via major histocompatibility complex (MHC) class I will activate cytotoxic CD8+ T cells that will eliminate transduced cells, whereas AAV antigen presentation via MHC class II will activate CD4+ T helper cells. Helper cells will amplify and expand both the cellular and humoral immune response (Fig. 1) and increase the anti-AAV antibody secretion by B cells. 108 Several assays have been adapted to detect AAV-specific T cell responses, such as lymphocyte proliferation assays in response to the AAV capsid or capsid-derived peptides, 109 IFN-ɣ ELISPOT, 110,111 or activated lymphocyte detection by flow cytometry. 13,110
Preclinical data and natural AAV infections in human population
Importantly, most of the AAV immunological studies performed in mice have shown that, upon AAV administration, mouse CD8+ T cells do not proliferate in vivo when re-exposed to AAV-derived antigens (capsid or transgene). 112,113 Furthermore, mouse CD8+ cells specific for AAV-derived epitopes fail to eliminate AAV-transduced cells. 114 Interestingly however, ex vivo expanded capsid-specific CD8+ T cells can kill AAV transducer murine and human hepatocytes in vitro (and to some degree in vivo in mice upon adoptive transfer). 115 –117 Despite the similarity of NHP to humans in many translational aspects, it has been also reported that AAV-specific CD8+ cytotoxic T cells obtained from NHP do not destroy AAV-transduced cells 77,118 despite most of them being effector CD8+ rather than memory T cells. 108 AAV-specific CD4+ T cells can also be detected in NHP and their prevalence rates are close to 50% in the NHP-screened population. 108
Regarding the healthy human population exposed to natural AAV infections, Li et al., reported detection of AAV capsid-specific CD8+ and/or CD4+ T cells in 50% of the screened peripheral blood samples. 108 Half of the responsive cells were identified as CD8+ memory cells, 25% as CD8+ effector cells, and the other 25% as effector memory cells, as confirmed for AAV1 in healthy donors. 81 Of note, different distributions were observed in NHP as humans have twice the proportion of CD8+ memory cells than primates. These comprehensive studies also revealed that human CD4+ T cells were mainly central memory cells. 108
Clinical trials and approved AAV therapies
The development of a cellular immune response to AAV was first described after FIX expression was lost in one subject enrolled in the high-dose group in the first AAV-mediated liver-directed clinical trial. 37 In this landmark study, the vector was administered through the hepatic artery. Unexpectedly, a loss of FIX expression in plasma was detected around 4 weeks after AAV dosing, overlapping with an asymptomatic rise in liver transaminases. For the second hemophilia B gene therapy trial, the AAV8 genome was changed to a self-complementary form and administered systemically. 82,119 Four out of six patients from the high-dose cohort (2 × 1012 vg/kg) showed the same transient increase of transaminases.
However, in this case, a short course of prednisolone treatment was able to control the cytotoxic CD8+ response allowing for long-term FIX expression. 119 Notably, this response was not observed at the lower vector doses tested in the trial, which suggested that the immune response was dose dependent. Data collected from a hemophilia A clinical trial did not evidence a consistent increase in capsid-specific T cells in peripheral blood, 120 although few patients dosed with Roctavian (Valoctocogene roxaparvovec) showed sporadic T cell-positive responses by IFNɣ ELISPOT. 111 Across trials, however, increase in liver enzymes has been consistently observed post-AAV infusion, in some cases requiring the administration of prolonged immunomodulatory regimens. 121 To date, no transgene-specific CD8+ responses have been reported upon liver-directed AAV administration. 122
The route of administration is an important factor in mounting a cell-mediated immune response. For instance, the muscle has the potential to initiate inflammation as well as promote infiltration of circulating immune cells, which has been widely exploited for efficient host vaccination, 123,124 including AAV-based vaccines. 125,126 Nevertheless, data from gene therapy clinical trials with intramuscular administration, including those for the treatment of lipoprotein lipase (LPL) deficiency, alpha-1 antitrypsin (AAT) deficiency, and Pompe disease and Duchenne muscular dystrophy, 127 –130 did not show a clearance of transduced cells despite the detection of capsid-specific T cells. This is in contrast with liver-directed clinical studies. Furthermore, CD4+ FoxP3 T cells were found both in the LPL and the AAT deficiency trials, suggesting an induction of tolerance in the administration site, preventing the elimination of transduced muscle cells. 131,132
To treat inborn and acquired neurological diseases AAV vectors are usually directly administered into the central nervous system.
133
–137
Because it is a local route of administration into a site long considered immunopriviledged, many trials have not included T cell response assays. Therefore, information in this regard is limited. In some of these trials, the anti-AAV antibody response in circulation was tested and an increase in NAbs was detected. This suggests that vector leakage from the injection site can trigger a peripheral B cell response and potentially T cell responses as well. Noteworthy, in a recent clinical trial for Tay-Sachs disease, mild T cells responses were observed upon AAV8 delivery to the cisterna magna and the thoracolumbar junction.
138
A different strategy was used for the treatment of SMA. Zolgensma, an US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved AAV9-based vector, is administered systemically together with prednisolone (
In the Clinical Review Document (
Modulation and mitigation strategies
Thanks to these valuable clinical experiences, many AAV-based clinical protocols include at least a corticosteroid regimen at the time of vector dosing, or in the event of a rise in transaminases (
Recently, nanoparticles loaded with rapamycin have shown a durable tolerogenic response to AAV vectors in preclinical studies, even allowing for vector readministration. 87,146 Other mitigation strategies focusing on Treg cell modulation include the incorporation of IgG-derived MHC class II epitopes into the therapeutic expression cassette to reduce AAV immunity 147 or the use of AAV-specific CAR-Tregs and polyclonal Tregs to allow for successful transgene expression in the presence of capsid-specific T cell responses. 148
In the recent studies described above, 88,89 the specific T cell response to AAV8 empty capsids (dose ∼2 × 1012 capsid particles/kg) was studied in healthy volunteers, as well as the potential of an immunotolerant treatment to control it (ImmTOR) as reported by Gordon et al., at the 25th ASGCT meeting. 149 As in previous studies on natural AAV infections, patient-specific differences in T cell induction were observed, together with asymptomatic alanine transaminase increases. Notably, a major contribution of CD4+ T helper cells was reported. In addition, an increase in IFNɣ levels was observed both in the group administered with the empty capsids only as well as in the cohort with ImmTOR. However, the increase of IFNɣ was delayed in the latter.
These results confirmed that cell-mediated responses in patients can be variable and suggest empty capsid removal from AAV preparations as a mitigation strategy to reduce T cell immunity to AAV. Finally, capsid engineering and alternate AAV serotype readministration have been proved as other promising strategies to evade T cell responses. 150
CONCLUSIONS
AAV vector-mediated gene transfer has shown great promise in the clinic in many applications of the technology, resulting in several regulatory approvals of gene therapy drugs based on the platform. Immunogenicity of AAV has limited the success of in vivo gene therapy affecting safety, efficacy, and durability of gene transfer and resulting in significant variability in clinical trials. Therefore, controlling host immune responses to AAV vectors is key to ensure long-term gene therapy efficiency and avoid undesired related toxicities. In this regard, insight from preclinical animal models has been instrumental for gaining understanding of the mechanisms of AAV immunogenicity, as exemplified, although not exhaustively, in Table 1. Nevertheless, there are non-negligible differences between these models and the human immune system prompting additional efforts to be put in gathering more data from clinical trials, including time-course studies of AAV immune responses, possibly also tracking of tissue infiltrating lymphocytes.
Immune responses against transgene product documented in preclinical studies of adeno-associated viral-mediated gene transfer
a-scg, a-sarcoglycan; ASM, acid sphingomyelinase; b-gal, b-galactosidase; CNS, central nervous system; DMD, Duchenne muscular dystrophy; EPO, erythropoietin; GAA, acid-a-glucosidase; GFP, green fluorescent protein; HA, hemagglutinin; hAAT, human a1-antitrypsin; hFIX, human clotting Factor IX; hFVIII, human clotting Factor VIII; HIV1, human immunodeficiency virus 1; HSV2-gB, herpes simplex virus type 2 glycoprotein B; IDUA, a-L-iduronidase; LGMD, limb-girdle muscular dystrophy; LPL, lipoprotein lipase; MPS I, type 1 mucopolysaccharidosis; NHP, non-human primates; Ova, ovalbumin; SM, skeletal muscle; TPP1, tripeptidyl peptidase 1; UGT1A1, uridine diphosphate glucuronosyl transferase family 1 member A1.
In summary, the immunology studies conducted in both preclinical models as well as in human trials over more than two decades have enabled the field to accumulate a significant body of knowledge that is being applied back to improve the AAV-based gene therapy technology. We now know that AAV vectors can be designed to engage less with the innate immune system that manufacturing should aim at reducing process-related impurities such as empty AAV capsids. We also learned how to manage immune responses in the clinic using various immunomodulatory regimens, and potential solutions to the issue of NAbs to AAV will be tested in the clinic in the near future. As the work continues to progress, more solutions will be added to the toolbox of gene therapy researchers, enabling the continuing success of this promising therapeutic modality.
Footnotes
AUTHOR DISCLOSURE
E.V. and A.K.-R. are inventors on pending and issued patents on lentiviral gene transfer filed by the Telethon Foundation and the San Raffaele Scientific Institute. F.M. is employee of Spark Therapeutics, a Roche company, and equity holder of Roche. He is also an inventor in pending and issued patents on the AAV vector platform and methods to overcome immunity to AAV vectors. G.G.A. is cofounder, shareholder, and employee of Vivet Therapeutics and issued patents on the AAV vector platform.
FUNDING INFORMATION
This work was supported by grants from the European Research Council (ERC-CoG 819815-ImmunoStem) and the Telethon Foundation (TELE22-AK) to A.K.-R.