
Abstract
Adenoviruses (AdVs) are being developed for oncolytic or vaccination therapy against existing and emerging conditions. Well-characterized replication-competent human and human primate AdVs expressing multiple payloads are desirable, but their replication in rodent models is limited. To score the timing of adenoviral gene expression in cell cultures, we developed fully replication-competent transcriptional reporter viruses for HAdV-C5, -B3, and -B35. The picornavirus-derived 2A sequence, which induces cotranslational peptide splitting and reinitiation (skipping), was linked to GFP and the fused sequence was inserted C-terminal of the early gene E1A, the intermediate early gene protein IX and the late fiber gene. The 2A peptide induced ribosomal skipping during translation of the messenger RNA (mRNA) and gave rise to GFP from the corresponding viral promoters, as shown by immunoblotting and flow cytometry analyses of human and rodent cells. In human cells, both species B and C AdV exhibited highest reporter expression for fiber, followed by protein IX and lowest for E1A. Inoculation with either HAdV-C5 or -B3/35 viruses encoding protein IX- or fiber-GFP gave rise to higher GFP levels in hamster than mouse cells. Remarkably, despite rather low 2A ribosomal skipping efficiency of ∼50% for E1A-2A-GFP, protein IX-2A-GFP, and fiber-2A-GFP, unprocessed protein IX-2A-GFP and fiber-2A-GFP fusion proteins were efficiently incorporated into HAdV-B3 virions, respectively. These data indicate that the B3 C-termini of protein IX and fiber can be considered for retargeting engineered oncolytic or vaccination vectors, or for antigen display. The variable expression levels of transgenes from different subviral promoters may be used to improve oncolytic AdV vectors expressing therapeutic genes.
INTRODUCTION
Adenoviruses (ADVS)
This is followed by the expression of the delayed early genes, E1B, E2, E3, and E4, whose promoters are under the control of the transactivating primary driver of viral gene expression, the E1A-289R, whereas E1A-243R can also have repressor functions on other promoters. 10 –12 The early genes are predominantly transcribed during the first 24 h of infection, and have functions important for promoting viral transcription and DNA replication, plus antagonizing the host cell immune response. Subsequently, expression of the intermediate early genes, IVa2 and protein IX, is initiated.
After the onset of viral DNA synthesis which starts between 6 and 30 h postinfection (pi) depending on the cell type, the transcription is switched to produce L1, a major transcript of ∼28,000 nucleotides downstream of the major late promoter. L1 gives rise to five families of RNAs (L1–L5), which are translated to viral structural and regulatory proteins with key roles in gene expression, and progeny formation.
AdV vectors are widely used in clinical gene therapy, mostly cancer treatment and vaccination boost regimens taking advantage of readily feasible engineering and biochemical modification of their genome and capsid, respectively. 13 –18 AdV vectors used in clinical oncolytic applications, so-called oncolytic AdVs (OAdVs) have shown an adequate safety profile in cancer immunotherapy. Tumor inflammation and immune recognition of tumors have promoted the concept that OAdVs can, for example, be used to sensitize tumors for checkpoint inhibitor treatment. 19 To exert multiple antitumor functions, OAdVs are armed with multiple cytotoxic and immunostimulatory payloads. In there, extent and timing of transgene expressions are pivotal for effective antitumor efficacy. 20
AdV are pseudo T = 25 icosahedral particles with limited genome packaging capacity, up to about 105% of their normal size, equivalent to ∼2 kilo base pairs of additional DNA in case of HAdVs. 21 This is a significant barrier to OAdVs, as removing viral genes from replication-competent OAdVs compromises OAdV efficiency. 22 To effectively use the genetic capacity of AdV, four general types of transgene insertion strategies have been applied to replicating OAdVs, including (1) insertion of autonomous mono-, bi-, or tricistronic expression cassettes, (2) linkage to viral genes via internal ribosome entry sequences (IRES), (3) gene fusion via translationally recoded 2A peptide sequences, and (4) insertion of exogenous splice acceptor (SA) sites. 20,23 –25
Both the 2A and SA strategies exploit viral mechanisms, give rise to efficient gene expression and, importantly, embed transgene expression into the viral replication cycle. For example, therapeutic or reporter genes inserted into the late AdV transcription unit are expressed late in infection, and reduce potential interference with early and intermediate processes, such as replication. Both SA sequences and 2A peptides are small, in the size range of ∼50 bp, 18–25 aa. Most commonly, the 2A sequence elements from the plus sense Foot and Mouth Disease Virus (FMDV) have been used to coexpress proteins, such as cytokines or antibodies. 26,27 For replication-competent AdVs, reporter or therapeutic transgenes have also been linked to viral genes, including E1A, E1B, E2A-DBP, protein IX, 23K protease, or fiber. 24,28 –34 Inserting the 2A peptide allows multiprotein expression from a single open reading frame by ribosomal skipping of one peptide bond resulting in two distinct proteins. 27,35
Ribosomal skipping gives rise to a 2A “scar” on the C-terminus of the leading protein and an additional proline on the following protein. For proteins targeted to the secretory pathway, insertion of a 4-aa furin protease cleavage site at the C-terminus of the leader allows to trim the 2A “scar.” 33,36
A considerable obstacle in the development of OAdVs expressing immune stimulators has been the lack of suitable immune-competent animal models. In addition, mice are essentially not permissive for HAdVs. Mouse cell cultures often lack the coxsackie and adenovirus receptor (CAR) for HAdV-C, 37 or have a block in late protein synthesis, 38 rendering murine models largely ineffective for productive HAdV replication. 39 –42 Likewise, species B HAdVs using CD46, desmoglein 2 (DSG-2), or both receptors 43 –45 encounter an even more severe replication barrier than HAdV-C in mouse cells. 46 –48 The chimeric HAdV-B11/3 OAdV enadenotucirev (EnAd) expresses E1A mRNA in mouse cell lines, but no other early or late viral mRNAs, resulting in a virtually complete lack of DNA replication and viral protein and progeny production. 48
Possible differences in the early stages of replication in murine cells between species C and B HAdVs, or a possible block of species B HAdVs in poor transactivation activity of HAdV-B11 E1A in murine cells have remained elusive. Hence, OAdVs are being tested in immune-compromised nude or SCID mice carrying xenografts, and immune response is assessed in separate immune-competent mouse tumor models. This dichotomy makes it difficult to determine the impact of oncolysis on immune stimulation and vice versa.
In this study, we report the generation of replication-competent HAdV-C5, -B3, and -B35 viruses containing a GFP reporter transcriptionally linked to the immediate early E1A, the intermediate early protein IX, and late the fiber gene. Four additional chimeric HAdV-B3 viruses contain HAdV-C5-derived inverted terminal repeat (ITR)-, E1A promoter-, and/or E1A gene-sequences. These viruses allow a comprehensive cross-comparison of the timing and efficiency of reporter expression between species B and C viruses in human cells, and a phenotypic characterization of the replication block in mouse and hamster cells. In addition, we provide confirmatory evidence that E1A from HAdV-B3 is efficiently translated in mouse cells, while E3B-RID-β, protein IX, and fiber linked reporters are not expressed. The expression of HAdV-C5-E1A is not able not rescue these deficiency of the species B viruses in mouse cells.
MATERIALS AND METHODS
Cells
Cells grown in Dulbecco's modified Eagle's medium (DMEM; D6429; Sigma-Aldrich) +8% fetal bovine serum (S0115; Biochrom AG, Berlin, Germany) included human embryonic retinoblast (HER) 911, 49 human lung carcinoma A549 (ATCC), and hamster BHK cells. 44,50 Mouse ovarian surface epithelium cell line ID8-Trp53−/− (here short ID8), kindly provided by Iain A. McNeish, University of Glasgow, was grown in DMEM supplemented with 1 × ITS (insulin-transferrin-sodium selenite media supplement, No. 11884; Sigma). ID8-Trp53−/− is a knockout derivative of ID8 cells 38 and was generated to closely resemble the ovarian cancer human disease. 51 ID8-DSG-2 cells, and BHK-DSG-2 were generated using lentiviral particles encoding human DSG-2. 44,52
Transduced cells were selected in medium containing 10 μg/mL blasticidin (ant-bl-1; InvivoGen), followed by fluorescence-activated cell sorting and single-cell cloning. BHK-CAR and BHK-CD46 cells were described previously. 50 ID8-CD46 was generated following stable transduction of ID8 cells with pcDNA3-hCD46 and processed as described elsewhere. 50 All cell lines were routinely screened for the absence of mycoplasma contamination.
Viruses
Human HAdV-C5-wt 300 was obtained from T. Shenk. 53 The other human prototype viruses HAdV-B3 and HAdV-B35 were kindly provided by the late T. Adrian (Medizinische Hochschule Hannover, Hannover, Germany) and were verified by DNA restriction analysis 54 and sequence analysis. 55 All human wild-type viruses were amplified in A549 cells. Generation of the 12 novel recombinant virus constructs (Table 1 and Fig. 1A–C, more detailed in Supplementary Fig. S1A–C) is described in detail in Supplementary Text S1 and Supplementary Table S1. HAdV gene region annotations were performed as described in Bieri et al. 29 The replication-deficient viruses were described earlier and included E1/E3B-deleted HAdV-C5-ΔE1-CMV-GFP 56 and E1-deleted HAdV-B3-ΔE1-CMV-GFP 55 and HAdV-B35-ΔE1-CMV-GFP-E4orf6-HAdV-C5. 29
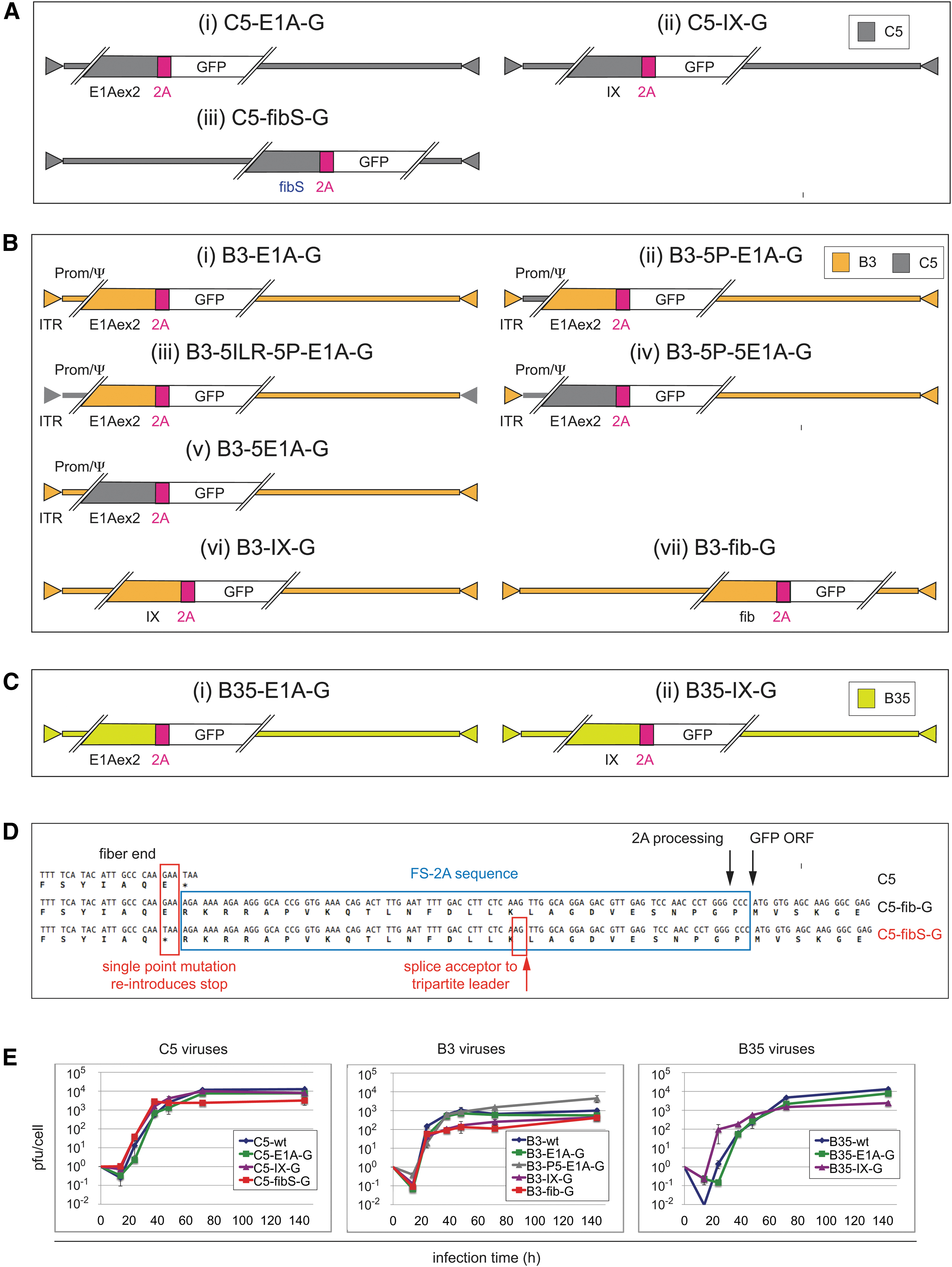
Schematic Genome maps and characterization of different transcription units-linked adenoviruses.
Recombinant adenoviruses used in this study
IV, infectious virus, titer was determined by plaque assay, infecting HER 911 cells.
HER, human embryonic retinoblast.
Recombinant HAdVs were plaque-purified in HER 911 cells, followed by amplification in either A549 or HER 911 cells, depending on their E1 status. The HAdV-derived vectors were concentrated and purified by standard double CsCl procedure, followed by titer determination using plaque assays in HER 911 cells. 56 For analysis of the GFP transcripts produced by HAdV-C5-fibS-2A-GFP, RNA was isolated from infected A549 cells 24 h pi and complementary DNAs (cDNAs) were generated using the 5′/3′ Rapid amplification of cDNA ends (RACE Kit, Roche) and anti-sense GFP primers SP1 (5′-CATGTGGTCGGGGTAGCGGCTGAAG-3′) and nested SP2 (5′-GCCCTCGCCGGACACGCTGAAC-3′), followed by cloning and sequencing.
Generation of recombinant HAdV-B3-E1A and antibodies
For production of recombinant HAdV-B3-E1A (short B3-E1A), the expression vector pET20b-A(H)6-AviTag-GST (kindly provided by Andreas Plückthun, Department of Biochemistry, University of Zurich, Zurich, Switzerland) 29,57 was used. For cloning, a DNA containing the B3-E1A ORF was polymerase chain reaction (PCR)-amplified using the forward and reverse primer 5′-GGA AGA TCT ATG AGA CAC CTG CGC TTC C-3′ and 5′-CCG GCT CGA GTC ATT GCC TTG GCA GTT TCC-3′ (Microsynth AG), respectively, in combination with template cDNAs reverse transcribed from RNA of HAdV-B3-infected A549 cells. 58 Sequence-confirmed pET20b-A(H)6-AviTag-GST-B3-E1A was used for transformation and protein production in E. coli (although this plasmid allows to express biotinylated protein, this was not applied here). The N-terminal His6-tagged protein was purified by single-step nickel-nitrilotriacetic acid (Ni-NTA)-agarose affinity chromatography.
Polyclonal rabbit antibodies against the purified His-tagged B3-E1A protein were generated following immunization of rabbits (BioGenes GmbH, Berlin, Germany).
For this purpose, one animal was first immunized with 200 μg of purified protein using complete Freund's adjuvant, followed by three booster immunizations using 100 μg and incomplete Freund's adjuvant. Final bleeds were done following a fourth booster. The obtained sera were mixed with thimerosal at 0.02% as preservative.
Polyacrylamide gel electrophoresis and Western blot
For western immunoblot analysis of GFP reporter protein and viral proteins, cells infected at a multiplicity of infection (MOI) of 10 were lysed in NETN (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5% NP40) supplemented with protease inhibitors (Mini-Complete; Roche). Analyses of cleared cell lysates obtained from equal fractions of plate wells were performed by polyacrylamide gel electrophoresis 59 followed by western blotting of electrotransferred protein to Immobilon-P membranes as described previously. 60 Membranes were saturated in TBS-T containing 5% dry milk and incubated with primary rabbit antibodies at 1:3,000. The mouse monoclonal antibodies were used at 1 μg/mL. The blots were stained with the novel rabbit anti-B3-E1A antibody, rabbit anti-HAdV-C2-E3B-RID-β (short C2-E3B-RID-β), 61 tubulin antibody (DM1A; Sigma), and the HAdV cross-reactive rabbit anti-HAdV-C5-IIIa (short anti-C5-IIIa) antibody (kindly provided by P. Hearing, School of Medicine, Stony Brook, NY) 62 or with mouse monoclonal anti-GFP antibodies (clone 7.1 and 13.1; Roche; all 1:2,000).
Additional primary antibodies included rabbit-anti C5-protein IX 63,64 and B3-protein IX, 65 rabbit anti C2-fiber 66,67 and B3-fiber (provided by J Chroboczek, Institut de Biologie Strucutrale, Grenoble, France), M58 mouse monoclonal anti-C5-E1A antibody (Thermo Fisher), and rabbit anti-B3 hexon. 32 Secondary HRP-conjugated donkey anti-rabbit IgG and sheep anti-mouse IgG (GE Healthcare) antibodies were used at 1:4,000, for 1 h at room temperature. For western stainings, replicate western blots containing the same samples were prepared to prevent multiple antibody-stainings on the same blot giving rise to overlapping protein bands. The immunoreactivity was determined using the Luminata Crescendo Western HRP substrate (Millipore) and scored using the ImageQuant LAS 4000 imager (GE Healthcare).
Flow cytometry, antibodies, and virus transduction
For cytofluorometric analysis of surface proteins, cells were detached with PBS-EDTA-20 mM, and 2 × 105 washed cells were subsequently incubated with 1 μg of specific antibody, followed by incubation with 1 μg of secondary conjugate antibodies. Specific antibodies included E4.3 for human CD46 detection (Pharmingen Switzerland, Basel, Switzerland), E1-1 for human CAR detection, 60 and 6D8 (Santa Cruz, Inc.) for human DSG-2 detection (Santa Cruz, Inc.). Secondary phycoerythrin labeled antibodies included goat anti-mouse (Ref. 103009), purchased from Bio-Rad.
For eGFP expression-based transduction analysis, triplicates of 105 cells were seeded in 12-well plates, incubated at 37°C in 5% CO2 for 8 h, and transduced with recombinant eGFP-expressing AdVs at various virus concentrations. Cells were harvested and processed for cytofluorometric analysis, including inactivation with 3% paraformaldehyde, at various time points pi.
Flow cytometry analyses were performed on a FACSCanto II with FACSDIVA software (BD Biosciences) at the Flow Cytometry Facility of UZH Irchel. Data analysis was performed with FlowJo 10.4. In all experiments, mean of fluorescence intensities (MFI) data represent the geometric mean of fluorescence intensities.
RESULTS
Generation of transcriptionally 2A-linked HAdV-C5, -B3 and -B35 viruses
We generated a set of 12 replication-competent reporter viruses containing GFP linked to specific virus transcription units, 3 of them derived from HAdV-C5 (short C5), 7 from HAdV-B3 (short B3), and 2 from HAdV-B35 (short B35) (Table 1 and Fig. 1A–C, for more details see Supplementary Fig. S1A–C). Viruses containing an E1A-2A-GFP (short E1A-G) carry a modified E1A gene whereby the GFP ORF was inserted at the end of the E1A exon 2 (Fig. 1A.i, B.i, C.i). Likewise, in viruses containing a protein IX-2A-GFP (short IX-G) and fiber-2A-GFP (short fib-G), the GFP ORF was inserted at the end of protein IX (Fig. 1A.ii, B.vi, C.ii) and fiber gene (Fig. 1A.iii, B.vii), respectively. Hence, while E1A represents the first early expressed viral gene, protein IX is expressed from its own intermediate early promoter, leading to low initial transcripts, and increased expression after DNA replication.
In contrast, fiber belongs to the genes that are expressed at the late phase of infection controlled by DNA replication and activation of the major late promoter. 2 –7 The FMDV-derived F2A24 sequence element used here consists of 5 aa of the FMDV upstream 1D gene, and 19 aa of the 2A sequence, including the N-terminal proline of the 2B downstream gene sequence. 68 –70 Further, the 4 aa residues of the furin cleavage site were added at the N-terminal site of F2A24-GFP, in anticipation that the GFP reporter could eventually be replaced with genes encoding secreted proteins. 33 The replication-deficient E1-deleted C5-ΔE1-CG, B3-ΔE1-CG, and B35-ΔE1-CG viruses carry a CMV promoter-controlled GFP expression cassette (CG) and were used in earlier studies. 29,55,56 C5-ΔE1-CG, but not B3-ΔE1-CG and B35-ΔE1-CG, also carries a deletion in the E3B region.
To address whether species B-derived E1A exerted poor transactivation activity in the early stage of virus replication in mouse cells, 48 we generated four additional chimeric B3-E1A-G viruses containing C5 ITR-/E1A promoter-/E1A gene-sequences (Fig. 1B.ii–v). To test whether low B3-E1A promoter activity contributes to poor gene activity in rodent cells, the promoter (P) and/or ITR (I) sequences were exchanged in two constructs. These included B3-5P-E1A-G, where the endogenous B3 E1A promoter containing the packaging sequence Ψ encompassing nucleotides 137–575 was replaced with the corresponding nucleotides of C5 (Fig. 1B.ii) 53,55 and B3-5ILR-5P-E1A-G, where both the left (L) and right (R) ITR sequences (I) were additionally swapped with the C5 ITR sequence (Fig. 1B.iii).
To test complementation with C5-E1A, the B3 E1A gene sequence consisting of two exons and one intron was exchanged in two further constructs. In B3-5P-5E1A-G, the promoter and E1A gene sequence were exchanged with the corresponding C5 sequence (Fig. 1B.iv), whereas in B3-5E1A-G, only the E1A gene sequence was exchanged, leaving the upstream promoter sequence unchanged (Fig. 1B.v).
Initial attempts to rescue the set of three fiber-2A-linked GFP viruses C5-fib-G, B3-fib-G, and B35-fib-G from the BACmid constructs yielded only B3-fib-G virus. HER 911 cells transfected with the BACmid containing the C5-fib-G construct initially gave rise to a slowly propagating virus. Upon repeated passaging on 911 cells for ∼1 month, it eventually yielded efficiently growing virus, of which plaque-purified clones were isolated. DNA analysis and RACE/PCR analysis on the GFP transcript from infected cells revealed that the last fiber codon for glutamic acid was mutated to a TAA stop (S) codon, which likely contributed to activation of a cryptic SA site within the downstream 2A sequence to the tripartite leader sequence of the MLP-controlled gene unit (Fig. 1D). Hence, in vitro evolution apparently selected for a novel MLP linked, cryptic splice site-activated C5-fibS-G fast growing virus variant.
To confirm these findings, the single point mutation was introduced into a C5-fib-G-BACmid. This construct rescued C5-fibS-G virus with similar growth properties as the above-mentioned evolutionary selected clone (not shown). The generation of a B35-fib-G variant remained unsuccessful.
We next analyzed the growth kinetics of the recombinant and wt AdVs. Human A549 cells were infected at MOI 1, and progeny virus collected from cells and supernatant was titered by plaque assays (Fig. 1E). A four log10 increase was found for C5 and B35 viruses, whereas B3 viruses increased titer by about 3 log10, compatible with results from the literature. 3,71 –73 Among the C5 viruses, yield for C5-fibS-G was about fivefold lower than for the other viruses. Note that the B35-wt and B35-E1A-G appeared to have a slightly slower onset of virus production when compared to the B35-IX-G and C5/B3 viruses, but they did not give rise to lower endpoint titers. The data show that growth of the C5 and B3/35 variants did not deviate more than about fivefold from the corresponding wt viruses, suggesting that the variants were not significantly impaired in growth.
2A-linked GFP expressing C5 viruses report early, intermediate, and late viral transcription
We next used time-resolved immunoblot analyses to characterize the expression kinetics of the GFP proteins under viral transcription control, as well as the viral proteins encoded in the corresponding joint mRNA. We compared infections of human A549 cells and mouse ID8 cells with C5-wt and recombinant C5-E1A-G, C5-IX-G, C5-fibS-G, and C5-ΔE1-CG. Replication-deficient C5-ΔE1-CG served as negative control for viral gene expression. A549 cells are known to express the C5 receptor CAR, 74 and ID8 cells are well infectable with C5 yielding GFP expression levels similar to HeLa cells suggesting that these cells express mouse CAR. 38 Lysates of infected cells were probed with antibodies against GFP, C5-E1A, C5 protein IX, C5 fiber, IIIa, and tubulin (Fig. 2A, B).
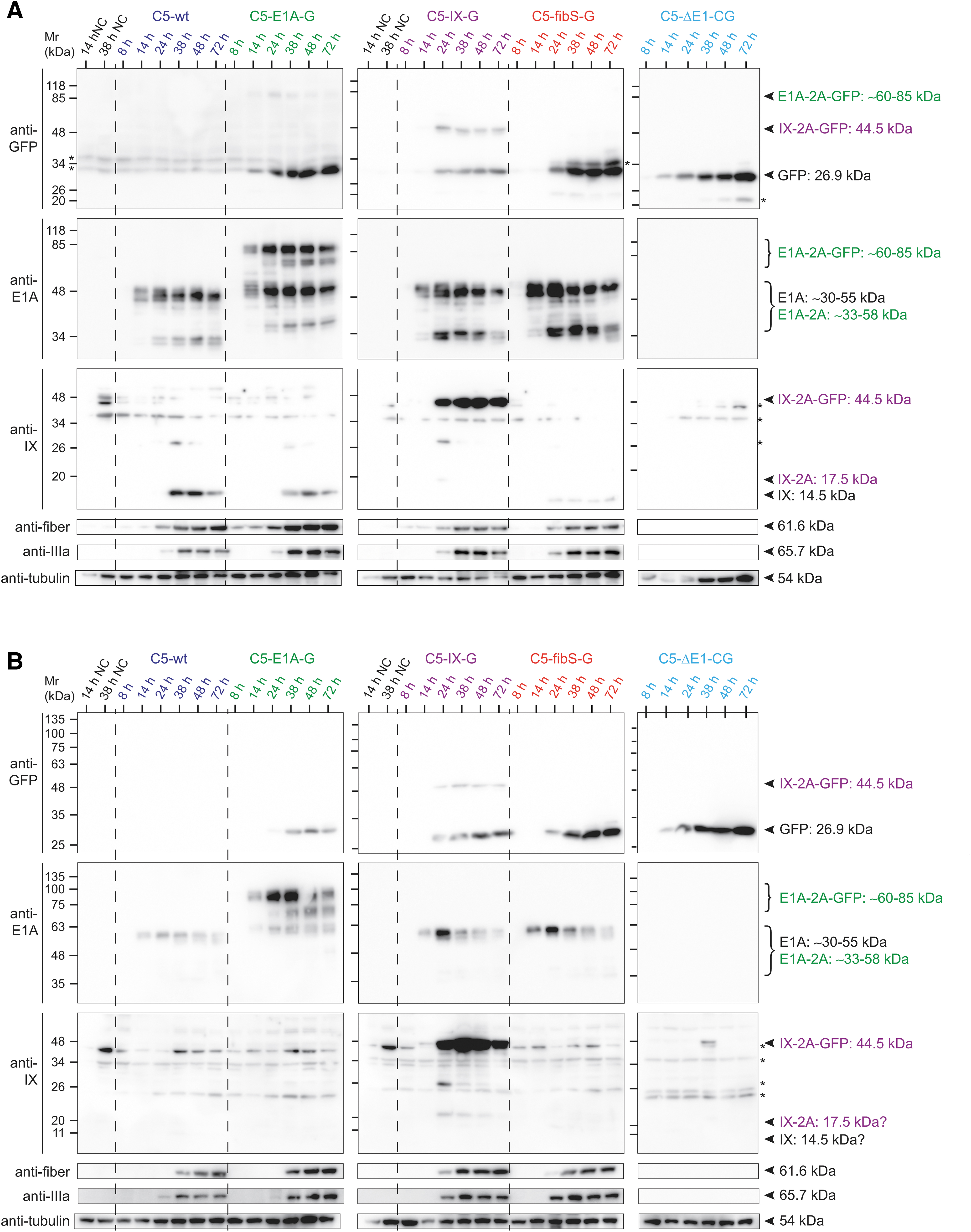
Infectivity kinetics analysis of transcriptionally linked C5 viruses in human A549 and mouse ID8 cells by immunoblotting. Immunoblot analysis of human A549
Importantly, the recombinant C5 GFP reporter viruses expressed the transgene similarly as the corresponding viral genes. In particular, E1A promoter-controlled GFP expression in A549 cells started ∼5–10 h before expression of intermediate genes mediated by the C5-IX-G and C5-fibS-G viruses. In general, C5-wt and all the recombinant viruses gave rise to comparable protein expression levels and kinetics in A549 and ID8 cells, although subtle differences may be noticed. For example, the GFP signal in ID8 cells infected with H5-E1A-G had a delayed and somewhat weaker expression of GFP than A549 cells. Second, the kinetics of E1A isoform expression from the four E1-positive viruses appeared similar, but expression levels and complexity were lower in ID8 cells compared to A549 cells.
Two protein bands in the range of 40–55 kDa had been ascribed to be from the 13S and 12S mRNA isoforms, whereas bands in the range of 30–35 kDa derive from 11S and 10S mRNA. 9 The detection of the E1A 9S isoform was not observed with the mab58 antibody, in agreement with the literature and with the notion from our vector design that the spliced E1A 9S mRNA is not directly linked to the GFP mRNA. 9,75
The observed changes in expression levels of the individual isoforms were time-dependent, with the two largest isoforms expressed first, followed by downregulation of the 13S encoded protein, intermediate stabilization of the 12S form, and finally an increase of the smaller isoforms later in infection. 2,9,12,76 While all these forms were readily detected in A549 cells, we noticed that ID8 mouse cells expressed a main protein band, likely corresponding to the larger 13S/12S-derived E1A isoforms, largely without the ∼30–35 kDa E1A isoforms.
Ribosome skipping of the bicistronic fusion proteins C5-E1A-2A-GFP and IX-2A-GFP was by far not complete, as shown by GFP immunoreactive proteins with higher molecular weight than GFP correlating to E1A-2A-GFP and IX-2A-GFP, respectively. A detailed analysis of 2A cleavage products was, however, complicated by the fact that some antibody stainings gave inconsistent results, for example, the strong bands around 40 kDa seen with antiprotein IX antibody staining in C5-IX-2A-GFP-infected cells was barely detected with anti-GFP antibodies. Analogous findings were made in C5-E1A-2A-GFP-infected cell lysates, where the upper 85 kDa E1A band was not or just barely visible by anti-GFP staining, suggesting that the anti-GFP antibody was less sensitive than the virus-directed antibodies used here.
In addition, some of the blots showed unexplained additional bands denoted by asterisks, such as GFP antibody-stained bands at 34 kDa above GFP in the C5-fibS-G lysates, and bands above protein IX in the C5-ΔE1-CG lysates stained with antiprotein IX antibodies. In addition, we saw a band at 20 kDa below GFP in C5-ΔE1-CG lysates, suggesting internal translation initiation or protein degradation, as suggested for other GFP-2A fusion proteins. 77 Curiously, staining of protein IX was mostly absent in ID8 cells, except for the C5-IX-G infections, where the unprocessed IX-2A-GFP form was stained with anti-GFP and antiprotein IX antibodies. C5-fibS-G infection yielded fiber lacking any 2A or GFP adducts in both ID8 and A549 cells. Expression of the late viral fiber protein was, however, delayed in ID8 cells, but not IIIa expression, suggesting that this was not a general observation for late viral proteins.
In summary, the C5 recombinants described here provide a viable strategy to map the gene expression profiles in different host cells, and might be useful to explore the cell states conducive for early, intermediate, and late viral gene expression.
2A-GFP expressing B3 viruses report early, but not intermediate and late viral gene expression in mouse cells, yet, in human cells, give rise to virions with GFP-chimeric protein IX and fiber proteins
We next analyzed infections with seven B3 reporter viruses, comprising protein IX- and fiber-linked B3-IX-G and B3-fib-G, as well as five E1A-2A-linked viruses with variable chimeric E1 promoter/gene composition, namely B3-E1A-G, B3-5P-E1A-G, B3-5ILR-5P-E1A-G, B3-5P-5E1A-G, and B3-5E1A-G. Viral gene expression was assessed in A549 and ID8-DSG-2 cells stably expressing human DSG-2 at a level of ∼40% compared to A549 cells (Supplementary Fig. S2A). B3-wt and B3-ΔE1-CG control viruses were included as reference for normal and replication-deficient infections, respectively. Lysates of B3-infected cells were probed with antibodies raised against GFP, B3-E1A, C2-E3B-RID-β, B3-IX, B3-hexon, C5-IIIa, and tubulin (Fig. 3A, B). In A549 cells infected with B3-wt, B3-IX-G, and B3-fib-G, the rabbit anti-B3-E1A predominantly stained a band of ∼50 kDa, and in some cases revealing a second band closely, corresponding to the 13S E1A and possibly the 12S mRNAs-derived proteins, akin to C5-E1A (Fig. 2A).

Infectivity kinetics analysis of transcriptionally linked B3 viruses in human A549 and mouse ID8-DSG-2 cells by immunoblotting. Immunoblot analysis of human A549
A weak signal probably representing the smaller 10S E1A mRNA isoform of ∼30 kDa was seen in B3-IX-G- and B3-fib-G- infected cells. Infection with all five E1A-2A-linked viruses revealed major bands of ∼85 and ∼53 kDa corresponding to unprocessed and processed E1A-2A-GFP and E1A-2A, respectively. The B3-5P-5E1A-G and B3-5E1A-G viruses containing the C5-derived E1A gene were probed with the C5-E1A-specific antibody. As expected, this gave rise to a C5-like complex pattern of E1A isoforms, distinct from the pattern with the B3-E1A-G, B3-5P-E1A-G, and B3-5ILR-5P-E1A-G samples probed with anti-B3-E1A antibodies. As noticed before with the C5 viruses, despite efficient staining of the unprocessed B3-E1A-2A-GFP by the anti-B3-E1A antibodies, the anti-GFP antibodies failed to stain the B3-E1A-2A-GFP sample, and rather weakly stained B3-protein IX-2A-GFP and B3-fiber-2A-GFP lysates. Akin to C5-E1A-G, the anti-B3-E1A antibody revealed ∼50% unprocessed/processed E1A-GFP fusion protein.
In contrast to the C5-IX-G-infected A549 cells, the anti-B3-protein IX antibody efficiently stained wt protein IX, IX-2A, and processed IX-2A, indicating that our B3-protein IX-directed antibody was more sensitive than the C5-protein IX antibody.
We were interested to analyze transcriptional transactivation mediated by B3-E1A for other early genes, such as E3, and conducted immunoblotting using the rabbit anti-C2-E3B-RID-β (former E3-14.5K) antibody. 61 The E3B-RID-β protein is present in all HAdV species and required for downregulation of the epidermal growth factor receptor and prevention of tumor necrosis factor cytolysis. 78 It is an integral membrane protein with a cytoplasmic C terminus. The E3B-RID-β antisera had been raised against the C2 C-terminal 15 aa proximal to the transmembrane domain, completely conserved in the species B AdVs. All replication-competent B3 viruses except B3-IX-G revealed strong expression of E3B-RIDβ in A549 cells.
In contrast, intermediate early protein IX, and the late proteins hexon and IIIa were expressed at similar levels and kinetics in all replication-competent B3 viruses. The E1-deleted B3-ΔE1-CG unexpectedly gave rise to pronounced expression of protein IX, hexon and IIIa at late times of infection (38–72 h pi), and very low levels of E3B-RIDβ. These results were distinct from E1/E3B-deleted C5-ΔE1-CG. For all recombinant B3 viruses, GFP expression started between 8 and 14 h pi, where B3-fib-G showed the most pronounced increase at the 24 h pi time point.
Interestingly, the B3 virus infections of mouse ID8-DSG-2 cells were different from the C5 infections (Figs. 2B and 3B). All replication-competent B3 viruses gave rise to E1A expression, but no transactivated downstream genes such as E3B-RIDβ, protein IX, hexon, or IIIa were expressed, as indicated by immunoblotting. E1A signals appeared 8–14 h pi, indicated by a ∼50 kDa band for B3-wt, and ∼85/53 kDa bands for the B3-E1A-2A-containing viruses. As seen before in C5-infected ID8 cells, E1A from C5-E1A-encoding viruses expressed the two larger E1A isoforms but not the smaller E1A isoforms of ∼30–35 kDa. Importantly, the replacement of the B3 promoter, ITR plus promoter, and/or the E1A gene by C5 sequences did not rescue the expression of the B3 genes downstream of E1A.
Replication-deficient B3-ΔE1-G gave rise to GFP expression 8 h pi, while GFP from the E1A-2A-GFP viruses appeared between 14 and 24 h pi, confirming efficient entry and nuclear gene delivery of the B3 viruses. Notably, E1A and GFP signals were lower in ID8-DSG-2 than A549 cells, possibly due to relatively low DSG-2 receptor expression in the mouse cells.
The rather low 2A processing in C5/B3-E1A-G, C5/B3-IX-G, and B3-fib-G proteins was in agreement with our previous results from mouse AdV (MAdV) expressing protein IX-2A-GFP genes. 29 We wondered whether unprocessed forms of protein IX-2A-G and fiber-2A-G were incorporated into virus particles. Immunoblots of CsCl-purified B3-IX-G and B3-fib-G virions as well as lysates stained with the anti-B3-protein IX antibody and anti-B3-fiber antibodies revealed both processed and unprocessed forms of protein IX-2A-GFP as well as fiber-2A-GFP proteins in B3-IX-G and B3-fib-G, respectively (Fig. 4). The band of protein IX-2A-GFP from B3-IX-G virions was much stronger than that of protein IX, but not fiber-2A-GFP compared to fiber in B3-fib-G virions, possibly due to the well-known low western blot transfer efficiency for particularly small proteins, such as protein IX. 64
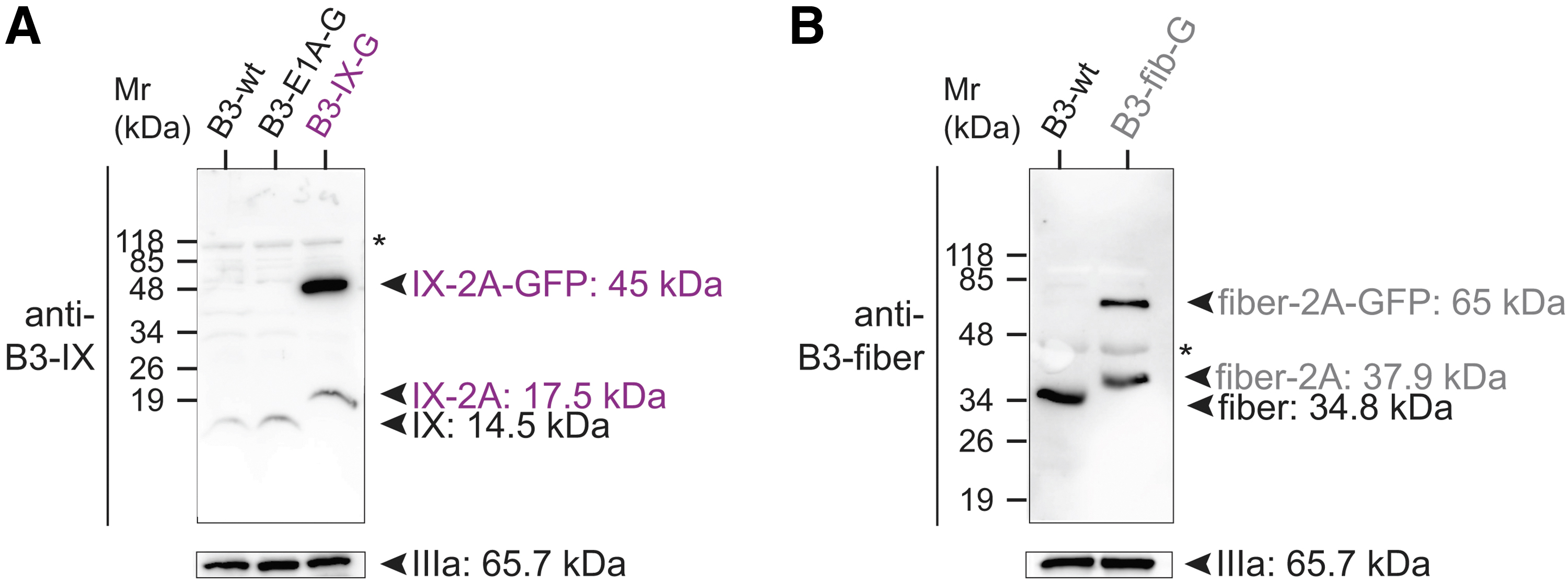
Analysis of processed and unprocessed protein IX and fiber proteins in purified B3-IX-G and B3-fib-G viruses. Immunoblot analysis of CsCl-purified viruses B3-IX-G and B3-fib-G viruses was performed using specific anti-B3-protein IX
Taken together, our data indicate that the robust expression of B3-E1A in ID8-DSG-2 cells does not suffice to give rise to significant production of proteins from B3-virus genes downstream of E1A. The swap of C5-E1A expression into B3 did not complement B3 gene expression downstream of E1A either. Intriguingly, the recombinant B3-IX-G and B3-fib-G viruses grown in human cells incorporated protein IX-GFP fusion proteins and fiber-GFP without adverse effects on virus growth.
Infectivity analysis of transcriptionally linked C5, B3, and B35 vectors in human and rodent cells
Replication-competent HAdVs encoding GFP reporter proteins are convenient to characterize the course of infection at the single cell and population levels in entry, replication, and assembly. 79 –81 We next compared the kinetics and levels of GFP reporter expression from C5, B3, and B35 transcriptional GFP reporter viruses in human A549, mouse ID8, and hamster BHK cells using sensitive flow cytometry analysis.
The hamster cells were included since they are reported to be more permissive for C5 replication than mouse cells. 39,82,83 The additionally included B35 viruses comprised B35-E1A-G, B35-IX-G, and B35-ΔE1-CG (Fig. 1C and Supplementary Fig. S1C). The C5 viruses were tested in A549, ID8, and BHK-CAR cells, the latter expressing about twofold higher CAR levels compared to A549 cells (Supplementary Fig. S2B). The B3 viruses were applied to A549, ID8-DSG-2, and BHK-DSG-2 cells, the latter expressing similar DSG-2 levels as A549 cells (Supplementary Fig. S2C). The B35 viruses were tested in A549, ID8-CD46, and BHK-CD46 cells stably expressing human CD46 both at a level of ∼20% compared to A549 cells (Supplementary Fig. 2D).
A549 cells were infected at MOI 2, 10, and 50 (Fig. 5A, D, G), whereas ID8 (Fig. 5B, E, H) and BHK cell types (Fig. 5C, F, I) were infected at MOI 50. Supplementary Table S2 summarizes maximal GFP values obtained from these viruses, the fold difference to E1A expression from the E1A-G viruses, and the time to reach 10%/25%/50% of maximal GFP expression. In A549 cells the replication-deficient C5-ΔE1-CG, B3-ΔE1-CG, and B35-ΔE1-CG viruses gave rise to dose-dependent GFP expression with highest levels at MOI 50. Similarly, these viruses gave highest GFP expression in rodent cells, except for C5-ΔE1-CG in BHK-CAR cells. In rodent cells, expression levels were lower than in A549 cells, possibly owing to lower expression of primary and/or secondary receptors.
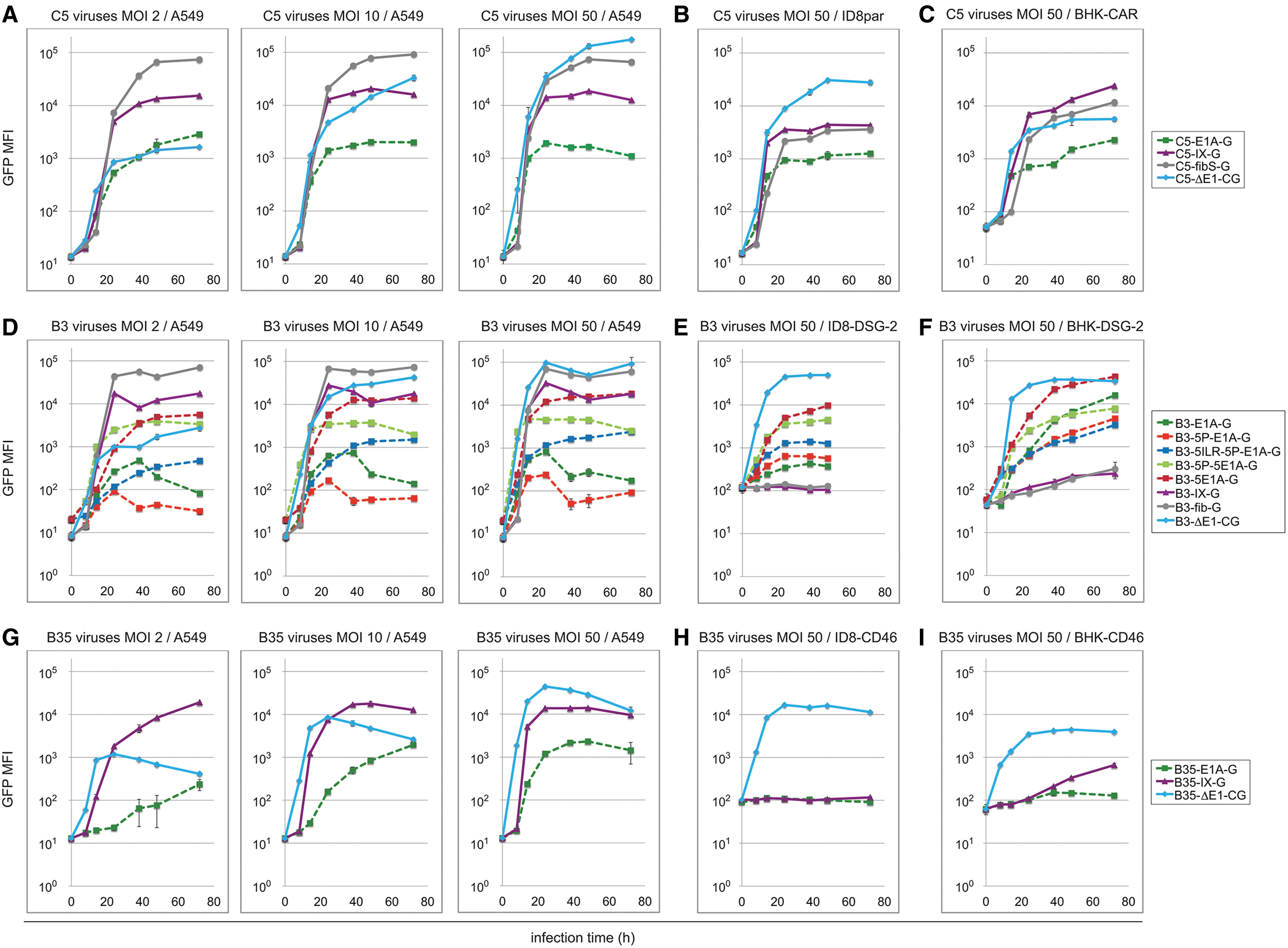
Kinetics and efficiency of GFP expression by transcriptionally linked C5, B3, and B35 viruses in human, mouse, and hamster cells by flow cytometry. Human A549 were infected with C5 viruses
In A549 cells, the replication-deficient C5, B3, and B35 viruses gave rise to robust dose-dependent expression kinetics (Fig. 5A, D, G and Supplementary Table S2). In contrast, the replication-competent viruses, and in particular the protein IX- and fiber-linked viruses already reached expression saturation at MOI 2, paralleled by shorter time points to reach 10%/25%/50% of maximal GFP expression levels. Highest levels were obtained from fiber-linked viruses, followed by protein IX- and E1A-linked viruses. For example, GFP expression from C5-fibS-G and C5-IX-G viruses was 37- and 8.3-fold higher, respectively, than C5-E1A-G levels.
Likewise, GFP expression from the B3-fib-G and B3-IX-G was 113- and 38-fold higher compared to the basal B3-E1A-G-mediated levels. To further characterize the kinetics of transgene expression of individual C5, B3, and B35 viruses relative GFP MFI was plotted as the fraction of maximal values for each virus individually (Supplementary Fig. S3). This analysis confirmed the ∼5–10 h delay of transgene expression onset from C5/B3 fiber- and protein IX-linked viruses relative to E1A-linked viruses, in particular at the lowest MOI of 2.
The GFP expression levels from the C5-E1A-G virus were similar in A549 and rodent cells, while for C5-IX-G and C5-fibS-G the expression increased by a moderate 3.5- and 2.9-fold in ID8 cells, and 10.6- and 5.2-fold in BHK-CAR cells, respectively, compared to basal C5-E1A-G-mediated expression (Fig. 5A–C). Note that these differences were more pronounced than the western blots for protein IX and fiber, which are semiquantitative. Whereas C5-E1A-G-, C5-IX-G-, and C5-fibS-G-mediated GFP expression continuously increased over time in BHK-CAR cells, this was not observed in ID8 cells. Maximal C5-IX-G-mediated GFP expression in BHK-CAR cells reached, with delayed kinetics, comparable levels as in and A549 cells, but was ∼10-fold lower in ID8 cells. The C5-fiber GFP expression reached ∼9-fold lower levels in BHK-CAR cells and ∼16-fold lower levels in ID8 cells, respectively.
Like for the C5-E1A-linked viruses, the five B3-E1A-linked viruses gave rise to lower expression levels in A549 cells compared to the protein IX- and fiber-linked viruses (Fig. 5D and Supplementary Table S2). However, among the five different B3-E1A constructs, GFP expression levels were strongly variable for unknown reasons, with lowest levels from B3-E1A-G and B3-5P-E1A-G, intermediate levels for the B3-5ILR-5P-E1A virus (up by 2.9-fold), and robust increase up to 23-fold from the C5-E1A chimeric B3-5P-5E1A-G and B3-5E1A-G viruses, compared to basal B3-E1A-G-mediated expression levels. The five B3-E1A viruses mediated similar GFP expression also in both rodent cell lines, with a trend to higher levels in the BHK-DSG-2 cells (Fig. 5E, F). Of note, GFP expression from the B3-IX-G and B3-fib-G viruses was completely abrogated in ID8-DSG-2 cells, although weak and continuous expression was noticed for both viruses in BHK-DSG-2 cells.
Analysis of the two replication-competent B35 viruses revealed a slow kinetics of the B35-E1A-G signal in A549 cells, but higher levels from B35-IX-G when compared to B35-E1A-G (Fig. 5G and Supplementary Table S2). No significant increase was observed in ID8-CD46 cells, but weak signals were seen in BHK-CD46 cells (Fig. 5H, I and Supplementary Table S2). The results with replication-deficient C5, B3, and B35 viruses confirmed the transduction susceptibility of these viruses in mouse and hamster cells.
In summary, flow cytometry analyses with a range of replication-competent C5, B3, and B35 viruses provided a wealth of comparative expression profiles of transcriptionally linked reporters applicable to different cell lines. The expression levels of C5 and B3 viruses were highest for fiber- and protein IX-linked GFP, and lowest for E1A-linked GFP in human cells. While E1A-linked GFP expression levels from C5/B3 were similar in human and rodent cell lines, fiber- and protein IX-linked GFP levels from C5/B3 were reduced in rodent cells, with higher levels in hamster cells compared to mouse cells.
DISCUSSION
Here, we developed a range of 12 replication-competent transcriptional reporter HAdVs expressing GFP in equimolar amounts as the respective viral gene linked to GFP. Reporter genes were expressed early, intermediate early, or late in infection depending on the gene-linkage. The observed transgene expression profiles from the fully replicating C5, B3, and B35 vectors were assessed in human and rodent cell lines. In human cells, highest GFP expression from both species B and C HAdVs was obtained with fiber, followed by protein IX and E1A linkage. In each case, expression of the transgene closely mimicked that of the linker gene, both in terms of time and quantity. Our study compares favorably to others, who had used different transcriptional control elements and/or different transgene insertion sites in the viral genome (for review see Farrera-Sal et al. 20 ).
Our replication-competent reporter viruses expressed the transgene under the control of HAdV early genes at lower levels than those linked to late genes, in line with reported transcriptome data. 6,7,84 In accordance, infection of A549 cells with OAdVs containing IRES-linked luciferase to E1A, E2B, and fiber give rise to highest luciferase expression for fiber linkage, 85 and L3-23K SA-linked granulocyte-macrophage colony-stimulating factor expression was approximately fourfold higher compared to expression from the early region 3 locus. 86 This may allude to the possibility that early genes of these replication-competent viruses are expressed from a lower copy number of genomes than late genes.
It is consistent with the notion that ∼100-fold higher transgene expression in the fiber downstream SA site was obtained compared to the E4 downstream SA-site, 87 or ∼10-fold higher transgene levels for fiber-IRES compared to the E4 downstream SA-site. 24 Splicing from the C5 tripartite leader to the transgene transcript in the fiber-SA is, however, crucial for high expression from the fiber downstream SA site, as shown by comparative studies with the E3B-SA site. 88
Our studies gave remarkably consistent transgene expression patterns, unlike others using different transcriptional control elements, where fiber-IRES-linked genes gave substantially higher expression than linkage to a fiber SA-, E4 SA-, or fiber 2A-site, although at the expense of cancer-specific replication, presumably due to transactivation of enhancer embedded in the transgene sequence. 24,89 Likewise, in the study by Farrera-Sal et al., the 100-fold higher expression from the fiber downstream SA site was at the expense of oncolytic potency, which the authors attributed to codon competition for translational cellular resources. 87 These studies show that different transcriptional control elements are difficult to compare, due to particular transgene and/or control element sequences, insertion site of the transgene, as well as virus cytopathic efficiency.
In contrast to the fiber downstream SA site (L6 SA site) used in other studies, the expression coupled to fiber L5 unit in our vectors was very consistent and efficient. The GFP splice event found in C5-fibS-G-infected cells is likely caused by activation of a cryptic splice site due to the stop codon mutation in the polypyrimidine tract. In this novel system, the transgene is part of the L5 unit and uses the same poly A site as the fiber gene. Fiber expression from C5-fibS-G-infected cells was not reduced compared to the other C5 viruses, and GFP levels were comparable to expression from B3-fib-G. Our finding that fiber SA-mediated expression in the C5-fibS-G virus and fiber-2A-mediated expression in the B3-fib-G gave rise to high expression levels strongly supports future considerations of fiber located transgenes for consistently high expression. It will be interesting to see if this system gives similarly high expression levels for other transgenes, and can be adopted to improve cytolytic activity and specificity of OAdVs.
Using the 2A system, a range of reporter and therapeutic transgenes had previously been transcriptionally linked to viral E1A, E1B, E2A-DBP, protein IX, or late 23K protease and fiber genes with variable degree of processing. In our study, processing efficiency at E1A, protein IX, and fiber linked by F2A24 to GFP was rather moderate, confirming our earlier findings with MAdVs encoding protein IX-2A-GFP. 29 Variation in processing has been attributed to the type and length of the 2A sequences, and for F2A in particular, the proximal upstream sequence context of ∼30 aa residues. 35,69,90 –95 Hence, the upstream sequences of the 19 aa “core” 2A cleavage sequence variably consist of FMDV 1D, synthetic spacer, furin cleavage and/or fused C-terminal target gene sequences.
This composite sequence corresponds to the length of the nascent peptide within the ribosome exit tunnel, 96 and it has been demonstrated that interactions between the C-terminal region of certain nascent peptides and the translocon complex can affect the structure of the C-terminus of 2A within the peptidyl-transferase center of the ribosome leading to inhibition of the 2A reaction. 27,97,98 Since all our 2A-linked genes had about the same rather moderate processing efficiency independent of the fused upstream sequence, it is tempting to speculate that the combination of furin cleavage sequence and F2A24 sequence may negatively affect cleavage efficiency. A more processive F2A58 sequence in the C5-derived vKM11 oncolytic virus reportedly had low amounts of unprocessed protein IX-2A-GFP fusion protein compared to our results. 30 Similar to our study, the authors found unprocessed protein IX-2A-GFP protein incorporated into virions.
However, the vKM11 virus had reduced plaque size, cytopathic effect, and viral burst size, unlike our B3-IX-G or C5-IX-G. We speculate that the long 2A sequence on protein IX may have interfered with vKM11 virion stability, the latter being known to be enhanced by the presence of normal protein IX in C5-wt. 99
The mix of unprocessed, full-length, and processed translation products in our viruses may thus turn out to be advantageous. In fact, a short and less efficient F2A16 sequence had been used in plant virus vectors to display antigens on rod-like virions with a mixture of processed and unprocessed F2A16. 100,101 It will be interesting to address whether GFP in our B3-fib-G and C5/B3-IX-G can be replaced with larger antigen sequences. For example, the protein IX surface-exposed C-terminus is known to allow incorporation of large antigen or redirecting fusion proteins in the range of 15–120 kDa, in stark contrast to the variable loops of hexon or fiber, and the C-term of fiber, which only accommodate small peptide sequences. 14,25,102 –104
Although virus stability was not affected by numerous protein IX-fusion proteins, inserting large proteins such as the malaria 41 kDa CS antigen failed to yield virus, 105 or reduced intrinsic capsid stability. 30,105,106 We propose that for potentially destabilizing large protein IX fusion proteins, a mixture of processed and unprocessed protein IX-2A proteins is advantageous compared to entirely unprocessed fusion proteins.
The finding that unprocessed fiber-GFP was incorporated into B3-fib-G viruses is surprising, and now considerably extends the scope of fiber-chimeric C5/B3 vectors, where peptide sequences of maximal ∼25–30 aa had been fused to the C-terminus of fiber (for review see Stepanenko and Chekhonin 104 ). With B35, the insertion of foreign peptides at the C-terminus of the fiber knob resulted in no viable progeny, 107 unlike B3-fib-G; whether this is related to the receptor usage of B3 (CD46 plus DSG-2) and B35 (CD46 only) is unknown. Interestingly, the binding geometry of trimeric fiber knob to CAR and CD46 is fundamentally different from that to DSG-2. While binding to CAR or CD46 engages all of the trimeric fiber knob subunits resulting in a 3:1 receptor to fiber knob ratio, binding of B3 fiber knob to DSG-2 was observed at a 1:1 and 2:1 ratios. 108,109
Since not all fiber knob subunits are occupied with unprocessed 2A-GFP, this might make the B3 knob trimer sterically less constrained for incorporation of 2A-processing mixes, compared to C5 and B35. We suggest that the B3 C-termini of fiber can be considered for both, retargeting of oncolytic or vaccination vectors, and insertion of particular antigens.
Yet, another potential application of transcriptionally linked reporter viruses are screens of human and nonhuman cells to determine their susceptibility to infection with particular AdV types. The strongly reduced C5 fiber- and protein IX-linked expression in mouse ID8 cells are in line with earlier reports on partial block in synthesis of late proteins in C2/5-infected ID8 cells. 38 Our finding of reduced C5-E1A isoform complexity in ID8 cells is not expected to affect virus replication in these cells, since a C2/5 mutant expressing only the single E1A 289R isoform did not affect replication in human IMR-90 cells. 12 The higher expression mediated by our C5/B3 fiber- and protein IX-linked viruses in hamster BHK cells compared to mouse ID8 cells, and in particular, a continuous expression increase over time in the hamster cells corroborate reports documenting more efficient C2/5 replication in hamster cells compared to mouse cells and extend this finding to B3/35 viruses. 39,82,83
Finally, immunoblotting analysis with anti-B3-E1A antibodies and additional B3-specific antibodies demonstrated efficient translation of the B3-E1A protein, and confirmed the lack of B3 protein expression in mouse cells, corroborating previous reports. 48 Based on the results obtained with our B3/C5 E1A-chimeric viruses, we exclude that low B3-E1A promoter activity contributes to poor gene activity in rodent cells, and reinforce that gene complementation with C5-E1A does not overcome the proposed poor transactivation activity of B3-E1A in rodent cells. It remains unclear why expression of C5-E1A or MAdV-1-E1A in EnAd 48 failed to complement the postulated poor transactivation activity of B3-E1A in murine cells. Earlier reports had shown that B3 and C5 E1A can interact homologously or heterologously with their respective transcriptional control regions and autoregulate E1A expression in human cells. 110
E1A does not directly bind to DNA, but interacts with cellular factors controlling transcription to exert its potent transcriptional activator function for viral and cellular genes. One approach to resolve the puzzling lack of E1A complementation in mouse cells might be to conduct transcriptome studies of B3/C5-infected mouse cells.
CONCLUSIONS
In this study, GFP transcriptional linkage to the early gene E1A, the intermediate early gene protein IX, and the late fiber gene allowed a comprehensive cross-comparison of the timing and efficiency of reporter expression between species B and C viruses in human cells, and a phenotypic characterization of the replication block in mouse and hamster cells. We found variable expression levels of transgenes from different subviral promoters, and this may help to improve OAdV vectors expressing therapeutic genes. Linkage by the FMDV-derived 2A sequence resulted in only partial processing of the targeted protein, and interestingly, yielded B3 virions carrying unprocessed protein IX-2A-GFP and fiber-2A-GFP fusion proteins. These findings indicate that the B3 C-termini of protein IX and fiber can be considered for retargeting engineered oncolytic or vaccination vectors, or for antigen display. We anticipate that our results have implications for the development of therapeutic adenoviral vectors for human gene therapy and vaccination.
ETHICS STATEMENTS
All data needed to evaluate the conclusions in the article are present in the article.
Footnotes
ACKNOWLEDGMENTS
We thank all providers of wt viruses, cell lines, antibodies, and plasmid reagents, in particular, Iain A. McNeish, University of Glasgow, for providing the ID8 cell line, and Hans-Gerhard Burgert, University Medical Center Freiburg, for contributing rabbit anti-HAdV-C2-E3B-RID-β antibodies. We also thank Leta Fuchs for technical assistance and the Flow Cytometry Facility of the University of Zurich Irchel for their help with flow cytometry and cell sorting.
AUTHORs' CONTRIBUTIONS
T.J.: methodology, investigation, writing—original draft, editing, formal analysis, and data curation. L.S.: methodology, investigation, writing—original draft, editing, formal analysis, and data curation. M.B.: methodology, investigation, editing, formal analysis, data curation, and funding acquisition. U.F.G.: conceptualization, writing—review and editing, supervision, and funding acquisition. S.H.: conceptualization, methodology, investigation, writing—review and editing, formal analysis, data curation, supervision, and funding acquisition. All authors reviewed the results and commented on the article.
AUTHOR DISCLOSURE
The authors declare that they have no competing financial interests.
FUNDING INFORMATION
S.H. was funded by the Swiss National Science Foundation (31003A_146286) supporting M.B.; U.F.G. was funded by the Swiss National Science Foundation (31003A_179256/1; 310030_212802); and M.B. was supported by the Novartis foundation for medical-biological research (16C222). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
SUPPLEMENTARY MATERIAL
Supplementary Text S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.