
Abstract
Inherited forms of blindness and deafness are highly prevalent and severe conditions that significantly impact the lives of millions of people worldwide. The lack of therapeutic options for these conditions poses a major socioeconomic burden. Over the last decades, gene therapy has proven to be a life changing treatment for hereditary and acquired forms of diseases, and extensive preclinical investigation in animal models of both retinal and inner ear disorders has highlighted promising translational opportunities for these disorders too. This led to dozens of clinical trials investigating the efficiency of gene therapy-based approaches, with some of the products for retinal conditions successfully reaching phase III of development or even market authorization. However, challenges remain for the use of gene therapy, which are related to both the features of the delivery vehicles currently available and characteristics of the retinal and inner ear disorders targeted. Therefore, further developments in gene therapy platforms' design, including exploitation of novel technologies such as genome editing, RNA-targeted therapies, and optogenetics, are actively ongoing, driving the field forward. In this study, we review the ongoing applications and achievements of gene therapy for treatment of inherited forms of blindness and deafness as well as the developments that are being pursued in the field to overcome the current limitations.
INTRODUCTION
The ability of an organism to sense and adapt to the surrounding environment is critical for its survival. As such, impairment in sensory organs' functionality, as it occurs in the most frequent sensory disorders—blindness and deafness—profoundly impacts the lives of affected people. Sensory deficits can be linked to genetic causes, environmental factors, or aging. 1 In developed countries, genetic causes account for more than 50% of all cases of vision or hearing loss. 2 Most commonly, loss of either sense manifests as an isolated symptom, however, they can also associate with other symptoms in syndromic forms of hereditary diseases. No curative option is currently available for these disorders, as sensory rehabilitation based on eyeglasses, hearing aids, retinal prostheses, and cochlear implants provides patients with only partial and temporary relief.
However, the treatment landscape is quickly changing. Over the last decades, progress in deciphering the genetic cause and pathophysiological processes underlying many hereditary forms of blindness and/or deafness has opened the way for the development of gene therapies based on replacement, modulation, or direct repair of defective genes. This has generated hope for the possibility to provide fundamental restoration of vision and hearing. In this study, we review the state of the gene therapy field and the current challenges for the treatment of inherited forms of blindness and deafness.
THE EYE AND EAR AS TARGETS FOR GENE THERAPY
Inherited forms of blindness and deafness are both characterized by a high degree of phenotypic and genetic heterogeneity, with relatively rare individual mutations across any one of the hundreds of genes underlying each of the specific sensory diseases, but altogether constituting a significant cause of blindness and/or deafness. 3,4
Inherited retinal diseases (IRDs) are a group of more than 300 forms of eye disorders, presenting with different patterns of inheritance. 3 Independently from the underlying causative gene, vision loss in IRDs results from functional impairment or loss of key retinal proteins and, consequently, progressive degeneration of retinal photoreceptors and/or retinal pigment epithelium (RPE) cells 5 (Fig. 1A).

Structure of the eye and ear and injection procedures used for delivery of gene therapies.
There is a similarly diverse spectrum of pathogenic genetic variants that cause hearing loss, with hundreds of causative genes that have been cloned and characterized. 4 Approximately 75–80% of monogenetic hearing loss (mHL) is nonsyndromic; the remaining 20–25% of mHL is syndromic, or associated with multisystem syndromes, including monogenic deaf–blind disorders, such as Usher syndrome. As with IRDs, these disorders can follow various inheritance patterns and can be congenital or progressive with delayed onset.
While historically no therapeutic option was available for either IRDs or mHL, gene therapy is dramatically changing the treatment landscape for these disorders. The eye and the ear are particularly suitable organs for gene therapy due to the several advantages they both offer. 6,7 These include their well-defined anatomy and relatively enclosed and small structure, which allow efficient delivery of the therapeutic agent to target cells at relatively low doses with little systemic exposure. In addition, the cells of the retina and inner ear are terminally differentiated and postmitotic, allowing for the potential of a single administration of gene therapy vectors to provide life-long benefits, even without integration of the viral DNA in the host cell genome. Furthermore, the reduced immune surveillance in these organs, as well as the availability of noninvasive techniques to monitor their functionality and/or morphology over time, makes these organs a unique environment for testing innovative therapeutics.
Over the years, the eye has been a leading organ for the development of gene therapies, achieving a major milestone in the field in December 2017, when the Food and Drug Administration (FDA) approved the first gene therapy product for an inherited disease in the United States, Luxturna®, which is a product being used to treat patients affected by RPE65-associated retinal dystrophy. 8 Clinical development of inner ear gene therapies has lagged compared with ocular gene therapies, in large part, because of the lack of an established inner ear route of administration. However, improved understanding of genetic contributions to hearing loss, recent advances in inner ear delivery, and learnings from decades of ophthalmic gene therapy development have enabled the initiation of inner ear gene therapy development.
The first Investigational New Drug application to receive FDA clearance for a genetic form of hearing loss due to mutations in the otoferlin (OTOF) gene occurred in 2022 (
As safe and effective gene therapy in the eye and ear requires precise delivery of the therapeutic agent both at the macro (surgical) and micro (cellular) levels, technological advances in the gene delivery are continuously evolving and continue to expand the retinal and inner ear gene therapy landscapes. A main determinant for effective gene therapy is the choice of the delivery vehicle. Nonviral vector-mediated gene therapy is being increasingly explored for several applications. 1,9 Advantages of nonviral vectors include their flexibility in terms of packaging capacities, potentially low immunogenicity due to the absence of viral components in the delivery particle, and ease of large-scale production. In addition, nonviral delivery results in temporary expression of the transgene, which can be particularly advantageous for applications where long-lasting expression of the transgene can lead to unwanted side effects, as in the case of genome editing tools (see “Conclusions and future outlooks”).
However, so far most nonviral vectors developed have shown low transduction efficiency, especially in large animal models. 1,6 Further developments of these platforms, which are being actively pursued in academia and industry, have the potential to change gene therapy development in the coming years. In the meantime, viral vectors are still the preferred vehicles to deliver transgenes to both the retina and the inner ear, and recombinant vectors based on adeno-associated viruses (AAVs) are considered the gold standard. Indeed, although adenovirus (Ad)- and lentivirus (LV)-based vectors were historically among the first vectors to be tested, with some LV-based products also reaching the clinical stage (Fig. 2), none of the Ad serotypes and LV pseudotypes tested thus far has matched the levels of transduction given by AAVs in the relevant target cells in the retina and inner ear. 1,6
Gene therapy approaches
AAVs are single-stranded DNA viruses, which, thanks to a size of ∼25 nm in diameter, can easily penetrate through tissues. 10 In addition, AAVs have a number of advantages including an excellent safety profile (lack of pathogenicity and low immunogenicity) and, as recombinant vectors, have an ability to provide long-lasting transgene expression after a single injection in postmitotic tissues. However, a fundamental limitation of AAVs is a limited packaging capacity of about 5 kilobases (kb), which impedes their use in several common forms of inherited diseases caused by mutations in genes with a larger coding sequence (CDS).
To date, 13 natural AAV serotypes and hundreds of different variants have been identified (
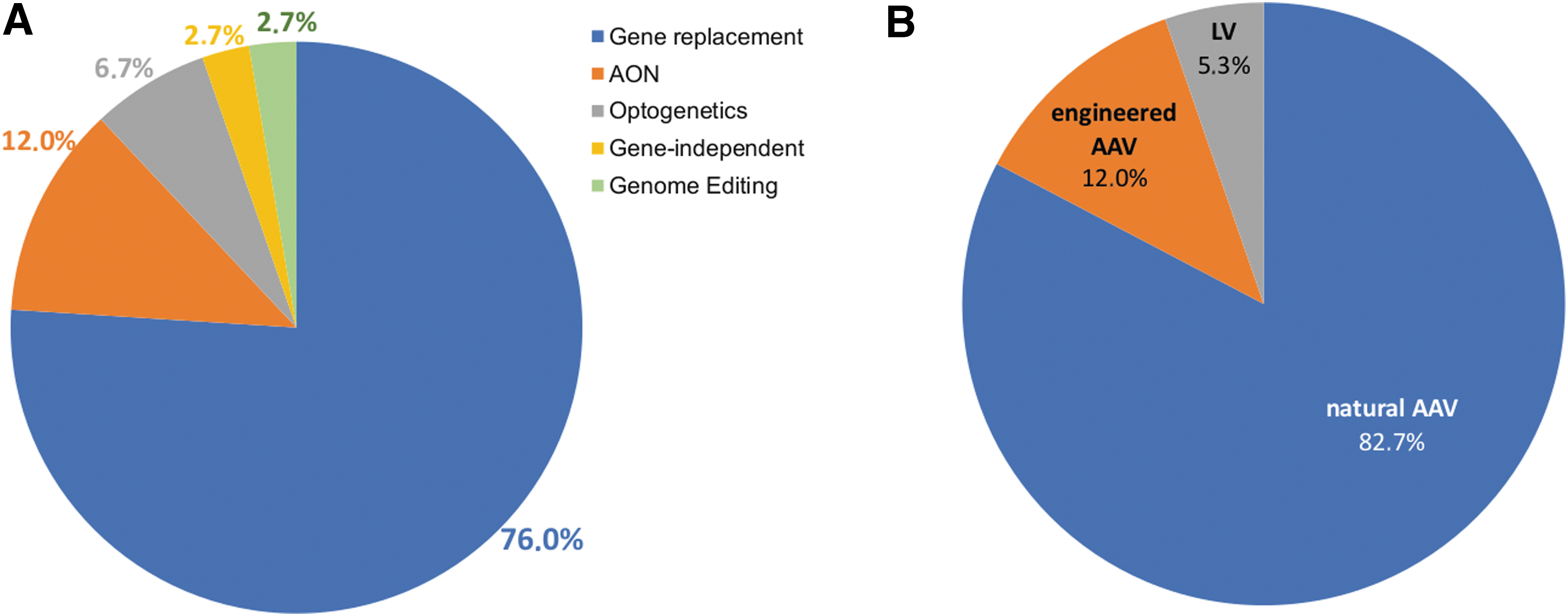
The investigation of different AAV serotypes and variants has enabled efficient access to specific retinal or inner ear cell types using various administration routes. 1,13,15 The findings that some variants, which resulted in improved tropism in small-animal models, have failed to achieve similar results in nonhuman primates, 13,16 have led to further refinement in the design and screening processes used for selection and isolation of improved variants. Several ongoing clinical trials are evaluating the ability of novel engineered AAV variants to deliver therapeutic genes to either the retina for IRD treatment (Fig. 2) or the inner ear (NCT05821959).
THE CURRENT GENE THERAPY SCENARIO IN THE EYE
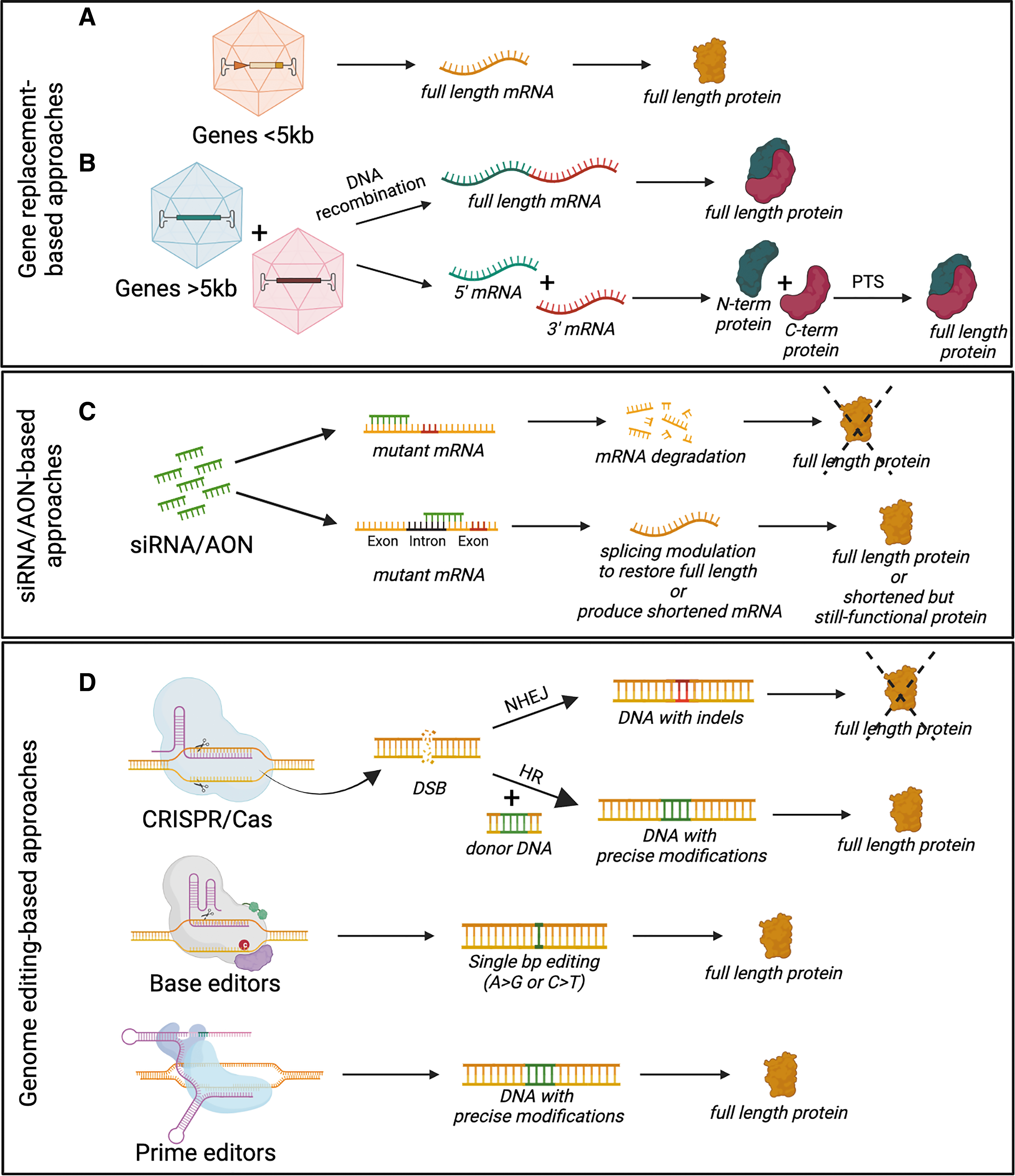
Gene replacement, which is based on the delivery of a correct copy of the mutated gene to diseased cells (Fig. 3A), and well suited to autosomal recessive disorders, has been the first and most extensively tested application to be investigated for treatment of IRDs. The two routes of administration of gene therapies to the retina most commonly used both in preclinical studies and clinical trials for IRD treatment are intravitreal and subretinal injections (Fig. 1A). Subretinal injection is currently considered to be the most efficient route for transducing RPE and photoreceptors, target cells for most IRD treatment, as upon intravitreal injections transduction is mostly limited to the ganglion and inner retinal cells. 6 There are now dozens of successful examples of AAV-based gene therapies leading to significant improvement of the retinal phenotype in IRD animal models. 17
Therapeutic approaches for treatment of inherited forms of vision and/or hearing loss.
Development of Luxturna, an AAV-based product for delivery of the RPE65 gene in RPE65-mediated IRDs, including the most common clinical phenotype associated with RPE65 mutations—Leber congenital amaurosis type 2 (LCA2) 18 —has represented the most successful example of retinal gene therapy. The characteristics of the disease, including the pattern of expression of the causative gene requiring transduction of the RPE, efficiently targeted from AAV, as well as the slowly degenerative nature of the disease, make LCA2 an ideal target for gene therapy and contributed to the outstanding results obtained. Importantly, the LCA2 trials also highlighted that translation from animal models to humans can be complex. 19 Efforts to target several additional common forms of IRDs, including other forms of LCA, retinitis pigmentosa (RP), choroideremia, Leber hereditary optic neuropathy, achromatopsia, X-linked retinoschisis, Stargardt disease, and other rod-cone and cone-rod dystrophies are actively ongoing across more than 40 clinical trials. 17,18
The collective data from clinical trials, spanning different vectors, routes of administration, indications, and therapeutic approaches (Fig. 2), have established the general safety of AAV vectors in the retina and, in some cases, early efficacy to support progression to phase III/pivotal trials. Clinical trial design, setting up of novel reliable trial endpoints, and refining surgical techniques to avoid retinal trauma and immunological responses are the current challenges that need to be overcome to expand the success of gene therapy to an increasing number of IRDs. 7,20 In addition, many IRDs remain untreatable with canonical AAV-based gene-replacement therapy given the underlying molecular mechanisms, disease features, and limitations of the currently available gene therapy vectors. Hence, alternative approaches are being developed, as further described below.
THE CURRENT GENE THERAPY SCENARIO IN THE EAR
In the ear, nonclinical data generated over the past decade demonstrate the potential to restore physiologic, high-acuity hearing in mHL, including some of the most common forms of congenital deafness. The most extensive and compelling nonclinical data to date are from mouse studies of gene replacement for OTOF-mediated hearing loss (ARO 2023 presentation—
OTOF-mediated hearing loss, also referred to as DFNB9, is a nonsyndromic condition typically identified in newborn hearing screening, with definitive diagnosis upon genetic confirmation. The physiologic defect that results from OTOF mutations is localized to the inner hair cells (IHCs) of the cochlea. Without functional OTOF protein, synaptic transmission between the IHCs and cochlear neurons is impaired, resulting in deafness. An important aspect of the OTOF-mediated hearing loss phenotype, from a gene therapy perspective, is that inner ear development and structural integrity are preserved in the absence of OTOF protein.
23
As a result, successful delivery of OTOF to IHCs has the potential to restore physiologic, high-acuity hearing in individuals born with profound hearing loss. Indeed, this has been demonstrated in multiple mouse models of OTOF-mediated hearing loss (ARO 2023 presentation—
These cases of auditory function restoration are especially impressive considering they successfully employ the use of dual-AAV gene replacement strategies. The OTOF protein is encoded by a 6 kb complementary DNA, which, as mentioned above, cannot be packaged in a single AAV vector. Previous work in multiple laboratories has shown that genes exceeding the AAV capacity can be delivered via dual-AAV vector methods, as further described below (Fig. 3B). OTOF-mediated hearing loss is the first inner ear condition to demonstrate the ability of the dual-AAV gene replacement strategy to restore function in vivo using a clinically relevant outcome measure.
That the first investigational gene therapies to begin clinical trials for any monogenic inner ear condition are targeting OTOF-mediated hearing loss, despite the challenges of OTOF's larger size, also highlights the importance of clear target biology, effective delivery, and strong animal models that recapitulate the human phenotype, in selecting target genes for inner ear gene therapy (see Hickox et al. 24 ). AAV gene therapy has been investigated in preclinical mouse studies targeting several other forms of mHL. These include Usher syndrome type 3A, and GJB2-mediated hearing loss, among others, and recent reviews have provided an overview of nonclinical gene therapy approaches investigated to date. 4,25 –27 In addition to the work addressing various forms of mHL, there are now multiple preclinical efforts ongoing to advance gene therapies for inner ear conditions that have a more complex etiology leveraging AAV as an efficient, one-time delivery vehicle. 28,29
Translation of inner ear therapies from early preclinical mouse studies to human therapies will require thorough understanding and further development of appropriate delivery, having historically posed some of the biggest challenges for inner ear gene therapy, as further discussed below.
GENE THERAPY BEYOND CANONICAL GENE REPLACEMENT
Besides canonical gene replacement, the current era of precision medicine is fueling the growth of several other genetic medicines. Indeed, for many diseases, gene replacement does not represent a straightforward option. First, in some autosomal dominant forms of inherited diseases, which account for about 15–20% of all IRDs 30 and 15% of nonsyndromic hearing loss cases, 31 gain-of-function or dominant-negative mutations in specific proteins result in toxic effects; in such cases, delivery of a correct copy of the mutated gene is ineffective, and silencing of the dominant allele is instead required. Second, several of the genes causative of IRDs and mHL exceed the packaging capacity of AAV vectors.
One approach that has gained increasing interest for overcoming both of the abovementioned limitations is RNA-targeting therapy, including RNA interfering-related approaches and antisense oligonucleotides (AONs). 1,32 Several RNA-targeting therapies are being investigated for IRDs, including both small-interfering RNA- and AON-based approaches. 32
Those in the most advanced phases of development are AONs and depending on the disease targeted rely on different mechanisms of actions 33 (Fig. 3C), including the following: (1) masking cryptic splice sites induced by mutations, to restore correct pre-mRNA splicing, as in the most common mutation in the CEP290 gene (c.2991 + 1655A>G), causative of LCA10; (2) masking of natural splicing signals to induce in-frame skipping of specific natural exons, which are mutational hotspots, to result in the production of shorter but still functional proteins, as in the case of exon 13 of the USH2A gene, whose mutations are among the most common causes of Usher syndrome type 2A; and (3) RNase H1-mediated knockdown of mRNA, including gain-of-function mutations, as is being explored for autosomal dominant RP due to common P23H mutation in the rhodopsin (RHO) gene.
Sepofarsen, targeting the c.2991 + 1655A>G mutation in CEP290 for treatment of LCA10, was the first in vivo application in the retina of AON, notably showing in phase I/II studies (NCT03140969) no severe side effects, improved vision at 3 months, and sustained visual gain at 15 months in one patient. However, the phase II/III trials (NCT04855045 and NCT03913143) did not meet the primary endpoint of improvement in best-corrected visual acuity or other secondary endpoints, despite post hoc analyses revealing a beneficial effect when comparing sepofarsen-treated eyes against the sham group if adjustments versus the contralateral eyes were considered. 34
While further studies are needed to conclusively establish the effectiveness of AON, the complex array of genetic mutations within each type and subtype of IRD brings an inherent challenge to the development of AON-based therapies for genetic eye diseases, given the sequence-specific nature of this approach. In addition, due to the temporary nature of AONs, repeated administration is required for sustained effect. This represents a major limitation for tissues where repeated injections are impractical or infeasible, such as the inner ear.
In the ear, promising nonclinical data in mice demonstrate the potential for AAV-mediated RNA-interference approaches to address autosomal dominant hearing loss. In one example, Smith and colleagues 35 assessed a mutation-agnostic approach targeting TMC1-mediated hearing loss. Mutations in TMC1 include both loss- and gain-of-function variants. In an effort to address all potential pathogenic variants of TMC1, researchers in the Smith laboratory delivered a single AAV vector encoding a microRNA to knock down both wild-type and mutant alleles, along with an engineered, knockdown-resistant Tmc1 gene. Using this simultaneous “knockdown plus replacement” approach, researchers were able to achieve robust and durable auditory brainstem response preservation across multiple auditory frequencies in mature mice.
Precise genome editing tools, such as CRISPR-Cas9, are emerging as an alternative to canonical gene therapy approaches. 36 Direct editing of mutated genes represents a valid approach to both tackle diseases due to mutations in genes whose CDS exceeds the AAV cargo capacity, and those resulting from gain-of-function mutations. In addition, many genetic diseases result from genes encoding proteins whose expression must be tightly regulated to function properly. In these, overexpression exceeding normal physiologic levels from strong promoters included in gene therapy vectors can lead to cytotoxicity. In these scenarios, use of genome editing tools may therefore be an appealing therapeutic possibility.
Yet, effective in vivo delivery represents an important hurdle to widespread clinical translation of CRISPR-Cas therapeutics as, most commonly, the sizes of CRISPR-Cas components require the use of two separate AAV vectors (i.e., one vector including Cas endonuclease expression cassette, while the other the gRNA expression cassette and, eventually, a repair donor DNA template). Both identification of Cas9 orthologs with reduced size, 37 –39 which allows packaging of both the Cas endonuclease and gRNA expression cassette in a single vector, and further development in delivery vehicles, including nonviral vector-based approaches, are actively ongoing to overcome this limitation.
Autosomal dominant RP due to mutations in RHO (RP4) has been largely used as a model for testing genome editing efficiency in the retina, with more than 100 dominant mutations reported in the RHO gene being amenable to Cas9-based treatments. 2 Cas9-based approaches were tested for the first time in the retina in 2016. 40 Initial efforts were focused on the development of mutation-specific approaches, relying on Cas9-mediated knockout of the mutant dominant allele, to reduce its toxic effect. 41 However, the high allelic heterogeneity of RP4 makes it impractical to develop mutation-specific genome editing approaches for each mutation. Consequently, mutation-independent methods have been tested, which rely on disruption of both alleles and concomitant delivery of a healthy copy of the gene. 42
However, this strategy presents several challenges, including the capability to reach appropriate levels of RHO replacement, as both haploinsufficiency can manifest if suboptimal levels of RHO expression are achieved and toxic effects if RHO is overexpressed. 43,44 Furthermore, if the CRISPR-Cas9 cut is in a coding region, the exogenous RHO replacement template should be modified to prevent its Cas9-mediated cut. Direct restoration of RHO wild-type sequence at the endogenous locus via Cas9-mediated editing would therefore be an ideal option. However, homologous recombination, which is usually explored to obtain precise genomic changes via integration of donor DNAs, only works in dividing cells. Hence, it is not a viable mechanism to induce DNA repair in postmitotic cells, as retinal photoreceptors affected in RP4.
As an alternative, both the nonhomologous end-joining and microhomology-mediated end-joining repair pathways have been used to achieve targeted integration of repair donor DNAs in the retina, achieving structural and function improvements in animal models of IRDs, including RP4. 45 –47
Correction of IRDs due to mutations in large genes represents the most advanced example of a Cas9-based therapeutic application in the retina, thus far. Mutations in CEP290 (CDS: 7.4 kb) are among the most frequent causes of LCA identified. As described above, the most common mutation in CEP290, c.2991 + 1655A>G, is a point mutation located within an intron that creates a novel splice donor site, resulting in the inclusion of 128 bp and creating a premature stop codon. In 2020, a clinical trial sponsored by Editas Medicine (NCT03872479) for LCA10, which explores a CRISPR-Cas9-based approach to specifically target this mutation by using two specific gRNAs to excise the abnormal splice donor generated by the mutation, 48 became the very first investigation of CRISPR-Cas9 safety in humans.
However, despite a good safety profile and preliminary efficacy signals in some participants, the trial has been paused (
CRISPR-Cas9 editing in the inner ear has also resulted in encouraging data spanning several gene targets. 2 Two studies targeting mouse Tmc1, one using a liposome vector 49 and other using AAVAnc80, 50 demonstrated partial preservation of hearing. The AAVAnc80-mediated delivery resulted in more durable preservation than the liposome approach, which may be due to more effective hair cell transduction by the AAV vector. CRISPR-Cas9 gene editing has also resulted in promising results in mouse models of KCNQ4-mediated hearing loss (also known as DFNA2A) 51,52 and PCDH15-mediated hearing loss (DFNB23). 53
Innovative Cas-based tools, such as base and prime editors (Fig. 3D), are also emerging as additional genetic medicine options, which can be used to induce precise mutations without generating double-strand breaks. Preliminary proof-of-concept studies both in the retina and inner ear have confirmed the potential of these strategies for applications targeting these tissues. 2,54 –58 However, despite interesting characteristics, these editing machineries represent an even greater challenge for in vivo delivery, as Cas proteins fused to effector domains are too large to be packaged in their entirety within AAV. To overcome this issue, dual AAV strategies, in which Cas fusion proteins are split into two AAV vectors and fused posttranslationally, 59 as described in the following section, as well as compact editor versions that can be delivered in a single AAV, 60,61 have been developed.
However, the critical issue of the vectorization of these platforms for in vivo delivery, as well as the high allelic heterogeneity of inherited forms of vision and hearing loss, has thus far left these approaches in a less advanced stage of development toward the clinic compared with Cas nuclease-based approaches.
CHALLENGES TO THE USE OF GENE THERAPY FOR EYE AND EAR DISORDERS
Effective targeting
As mentioned earlier, subretinal injections are the preferred way to transduce photoreceptors and RPE. Indeed, although intravitreal injections result in a pan-retinal distribution of vector, transduction is mostly limited to the ganglion cells and inner retinal cells. In addition, there is a higher probability of immune response due to the presence of immune cells in the vitreous. 13 However, subretinal injection releases the gene therapy product only in a limited region of the retina and is more invasive than an intravitreal injection, since it requires transient detachment of the RPE from the underlying photoreceptors (Fig. 1A). This makes it a less attractive option in diseases where the retina is already rendered fragile by ongoing degenerative processes. Therefore, identification of engineered AAV variants with better transduction profiles upon intravitreal delivery and the refinement of surgical techniques to avoid retinal trauma are crucial steps for improving clinical translatability of gene therapies for IRDs.
For translation of inner ear therapies from early preclinical mouse studies to human therapies, delivery is also among the most important development considerations. Because there are currently no drugs, biologics, or fluids approved for administration to the inner ear in most geographies around the world, the development of inner ear gene therapy is reliant on a novel route of administration. Although inner ear gene therapy in mouse models has been attempted via delivery to the endolymphatic and perilymphatic spaces (see Petit et al. 4 for recent review), the prevailing view of otologists and other auditory specialists is that delivery to the endolymphatic space requires disruption of the sensory epithelium's structural integrity, and intracochlear administration via the perilymph is more likely to be safely translated to humans.
Indeed, all intracochlear deliveries previously and currently being evaluated in human clinical trials rely on the delivery of viral vector to the perilymph.
The intracochlear route of administration to the perilymph can consist of several surgical delivery approaches and can be designed to allow for targeted local delivery of vector across the entire frequency range of the cochlear epithelium. To achieve this, a vent hole at a distal point of the perilymphatic space relative to the administration site provides an outlet for displaced perilymph during intracochlear infusion, avoiding a concentration gradient with relatively high levels of vector proximal to the administration site, and potentially subtherapeutic levels at regions further from the administration site. The location of vent hole is, in turn, dependent on other aspects of the surgical approach. A vent hole in the stapes footplate has been shown to enable nearly 100% transduction of hair cells across the entire auditory frequency range in nonhuman primates.
Fenestration of the stapes footplate is well established in clinical practice, can be achieved in association with a minimally invasive transcanal approach (Fig. 1B) and, as a result, is potentially conducive to future applications in more prevalent inner ear conditions, for which bilateral administration with potential for reduced anesthesia will be important to achieving broadest access for individuals who can benefit. Alternatively, if the more invasive transmastoid surgical procedure via facial recess approach is selected rather than the transcanal approach, the stapes footplate is not accessible for fenestration by the surgeon. For intracochlear AAV administration, the transmastoid approach can be accompanied by a vent hole in the semicircular canal (Fig. 1B). Ongoing and future clinical trials will inform the safety, efficacy, and scalability of these approaches.
Delivery to the inner ear compartment pairs considerations at the surgical level together with those at the cellular and molecular levels. Cellular and molecular delivery is informed by data on the type and percentage of cells that express the transgene of interest at various vector doses. In the case of OTOF-mediated hearing loss, the goal of gene therapy is to restore hearing by achieving sufficient expression of full-length OTOF protein in the IHCs. Although transgene expression in as few as 20% of IHCs has been shown to restore auditory function in mouse models (ARO 2023 presentation—
Multiple independent studies with several different capsids have shown that even broadly tropic capsids with ubiquitous promoters result in OTOF protein expression that is observed only in the IHCs; data also show that IHCs can tolerate overexpression of OTOF to levels that exceed wild-type expression levels (by at least 150% of wild-type expression levels). 21 In mouse models, durable restoration of auditory function to wild-type levels has been demonstrated in these cases, indicating that IHCs can tolerate OTOF overexpression well above wild-type levels. Translation of these early results to hearing restoration in humans will be dependent on the selection of dose and delivery approach.
In the case of GJB2-mediated hearing loss, multiple independent studies suggest that connexin 26 (Cx26), the protein encoded by GJB2, is not well tolerated when expressed in IHCs, highlighting that the need for and degree of cell-type selectivity are highly transgene dependent. Furthermore, the precise supporting of cell populations that require continuous expression of Cx26 to maintain and potentially restore hearing in humans is not known to the same degree of confidence as with OTOF in IHCs for OTOF-mediated hearing loss. As a result, early efforts to advance potential gene therapies for GJB2-mediated hearing loss deploy a wide range of therapeutic strategies based on different delivery strategies, which combine the use of specific regulatory elements and/or AAV capsids, and including editing approaches, to avoid expression in hair cells.
Delivery of large genes
As discussed above, AAVs are the currently preferred vectors for retinal and inner ear gene therapy. The small size of AAV vectors favors efficient diffusion of the particles across tissues and physical barriers, therefore providing high levels of transduction. However, the small particle size also represents one of the main drawbacks of AAV, as it limits its applicability to expression cassettes <5 kb. Thus, considerable efforts have been made to identify strategies to expand AAV transfer capacity.
The strategy that has been most extensively studied and tested over the past years is to coadminister two or more AAV vectors that carry separate parts of a gene (Fig. 3B). Joining of the separate parts at the DNA (dual AAV vectors), pre-mRNA (mRNA trans-splicing), or protein level (AAV inteins) is then explored to reconstitute the expression of the large therapeutic protein (see Tornabene and Trapani 62 for review). Each of these strategies has some advantages as well as drawbacks for successful application, which have been reviewed in detail elsewhere. 62 Overall, the several studies that have exploited these strategies have highlighted that there is not one that fits every need. Dual AAV vectors based on DNA recombination have an easier design, and, as such, have been successfully applied to deliver different large genes causative of IRDs and/or inner ear conditions. As a confirmation of the efficiency of the platform, and as mentioned above, the first AAV gene therapies to enter clinical development for inherited forms of hearing loss are deploying dual AAV-mediated gene replacement strategies to treat OTOF-mediated hearing loss.
These first clinical trials are now beginning to enroll participants, and initial clinical data are expected in the coming months. Similarly, a phase I/II clinical trial testing the safety and efficacy of dual AAV for delivery of the large MYO7A gene in the retina of Usher 1B patients is planned. 16
Alternative strategies relying on reconstitution at the protein level, via the process of protein trans-splicing (PTS) mediated by inteins, 63 have shown higher efficiency of reconstitution compared with other platforms in side-by-side comparisons in the retina. However, the design of AAV-intein constructs can be challenging, making some proteins more amenable than others to reconstitution via PTS-based approaches. Therefore, while several different strategies for AAV-mediated large gene reconstitution are currently available, the choice of which to explore will most probably depend on the protein to be reconstituted, the target cells, and the nature of the disease. Further data on the safety and efficacy of these platforms, which are expected in the near future from the planned clinical trials, will allow to finally define whether the levels of expression achieved are therapeutically relevant in humans.
Intervention windows and disease progression
The intervention window at which gene therapy is applied is also an important consideration for gene therapies beginning clinical development. Indeed, for conditions caused by mutations in genes with crucial roles in organ development, postnatal intervention might be unable to restore the structure or function needed for therapeutic benefit. Also, while gene addition strategies aim to halt/slow down the rate of degeneration of target cells, during the later stages of the diseases the damage to the structures may be irreversible, rendering gene therapy futile. 4,5
To address this issue, several strategies are being developed. In the eye, gene therapy is being explored to convert the surviving retinal neurons into photosensitive cells via optogenetic therapy. 1,64,65 Optogenetics is a mutation-independent approach in which light-sensitive proteins are expressed in the remaining nonlight-sensitive neurons (most commonly ganglion or bipolar cells) in the inner retina, to confer them sensitivity to light. Microbial opsins (as the light-gated ion channel channelrhodopsin-2, ChR2) are historically the first tools developed for optogenetic purposes, 66 and variants with improved characteristics (the fast gating Chrimson mutant, ChrimsonR) have successfully reached the clinical stage, showing successful outcomes in an RP patient (NCT03326336). 67
However, limitations of this platform are both the slow kinetics and the low light sensitivity of the microbial-derived opsins initially used for optogenetic purposes. 1 These do not operate at physiological light levels and require light intensities that are provided through an external light-amplifying device (goggles), making the system more complex and potentially toxic in an already degenerated retina. 5 In addition, microbial opsins are not native mammalian proteins, raising the possibility of adverse immune responses. 65 Novel nonmicrobial optogenetic tools with improved light sensitivity, including vertebrate opsins coupled to signal amplification cascades (such as rhodopsin, cone opsin, and melanopsin) and genetically engineered photoswitches, have been developed to overcome these limitations and are being tested in additional clinical trials (Fig. 2A).
Another main challenge of optogenetics for vision restoration is the efficient targeting of the outermost surviving retinal cells, before major remodeling of the retina occurs due to the degenerative process. This would allow to reproduce the most “natural” processing of images, which normally starts in photoreceptors and is then integrated by bipolar cells and passed to ganglion cells. Sensitization of ganglion or bipolar cells bypasses several of the signaling steps. In the near future, targeting of “dormant cones” (i.e., residual cones found in the fovea of RP patients, which have intact cell bodies but are devoid of the light-sensitive outer segments, becoming insensitive to light) with more sensitive G protein-coupled opsins could lead to better visual outcomes. 64
Lastly, application of optogenetics in clinical practice requires the establishment of efficient, cell-specific, and safe means for ectopic opsin expression, which is dependent on further development of both cell-specific promoters and potent vectors and efficient delivery routes.
The inner ear begins forming early in fetal development, and in humans is typically fully functional before birth. This confers advantages for the potential treatment of some forms of hearing loss, as it allows for administration earlier in infancy; because the human inner ear is fully developed at birth, an intracochlear administration is feasible within the first year of life, when inner ear integrity and central neural plasticity are highest, providing opportunity for greatest benefit. However, a challenge associated with this early development is that mutations in genes important to the development and maturation of the inner ear can lead to loss of sensory cells and degradation of cellular architecture. This loss of cellular substrate reduces the potential for gene therapy in these individuals.
For genetic hearing loss that involves genes required for cochlear development, successful therapeutic approaches may require in utero delivery approaches that have been explored in mouse models 68 but not yet translated to human use. Alternatively, the pairing of regenerative medicine approaches, currently being advanced by multiple laboratories in academia and industry, with gene replacement using intracochlear delivery approaches currently being investigated in clinical trials could open future opportunities to address all forms of sensorineural hearing loss, regardless of etiology.
CONCLUSIONS AND FUTURE OUTLOOKS
The last decades have been characterized by an extensive development of gene therapy-based approaches for treatment of hundreds of acquired and inherited diseases, including inherited forms of vision and hearing loss. The availability of AAV as a safe and effective vector for transducing target cells in the retina and inner ear has fueled such development. However, the heterogeneity of both IRDs and mHL complicates the development of therapeutics, as the gene therapy approach needs to be adapted based on the specific challenges each specific disease/target gene posseses. Further technological improvements are therefore required to expand gene therapy applicability to a growing number of diseases.
Strategies to optimize delivery vehicles are focusing on both engineering of AAV vectors to increase their efficiency and/or reduce immunogenicity, and identification of more efficient nonviral vectors. Nonviral vectors are receiving increasing interest for their potential to deliver large genes, while avoiding introduction of viral components in target cells, having the possibility to change the gene therapy paradigm. However, the current transduction efficiency of nonviral vectors in both the retina and the inner ear is still low, limiting their applicability for treatment of hereditary forms of vision and hearing loss. Therefore, gene therapy approaches for treatment of diseases due to mutations in large genes mainly focus on developments of AAV vectors and the recently described strategies relying on co-administration of multiple AAV vectors, which have achieved therapeutically relevant levels of protein expression in several models of IRDs and hearing loss, and are now in clinical investigation.
The advent of the CRISPR/Cas9 genome editing tool has also reshaped the way genetic diseases, including those affecting the retina and inner ear, can be approached. However, current limitations of the CRISPR/Cas9 system include safety concerns related to prolonged expression of the gene-editing components, which could potentially result in genotoxicity, toxicity associated with off-target effect and immune responses against microbial Cas proteins, as reports have shown humoral and cell-mediated adaptive immune responses against Cas9 derived from Streptococcus pyogenes and Staphylococcus aureus in humans. 69 –71 Therefore, further developments in both editing tools and delivery technologies are required before Cas-based technology becomes a clinical reality for many retinal and inner ear diseases.
In the eye, gene-independent strategies, as those based on optogenetic tools (NCT02556736, NCT03326336, NCT04919473, NCT04945772, NCT05417126) or molecules with the potential to prolong photoreceptor cell survival in a gene agnostic manner (NCT05203939, NCT05748873), have also reached the clinical stage (Fig. 2B). While the gene agnostic nature of these approaches would represent a major advantage given the heterogeneous nature of IRDs, the sophisticated mechanism of action of these strategies complicates their design, potentially impacting their efficacy. The results of the ongoing clinical trials will define the efficacy of these approaches in humans.
In conclusion, despite the number of challenges that still need to be overcome, the actively ongoing research around all these technological improvements provides optimism that new therapeutic avenues will be made available to patients affected by many retinal and inner ear diseases in the coming years.
Footnotes
ACKNOWLEDGMENTS
Figures were created with BioRender.com.
AUTHORs' CONTRIBUTIONS
I.T. developed the concept of the article, defined the structural content, and used contributions to write the original draft. E.J.S. and I.T. analyzed the underlying literature, contributed to the original draft, edited and approved the final version of the article.
AUTHOR DISCLOSURE
I.T. is coinventor on patents related to both dual AAV and AAV intein vectors. E.J.S. is a cofounder of Akouos, Inc., a wholly owned subsidiary of Eli Lilly and Company, and coinventor on patents related to inner ear gene therapy.
FUNDING INFORMATION
I.T. reports grant from Fondazione Telethon (grant number: TGM22MT02), the Foundation Fighting Blindness (grant number: TA-GT-0920-0791-FED), and the Italian Ministry of Research (MUR) under the EJP RD program (grant number: DEC 790).