
Abstract
Advanced gene transfer technologies and profound immunological insights have enabled substantial increases in the efficacy of anticancer adoptive cellular therapy (ACT). In recent years, the U.S. Food and Drug Administration and European Medicines Agency have approved six engineered T cell therapeutic products, all chimeric antigen receptor-engineered T cells directed against B cell malignancies. Despite encouraging clinical results, engineered T cell therapy is still constrained by challenges, which could be addressed by genome editing. As RNA-guided Clustered Regularly Interspaced Short Palindromic Repeats technology passes its 10-year anniversary, we review emerging applications of genome editing approaches designed to (1) overcome resistance to therapy, including cancer immune evasion mechanisms; (2) avoid unwanted immune reactions related to allogeneic T cell products; (3) increase fitness, expansion capacity, persistence, and potency of engineered T cells, while preserving their safety profile; and (4) improve the ability of therapeutic cells to resist immunosuppressive signals active in the tumor microenvironment. Overall, these innovative approaches should widen the safe and effective use of ACT to larger number of patients affected by cancer.
INTRODUCTION
Among therapeutic options available to increase the efficacy of anticancer T cells, genetic engineering can deliver intrinsic antineoplastic responses while simultaneously increasing therapeutic benefit through next-generation genome editing. The original concept, developed in the late 1980s, aimed to confer cancer cell recognition and killing by autologous or allogeneic T cells. Early pioneering approaches of adoptive T cell transfer relied on naturally occurring autologous tumor-infiltrating lymphocytes (TILs) obtained from resected solid tumors.
TILs include T cells capable of recognizing tumor-associated antigens, mainly those resulting from tumor-specific mutations or “neoantigens.” These T cells can be selected and expanded ex vivo, and reinfused into patients after lymphodepletion to create a favorable niche for their expansion. 1 TIL T cell therapy proved particularly effective against metastatic melanoma. Because TILs recognize the products of cancer mutations, identification of neoantigen-reactive T cells can now be facilitated by next-generation sequencing, easing their stimulation and selection before infusion into patients with solid tumors. 2 –4
In the allogeneic setting, cellular therapies against relapsed hematological malignancies first used donor lymphocyte infusions (DLIs) after allogeneic hematopoietic stem cell transplantation (allo-SCT), to exploit the alloreactive graft versus leukemia effect and to enhance post-transplant immune reconstitution. DLI has historically been useful against chronic myelogenous leukemia but has been less effective against other hematological malignancies. 5 –7
With the advent of cell engineering, antitumor T cells have been taken to the next level, eventually proving effective against difficult-to-treat malignancies and revolutionizing the field of cancer immunotherapy. 8 By using viral vectors, T cells have been genetically engineered for enhanced persistence, 9 improved safety, 10 and, more recently, to express an artificial receptor that provides tumor-specific antigen recognition. Isolation and sequencing of T cell receptors (TCRs) specific for tumor-specific or tissue-restricted antigens enable the design and cloning of artificial TCRs.
Adoptive transfer of TCR-engineered T cells avoids the challenges associated with expanding TILs in vitro and is currently being applied to target multiple tumor types, including melanoma, synovial sarcoma, acute myeloid leukemia (AML), and myeloma. 11 To circumvent human leukocyte antigen (HLA) restriction, chimeric antigen receptor (CAR) molecules have been designed for both cellular and humoral immunity, by combining the antigen-recognition ability of antibodies with the effector functions of immune cells. Engagement of an antibody single chain fragment variable (ScFv) region redirects T cell antigen specificity to surface molecules expressed by the tumor.
The first generation of CAR T cells included only the CD3ζ chain as the intracellular signaling domain 12 and proved safe but with limited persistence. 13 The use of CAR constructs based on ScFv linked to the signaling domain of the Fc receptor γ chain was also explored, resulting in suboptimal efficacy. 14 Costimulation domains added to subsequent generations of CAR T cells improved T cell function, proliferation, and persistence, producing remarkable and long-lasting clinical responses against B cell malignancies. 15 –18
Since 2017, the U.S. Food and Drug Administration and European Medicines Agency have approved six CAR T cell products, including four against CD19 expressed in B cell lymphoma 19 –22 and acute lymphoblastic leukemia, 23 and two anti-B-cell maturation antigen CARs against multiple myeloma. 24,25 We are now witnessing a progressive shift toward approved adoptive cellular therapies (ACTs) performed in earlier lines of treatment. In addition, we are also seeing the emergence of therapies aimed at new targets with evidence of clinical efficacy not only for liquid but also for solid tumors. 26
Current research is focused on using advanced engineering approaches to increase efficacy and overcome mechanisms of resistance to engineered T cells. Although efficient transgene insertion has commonly been achieved using viral vectors, nonviral technologies are only recently emerging as sustainable alternatives. 27 –29 In this context, genome editing is progressively gaining momentum as the choice technology to precisely introduce artificial receptors and replace endogenous TCRs to facilitate applications in allogeneic settings. In addition, genome editing is being exploited to generate engineered T cells with increased potency, persistence, and resistance to inhibitory signals and fratricidal activity (Fig. 1).
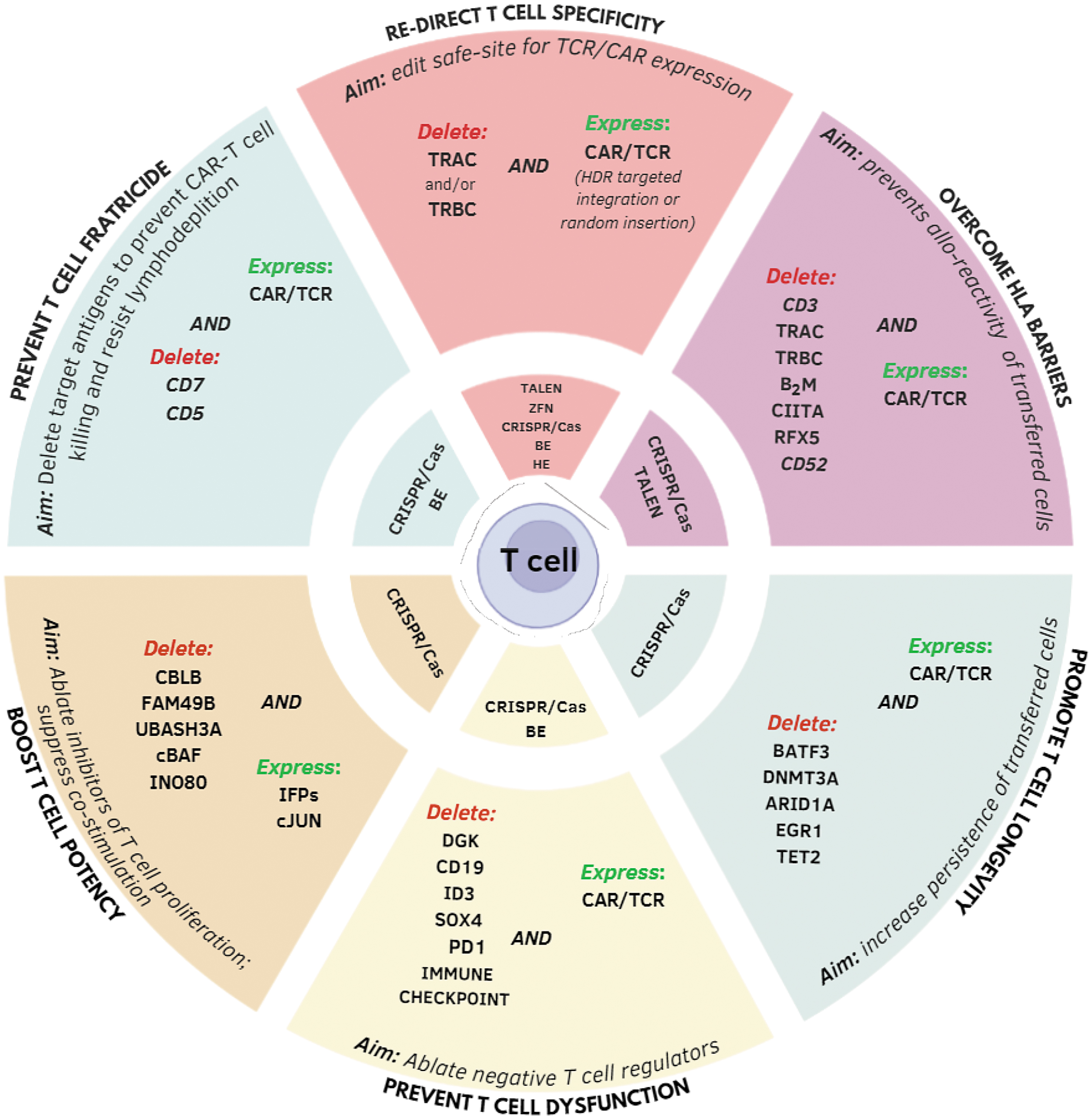
Genome editing applied to T cells for cancer immunotherapy: applications. TALENs, HE, ZFNs, and CRISPR/Cas9 platforms, including BE system can be used to obtain permanent gene disruption and/or to promote targeted integration of a selected transgene in a preselected genomic region. A summary of some of the genes edited to date in the context of adoptive T cell therapy is reported, together with the specific nuclease system used. ARID1A, AT-Rich Interaction Domain 1A; B2m, B2-microglobulin; BATF3, basic leucine zipper ATF-like transcription factor 3; BEs, base editors; CAR, chimeric antigen receptor; cBAF, canonical BAF complex; CBLC, Cbl Proto-Oncogene C; CIITA, Class II Major Histocompatibility Complex Transactivator; DGK, diacylglycerol kinase; DNMT3A, DNA methyltransferase 3 alpha; EGR1, early growth response protein 1; FAM49B, family with sequence similarity 49, member B; HE, homing endonucleases; ID3, inhibitor of DNA binding 3; IFPs, immunomodulatory fusion proteins; INO80, chromatin-remodeling ATPase INO80; RFX5, regulator factor X5; SOX4, SRY-box transcription factor 4; TALENs, transcription activator-like effector nucleases; TCR, T cell receptor; TET2, tet methylcytosine dioxygenase 2; UBASH3A, Ubiquitin Associated and SH3 Domain Containing A; ZFNs, zinc finger nucleases.
As we recently celebrated the 10-year anniversary of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology, we now critically review the results and emerging applications of T cell genome editing for cancer immunotherapy, including progress to date, clinical trials (Table 1) and challenges for the future.
Completed and active clinical trials with genome-edited T cells
AAV, adeno-associated virus; B2M, B2-microglobulin; BCMA, B-cell maturation antigen; BEs, base editors; CAR, chimeric antigen receptor; CCR5, C–C chemokine receptor type 5; CISH, cytokine inducible SH2 containing protein; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; EGFR, epidermal growth factor receptor; GOSH, Great Ormond Street Hospital; HE, homing endonuclease; HLA-E, human leukocyte antigen E; HPK1, hematopoietic progenitor kinase 1; LV, lentiviral vector; M/R, myxoid and round; mRNA, messenger RNA; MUC1, mucin 1; n.a., not available; NY-ESO-1, New York esophageal squamous cell carcinoma 1 antigen; PD-1, programmed cell death protein 1; PDCD1, programmed cell death 1 gene; R/R, relapsed and refractory; RNP, ribonucleoprotein; sgRNA, single guide RNA; SLAMF7 (alias CS1), signaling lymphocytic activation molecule 7; START, synthetic TCR and antigen receptor; TALEN, transcription activator-like effector nucleases; TCR, T cell receptor; TGF-βRII, TGF-β receptor II; TRAC, TCRα constant chain; TRBC, TCRβ constant chain; WT-1, Wilms tumor antigen 1; ZFNs, zinc finger nucleases.
GENOME EDITING PLATFORMS APPLIED TO T CELLS
The T cell genome editing tools most widely tested in human studies can be broadly divided into those with protein-based DNA recognition moieties, fused to endonuclease domains, and RNA-guided nuclease enzymes that are designed to create highly specific double-strand DNA (dsDNA) breaks. Subsequent repair by nonhomologous end joining can efficiently disrupt coding sequences by creating gene knockout effects, such as insertions and deletions (indels). Alternatively, using a suitable DNA template for repair by homologous recombination enables an opportunity for site-specific gene insertions.
Replacing nuclease activity with alternative enzymes, including deaminases for nucleotide conversion, and reverse transcriptase for localized generation of repair template DNA, is providing an ever increasing toolbox for T cell engineering. These developments have evolved alongside techniques for delivery of molecular tools into T cells, including viral and nonviral platforms, as well as strategies for using autologous and allogeneic starting populations. The earliest reported experimental knockout studies in humans used zinc finger nucleases (ZFNs), which comprised a Fok1 DNA cleaving domain and dual DNA-binding finger-like modules for targeted DNA binding.
Autologous T cells from subjects with HIV infections were edited with adenoviral vector-delivered ZFN to disrupt the C–C chemokine receptor type 5 (CCR5) virus entry receptor. 30 ZFN enabled exploitation for the first time of homologous recombination for targeted transgene integration in primary T cells, 31 and reprogramming of T cell specificity through TCR gene editing. 32
Subsequently, transcription activator-like effector nucleases (TALENs) were explored. TALEN are delivered as messenger RNA (mRNA) by electroporation and could incorporate longer programmable DNA-binding domains for high specificity to the target site. TALENs were used for highly efficient disruption of the TCRα constant chain (TRAC) gene to prevent graft versus host disease (GVHD), and knockout of CD52 to evade lymphodepletion by the CD52-binding monoclonal antibody (alemtuzumab), in the context of a universal allogeneic, CAR19-targeting CAR therapy. 33
Similar products were generated using homing endonucleases (meganucleases), integrating a CAR19 template into the TRAC locus that was delivered through adeno-associated virus (AAV). 34 Over the past decade, RNA-guided CRISPR/Cas9 technology 35 –38 has been widely translated from the laboratory 39 –42 to clinical T cell studies, that is, for programmed cell death protein 1 (PD1) disruption in recombinant TCR- 43 and CAR-redirected T cells. 44,45 The platforms rely on a carefully designed single guide RNA (sgRNA) sequence to direct Cas9 (or related enzymes) to specific DNA loci close to particular protospacer adjacent motif (PAM) sequences.
Improved electroporation systems, and the availability of highly purified recombinant Cas9 protein, sgRNAs and synthetic DNAs, have enabled nonviral guide-dependent CAR transgene insertions, at efficiencies suitable for clinical manufacture. 27 There are also hybrid delivery systems employing lentiviral delivery of a CAR transgene and guide RNAs for use in combination with Cas9 mRNA. 44 Targeted AAV-mediated delivery of CAR and nonpolymorphic HLA has been reported using CRISPR/Cas12a and chimeric RNA–DNA guides after multiplexed editing. 46
Thus there are now available numerous natural and synthetic Cas systems, with a variety of PAM dependencies and evermore stringent windows of operational activity. Although the CRISPR/Cas9 technology is bacterial in origin, CRISPR/Cas9-based ex vivo engineering strategies have not been particularly hampered by immunogenicity or toxic effects during T cell manufacture.
Additional advances have substituted Cas9 with deactivated derivatives that localize in a guide-dependent manner but operate in conjunction with deamination enzymes for highly targeted base conversion, including C > T and A > G editing within a specified distance from the PAM sequence. CRISPR-guided cytidine deamination base editors 47 have thus been used to introduce premature stop codons for knockout effects, 48 and targeted adenosine deamination 49 has been used to disrupt gene expression through modification of critical splice sites. These alternative platforms have the advantage of not causing dsDNA breaks and are ideal for multiplex editing where there is otherwise a risk of translocations and other chromosomal aberrations in T cells. 50 –52
Multiple base-editor variants have now been synthetically engineered, including highly efficient compact versions with narrow windows of activity. 53,54 A further alternative to nuclease-mediated DNA breakage and repair by homologous recombination has been prime editing, which incorporates an impaired Cas9 fused to a murine reverse transcriptase element and a prime editing guide RNA, enabling localized insertion, deletion, or base conversion without dsDNA breaks. 55
The platforms are still evolving; there are also untethered versions with split Cas and reverse transcriptase architecture. 56 For nonviral delivery of larger transgene cargos, or multicistronic cassettes, hybrid engineering platforms can support CRISPR-guided site-specific transposition. 57 –59 Finally, the transient expression of engineered transcriptional repressors, which bind at a target genetic locus, now enables mediation of repressive histone marks and DNA methylation for long-term targeted epigenome editing. 60
GENOME EDITING TO REDIRECT T CELL SPECIFICITY: TCR/CAR GENE EDITING
In general, T cells respond to a single antigenic epitope that is presented by an HLA molecule, by way of a specific TCR, which is a heterodimeric glycoprotein composed of an α and β chain associated with the CD3 complex. A critical step for effective ACT is the redirecting of T cells to recognize and kill tumor antigens. This aim can be achieved using tumor-specific TCRs isolated from patients or healthy donors, or with CARs.
One of the first applications of genome editing tools in cancer immunotherapy was TCR gene editing. 32 By combining permanent genetic disruption of the endogenous TCR repertoire with transfer and expression of a tumor-specific TCR, gene editing first enabled redirection of T cell specificity and overcame some of the limitations encountered with conventional gene transfer of a tumor-specific TCR into mature lymphocytes.
TCR gene editing avoids the dilution of a tumor-specific TCR with endogenous TCRs and maximizes the expression level of the transgenic TCR in T cells, thus promoting the generation of high-avidity cellular products. Furthermore, this procedure eliminates the risks of unwanted antigen recognition through mispairing between the tumor-specific and endogenous TCR chains. Overall, TCR gene editing proved superior to TCR gene transfer in vivo and in vitro. 32
Although targeted transgene integration can be achieved in T cells with ZFNs, 31 efficiency is low. Consequently, the first manufacturing protocols for TCR gene editing were based on sequential steps, including gene disruption and lentiviral-mediated TCR gene transfer. 32,61 The advent of highly efficient CRISPR/Cas9 editing has significantly increased the efficacy of gene disruption and of homology-directed repair, while also permitting simultaneous multiple gene editing. TCR-edited T cells proved safe in a pilot study of cancer patients. 43 Using CRISPR/Cas9, efficient orthotopic TCR gene replacement produced high levels of endogenously regulated TCR expression, similar to that observed with unmodified T cells, and with improved T cell function. 62 –64
TCR genetic disruption has been also applied to CAR T cell manufacture. In this setting, the TCR repertoire can be abrogated by disrupting either TCR α or β chain, and represents a major advance for generating allogeneic universal CAR T cells that are unlikely to mediate detrimental GVHD. Initially modeled with ZFNs, 65 TRAC-disrupted CAR T cells were then efficiently also obtained with TALENs. Qasim et al. were the first to report complete remissions in infants with B-cell acute lymphoblastic leukemia (B-ALL) infused with universal TALEN gene-edited CD19-redirected CAR T cells. 33
Similarly in the field of CAR T cells, the introduction of CRISPR/Cas9 allowed efficient targeted CAR gene integration. Eyquem et al. showed that the integration of a CD19-specific CAR into the TRAC locus of human peripheral blood T cells resulted in uniform CAR expression and enhanced T cell potency, possibly due to reduced tonic CAR signaling, effective internalization, and re-expression of the CAR after antigen encounter. 66
GENOME EDITING TO OVERCOME HLA BARRIERS
The majority of CAR T therapies to date have employed autologous T cells, but rely on complex harvest and manufacturing logistics, and poor-quality substrate cells from heavily pretreated patients. The use of donor-derived premanufactured T cells would address some of these limitations. A handful of studies using allogeneic T cells derived from HLA-matched hematopoietic stem cell donors showed feasibility. 67,68 A more ambitious option of generating banks of donor-derived T cells from nonmatched donors is now in development, but must address two major issues related to HLA barriers. First, the threshold of risk of GVHD from donor-derived cells is well characterized in the allo-SCT setting. 69,70
Second, host-mediated rejection of mismatched infused T cells mediated by host antibodies and cellular compartments, through recognition of HLA classes I and II molecules on incoming T cells. Both of these limitations can be addressed through genome editing approaches.
Genome editing to prevent alloreactivity
Disruption of either the TCR α or β chain, or the parts of the CD3 complex can be used to prevent cell surface assembly of a functional multimeric TCR. Residual T cells with TCRα or β expression can be further depleted using magnetic bead depletion systems, providing stringently enriched allogeneic T cell products that are unlikely to cause significant alloreactivity. Thresholds for how many TCRα or β T cells a product can carry without causing GVHD can be estimated from the mismatched SCT experience and is usually set at around 5 × 104/kg, 69,70 although this number may be variable for T cells that have been manipulated and cultured to express CARs.
For removal of TCRα or β from allogeneic CAR T cells, a number of studies have reported the use of TALENs in therapies for B-ALL, 33,71,72 lymphomas, 73,74 and AML 75 ; homing endonucleases for B cell malignancies 76 ; CRISPR/Cas9 for B cell 45,77 ; and T cell cancers 78,79 ; and base editing for T-cell acute lymphoblastic leukemia (T-ALL). 80,81 In these studies, GVHD has generally been absent or limited in severity, or readily managed with immunosuppression.
Genome editing to address host-mediated rejection
Host-mediated immune rejection is a major barrier to allogeneic immune effector cells, even in immunosuppressed patients. Circulating anti-HLA antibodies can meditate rapid clearance and generally contraindicate administering the products, if directed against donor HLA. In addition, cellular responses mediated by host lymphocytes can reject mismatched cells within a period of days or weeks. Lymphodepletion can limit such responses and, compared with the autologous setting, may need to be augmented with, for example, the inclusion of alemtuzumab as serotherapy targeting CD52 to provide an advantage to CD52 knockedout CAR T cells for a period of ∼4 weeks. 33,71,77
Other strategies propose conferring an advantage to infused cells by reducing the reactivity of host CD3+ T cells with anti-CD3 monoclonal therapy (while infused TCR/CD3-disrupted cells remain unaffected) 82 or disrupting deoxycytidine kinase in CAR T cells to confer fludarabine resistance. 83 Engineering approaches to express alloimmune defense receptors 84,85 have also been investigated for targeting host alloreactive cells. Clinical studies are underway to explore CD7-redirected CAR T cell-mediated lymphodepletion strategies for use in combination with CAR T cells targeting CD19 (CAR19). 86
Alternative approaches to directly remove HLA molecules on infused cells, targeting the conserved β2 microglobulin (B2m) domain of HLA class I molecules that interact with CD8 T cells, are also in early clinical phase testing. 87 For example, CRISPR/Cas9-edited universal site-specific CAR19 devoid of B2m has been used in patients with B cell non-Hodgkin lymphoma (B NHL), with 38% achieving remission without GVHD. 45 There had been concerns about triggering natural killer (NK)-mediated “missing-self” immunity against HLA class I negative cells, but it is unclear how relevant these effects may be, and mitigation strategies such as the inclusion of nonpolymorphic HLA-E are available, if required. 88
Also unknown is the extent to which additional removal of HLA class II, expressed on activated CD4 T cells, is required to avoid CD4 T cell-mediated responses. Class II molecules are highly polymorphic, without readily targeted conserved domains suitable for editing across multiple donors, but are dependent on the activity of critical transcription factors upon T cell activation.
CRISPR/Cas9 disruption of DNA-binding regulator factor X5 protein, part of a multifactor HLA control complex, has enabled the generation of universal anti-CD7 CAR T cell approaches that are now being studied. 72 These immune-stealth approaches are at early experimental stages, mindful of the heightened potential risks associated with creating viral reservoirs or bypassing immunological surveillance in the event of transformation conferred by cells depleted of all antigen-presenting pathways.
GENOME EDITING TO INCREASE PERSISTENCE OF CAR T CELLS
Increasing persistence of autologous and allogeneic engineered T cells
CAR T cell persistence has corroborative efficacy in several clinical trials. 89,90 Persistence may be affected by both intrinsic factors (e.g., fitness, exhaustion, and memory formation) and extrinsic factors (e.g., immune rejection and homing to lymphoid organs). Therefore, various attempts have been undertaken to increase the persistence of CAR T cells by modifying the manufacturing process, CAR design, or genetic-engineering approaches. In particular, specific T cell subpopulations have been associated with improved immune memory formation and long-term remission in patients, making premanufacture selection of T cell populations critical for optimal cell products 91,92 and in particular for CAR T cells. 93
CAR construct intracellular domain selection (e.g., CD28, 4-1BB) has been shown to influence the fate of transferred cells, such that sophisticated approaches to CAR design or even combinations of different intracellular CAR domains may better sustain persistence. 94 In addition, a genome-wide T cell screen identified exhaustion-specific factors that can also negatively affect T cell persistence; the specific deletion of these genes led to an improvement in human T cell persistence and tumor control in vitro and in vitro. 95
Also being considered is the overexpression of specific genes that have been identified as beneficial for memory formation and persistence. For example, the basic leucine zipper ATF-like transcription factor 3 (BATF3) is induced shortly after CD8+ T cell activation and has long-lasting effects on contraction, cellular/mitochondrial fitness, and longevity, and thus impacts CD8 T cell memory. BATF3 overexpression further enhanced T cell persistence in vitro, making it a suitable target for CAR T cell optimization. 96
In contrast to the TCR replacement strategy mentioned above, Stenger et al. have shown that the endogenous TCR may contribute to the persistence of CAR T cells. 97 However, for the use of “off-the-shelf” allogeneic T cells, it is mandatory to address extrinsic factors, such as immune rejection by deletion of the endogenous TCR. 98 The introduction of an alloimmune defense receptor, such as the 4-1BB extracellular domain, HLA-E/HLA-G, or sialic acid-binding immunoglobulin-type lectin (Siglec)-7/9, 84,88,99 was able to prevent cytolytic reactions against CAR T cells by endogenous T or NK cells.
In addition, triple knockout of B2m, Class II Major Histocompatibility Complex Transactivator, and TRAC did not induce GVHD in xenograft mouse models, but improved the persistence of CD19 CAR T cells and maintained antitumor efficacy. 100 In some cases, CAR T cells do not reach the targeted tumor site. Therefore, specific homing markers (e.g., CCR4 or CXCR1/CXCR2) have been identified and modified in allogeneic CAR T cells to enhance extravasation to either target tissues or lymphoid organs. 101,102
Increasing persistence of inducible pluripotent stem cell-derived engineered T cells
As another option for an “off-the-shelf” solution, inducible pluripotent stem cells (iPSCs) are considered promising in terms of scalability and efficiency. 103 iPSCs can be generated from a single donor and differentiated into any immune cell type. 104,105 In addition, arbitrary gene modifications can be introduced during the manufacturing process to provide a hyperindividualized solution for patients. However, the production and maintenance of an iPSC culture are a highly complex and labor-intensive process. The iPSC production systems are mainly based on animal cell products, resulting in high variability and the risk of species cross-contamination. 106
Furthermore, CAR T cell persistence is positively influenced by a balanced ratio of CD4 and CD8 CAR T cells, 107 but the majority of iPSC-derived CAR T cells correspond to a CD8+ phenotype. 108 The factors responsible for a CD4-specific lineage have been identified, but these production systems still rely on murine feeder systems, which precludes clinical application. 105 Regardless of limitations of the manufacturing process, extensive studies have already aimed to improve the persistence of iPSC-derived CAR T cells.
Deletion of the diacylglycerol (DAG)α and the DAGζ molecules, and introduction of a membrane-bound interleukin (IL)-15/IL-15ra fusion protein, improved proliferation and persistence of iPSC-derived CAR T cells in vitro. 109 The induction of modifications before phenotypic lineage induction also has potential for hypoimmunogenic CAR T cells. Wang et al. demonstrated in an in vitro model that disruption of CD155 expression in HLA-2, HLA-I, and HLA-II null iPSCs enabled iPSC-derived CD20 CAR T cells to largely escape NKG2A+ DNAM-1+ NK cell-mediated cytolysis, as well as CD8 and CD4 T cell-mediated cytolysis, paving the way for off-the-shelf hypoimmunogenic cell products for CAR T cell therapy. 110
PREVENTING T CELL DYSFUNCTION WITH GENOME EDITING APPROACHES
After antigen recognition, negative regulators of T cell activation are transiently upregulated, preventing T cells from becoming overly stimulated. However, chronic antigen exposure can lead to sustained overexpression of these negative regulators, leading to T cell exhaustion and/or dysfunction. 111 Significant effort has focused on preventing T cell dysfunction by inhibiting immunosuppressive signals in T cells.
As the PD1/programmed death ligand 1 (PD-L1) axis is considered key in driving T cell exhaustion, antibody-mediated blockade of the PD-1/PD-L1 axis has been combined with engineered T cells in preclinical studies and in clinical trials, with some signs of efficacy. 112,113 Antibody-mediated blockade of the PD-1/PD-L1 axis can control the timing and duration of inhibition, and allow the reinvigoration of neoantigen-specific tumor-infiltrating T cells. However, these therapies can be associated with loss of peripheral tolerance and severe toxicities.
An alternative is to use genome editing to ablate the gene encoding PD-1 (Pdcd1). This strategy has the advantage of specifically inhibiting the PD1–PD-L1 axis only in engineered tumor-specific T cells, perhaps mitigating toxicities related to autoreactivity. However, the consequences of long-term PD-1 elimination remain poorly understood, especially in the context of CAR T cells. Although some studies suggest that genetic absence of PD-1 can accelerate T cell exhaustion and impair T cell functionality, 114,115 others suggest the opposite. 41,116
PD-1 has also been described as a haplo-insufficient suppressor of T cell lymphomagenesis, 117 although a recent study using CD19-CAR T cells in immunocompetent mice suggests that PD-1 knockout CAR T cells can differentiate, form memory, and persist in the presence of constant antigen exposure provided by continuous B cell renewal, with no evidence of malignant transformation. 118
Early clinical trials with PD-1-disrupted T cells have shown feasibility and safety in the absence of unconstrained proliferation or persistence, but also in the absence of objective antitumor responses. 43,110,119 Missing consensus on whether PD-1 knockout is beneficial or detrimental may be due to the lack of clinically relevant preclinical models, 120 different levels of PD-L1 expression across treated patients, and different sensitivities of the engineered T cells to PD-1/PD-L1 axis signaling. In this regard, we and others have observed that CARs containing different ScFv and costimulatory domains can respond differently to inhibition of the PD-1/PD-L1 axis. 121
An important observation from studies that knocked out PD-1 in tumor-specific T cells is that T cell exhaustion can occur in the absence of PD-1 expression. Expression levels of T-cell immunoglobulin and mucin-domain containing 3, lymphocyte activation gene 3, T-cell immunoreceptor with Ig and ITIM domains, and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) are also hallmarks of T cell exhaustion, and inhibition of these markers is being actively investigated to promote antitumor responses. Antibody-mediated blocking or genetic ablation of any one or of a combination of these immune checkpoint can boost the therapeutic efficacy of tumor-directed T cells, although further studies are required to better understand their role in CAR T cell function. 122,123
Other pathways have also been targeted with genome editing to enhance CAR T cell function. For example, gene disruption of TGF-β receptor II (TGF-βRII) in CAR T cells was reported to reduce the induced Treg conversion, prevent the exhaustion of CAR T cells, and increase their antitumor activity. 124 In addition, gene disruption of Fas reduced activation-induced cell death in engineered T cells, and increased the antitumor effect of CAR T cells. 41
In a different approach, elimination of the DAG kinase, to increase TCR signaling, rendered CAR T cells resistant to soluble immunosuppressive factors, preventing T cell dysfunction driven or exacerbated by the tumor microenvironment. 125 CD8+ T cells from humans' cancer express high levels of nuclear receptor transcription factors (NR4A) and display enrichment of NR4A-binding motifs in accessible chromatin regions. Triple gene disruption of NR4A transcription factors in CAR T cells promotes tumor regression and prolonged the survival of tumor-bearing mice. 126
Typically, these knockout strategies have been used to identify candidate genes, based on their previously described roles in inhibiting T cell activation or function. With the recent development of next-generation sequencing, high-throughput genetic perturbation technologies, and genetic screens, new candidate genes have been identified for roles driving T cell exhaustion. Examples include CD39, identified as a major driver of T cell exhaustion in both primary and metastatic colorectal tumor models testing HER-2-specific TCR-edited T cells for eliminating human epidermal growth factor receptor 2 (HER-2) expressing patient-derived organoids in vitro and in vitro. 127
Another model of continuous antigen exposure, which is present in many solid tumor settings, used single-cell RNA sequencing to identify transcription factors associated with CAR T cell dysregulation associated with a CD8 T to NK-like T cell transition and identified SRY-box transcription factor 4 (SOX4) and inhibitor of DNA binding 3 (ID3) as key regulators of T cell exhaustion. 128 CRISPR/Cas9-mediated ID3 and SOX4 knockouts prevented or delayed T cell dysfunction and improved CAR T cell efficacy against solid tumors.
Signatures of T cell exhaustion and dysfunction include global chromatin remodeling and epigenetic changes, and genome-wide CRISPR screens have also identified genes involved in epigenetic regulation as candidates for therapeutic editing. Genome-editing disruption of regulators, such as DNA methyltransferase 3 alpha (DNMT3A) and AT-rich interaction domain 1A, has reversed exhausted epigenetic states in vitro and in animal models. 129,130 In addition, chronic CAR T stimulation can coordinate an early growth response protein 2 (EGR2)-dependent epigenomic resistance program in T cells, and knocking out the gene encoding the EGR2 transcriptional regulator renders CAR T cells impervious to dysfunction and improves memory differentiation. 131
Finally, a recent study used an in vitro model of T cell exhaustion mediated by tonic signaling to identify the Mediator Complex Subunit 12 (MED12) and Cyclin C genes, which encode proteins in the kinase module of the multiprotein mediator complex, as regulators of CAR T cell effector functions. Indeed, MED12-deficient T cells showed enhanced potency in mediating antitumor effects. 132 Although challenging in primary T cells and in vitro models, these types of CRISPR screenings can enable the unbiased discovery and functional characterization of gene targets and pathways with key roles in T cell function. 133
Whether these candidate genes or pathways can be targeted with small molecules, antibodies, or gene-editing approaches will require further preclinical studies. Any concomitant knockout of tumor-suppressor activity would have to be balanced against heightened risks of genotoxicity, even though the clinical experience with thousands of engineered and infused T cells indicates that these cells are particularly resistant to transformation.
GENOME EDITING TO BOOST THE POTENCY OF ADOPTIVELY TRANSFERRED CELLS
Genome-wide CRISPR/Cas9 editing offers further avenues to enhance T cell activation, proliferation, and survival, thereby overcoming the inhibitory tumor environment. In vitro screens have successfully identified potentially targetable genes involved in T cell function, including inhibitors of T cell proliferation (CBLB, CD5, FAM49B, RASA2, MAPK14), 134 –136 suppressors of the Jak/STAT signaling pathway (SOCS1, TCEB2, RNF, CUL5), and inhibitors of nuclear factor kappa B (NF-κB) signaling (UBASH3A, TNFAIBS, TNIP1). 133
Deletion of genes affecting T cell chromatin remodeling related to exhaustion (canonical BAF complex), metabolism (chromatin-remodeling ATPase INO80), and methylation-associated plasticity (DNMT3A) impact on cell fitness and antitumor activity, 95,130,137 with more efficiency compared with deleting individual genes specifically associated with activation/exhaustion such as PD1. 43 These approaches hold translational potential for human T cell products, but selection of the pathway/individual gene may be dependent on the transferred T cells' protein target and type of tumor targetted, highlighting the validity and rationale for in vitro screens. 137
In addition, many targeted pathways have been associated with tumorigenesis in different contexts, either by ablating negative regulation of oncogenes (e.g., CBLB, RASA2, FAM49B) or suppressing tumor-suppressor genes (e.g., MAPK14), underscoring the necessity of thorough safety screening and monitoring.
There is evidence that ablation or perturbation of inhibitory genes in substrate cells can enhance function in transferred cells, as in the unusual experience of tet methylcytosine dioxygenase 2 (TET2) gene inactivation by CAR construct integration in a patient with an additional hypomorphic TET2 allele. 138 Although TET2 is a tumor-suppressor gene and a master regulator of blood cell formation, its disruption alone has not been associated with overt oncogenesis. 139
Disrupting TET2 in a patient altered the differentiation state and proliferative capacity of CAR-expressing T cells, resulting in considerable and durable therapeutic effects without transformation during >4 years. However, further studies have indicated that biallelic TET2 ablation also enables antigen-independent CAR T cell clonal expansion, raising potential safety concerns associated with disruptions of genes linked to broad epigenetic changes. 140
In addition to gene ablation, the overexpression or knockingin of natural or synthetic genes holds promise for enhancing cell therapy. Although technically challenging, CRISPR activation screens are increasingly employed to identify genes that confer advantages to T cells through overexpression. 141 As proof of principle, overexpressing the activator protein 1 (AP-1) factor c-Jun in CAR T cells driven by tonic-signaling demonstrated the ability to prevent exhaustion and enhance potency in solid tumor models. 142 This strategy is now being implemented in patients. 143 Although c-Jun may not be implicated in mature T cell transformation, concerns regarding potential oncogenesis remain as c-Jun expression has been described in cancer settings. 144
To maintain T cell efficacy in the face of repeated antigen stimulation, T cell activation with associated proliferation and survival requires a costimulatory signal concurrent with TCR triggering. 145 Unlike CARs that already include a costimulatory domain, TCRs and other newer constructs, such as TCR fusion (T cell receptor fusion constructs [TRuC]) and HLA-independent T cell receptors, may require independent costimulatory receptor triggering for maximal efficiency. 146,147 Ligands for these coreceptors are seldom present on tumor cells, which further commonly upregulate inhibitory ligands that interfere with T cell activation. 148,149
To overcome both of these challenges, synthetic immunomodulatory fusion proteins (IFPs) that combine an inhibitory ectodomain with a costimulatory endodomain are being pursued, introduced either by gene knockins or lentiviral-mediated insertions. These constructs are geared to minimize/ablate the negative effects of suppressive ligands and transmit a positive cosignal to the binder (TCR/CAR/TRUC/Other). 150 –152 Such constructs fall into broad categories that include “Signal 2” CD28 family of receptors (CD28, ICOS, CTLA-4, and PD1), TNF superfamily (CD27, CD30, CD40, OX40, and 4-1BB), and fibronectin type III domains typically constituting “Signal 3” cytokine receptors (e.g., IL-2, IL-7, IL-15, and IL-21).
IFP choice may be target and tumor context dependent. For the IFPs to signal, they must first find their inhibitory receptors either on tumor cells or cells present in the tumor microenvironment. For example, not all negative regulators are consistently expressed across tumor types, emphasizing the need for careful selection of IFPs for a particular tumor type. 153 The type of intracelluar signal transmitted by the IFP also has to be considered. For example, upon cross-linking, the cytoplasmic CD28 domain binds to phosphaditylinositol 3-kinase that has been phosphorylated on tyrosine by lymphocyte-specific protein tyrosine kinase, enhancing NF-κB, nuclear factor of activated T cell and AP-1 signaling pathways that in turn mediate survival, proliferation, cytokine production, and differentiation. 154
4-1BB instead promotes the activity of NF-κB through the activation of the inhibitor of NF-κB (IkB) kinase complex, induces the antiapoptotic protein B cell lymphoma-extra large (Bcl-xL), proliferation, and promotes memory differentiation and mitochondrial biogenesis. 155 In CAR designs, CD28 signaling elicited faster larger magnitude changes in protein phosphorylation, which correlated with effector functions, compared with the slower memory-type activities of 4-1BB that produce more sustained antitumor activity against established tumors in vitro. 156
Another strategy being explored to boost transferred cell function is to express cytokine receptors' intracellular tails as a means for T cells to also secrete supportive cytokines either constitutively or upon synthetic cytokine triggering. 157,158 As most costimulation signals do not function in the absence of binding of the primary construct to the target, these approaches may offer modulable and safe translation strategies.
PREVENTING CAR T CELL FRATRICIDE USING GENOME EDITING APPROACHES
Treating T cell malignancies remains one of the key challenges for CAR T cell therapy. T cell neoplasms have a very poor prognosis, leading to high rates of relapse and mortality in both children and adults, which represents a significant unmet medical need. However, most T lineage antigens, which could potentially be targeted with CAR T cells, are similarly expressed in both normal and malignant T cells. Thus, the introduction of a T cell surface protein-redirected CAR into a normal T cell can result, as the product is being generated, in CAR-mediated killing—a phenomenon known as “fratricide.”
Fratricide of CAR T cells during the manufacturing process limits ex vivo T cell expansion, accelerates T cell differentiation and exhaustion due to constant CAR signaling, and results in impaired antitumor efficacy. Fratricide has been observed in CAR T cell product development for T cell and non-T cell malignancies targeting CD3, TCRβ, CD7, CD38, CD70, and NKG2D ligands. To avoid fratricide, one solution is to use genome editing to eliminate the targeted antigen from the surface of CAR T cells. Functional CAR T cells targeting CD3 or CD7 have been successfully generated by knocking out CD3 or CD7, respectively, using TALENs, 159 CRISPR/Cas9, 160 or base editors. 51,52
Collecting enough healthy autologous T cells from patients with T cell malignancies constitutes another limitation for the generation of CAR T cells. As a consequence, significant efforts are under way to develop allogeneic approaches to treat these patients. To generate off-the-shelf CAR T cells, genetic disruption of the CAR target is typically combined with the disruption of the TCR gene. In addition, the ablation of the CD52 gene can be incorporated into this multiple-target editing strategy to confer resistance to depletion by alemtuzumab.
The feasibility and potential of this approach have already been demonstrated in some clinical trials. Off-the-shelf anti-CD7 CAR T cells, which are resistant to fratricide through genome-editing 161 or base-editing 81 approaches, have induced deep and durable responses in a small number of selected patients with relapsed or refractory T-ALL.
Alternatives to mitigate fratricide during T cell manufacturing without genome editing are also being explored, especially in the context of autologous CAR T cells. This includes targeting T lineage antigens that are downregulated after CAR expression (such as CD5), 162 blocking CAR signaling with pharmacological inhibitors of key signaling kinases, 163 or selecting a subpopulation of T cells for engineering that do not express the targeted antigen. 164 Finally, the expansion of naturally selected CD7 CAR T cells during manufacturing has also been proposed and tested in clinical trials, with impressive clinical results. 165
LONG-TERM SAFETY WITH GENOME-EDITED T CELL PRODUCTS: CONSIDERATIONS FROM CLINICAL TRIALS
Engineered T cells were first reported in a clinical study in 1990, using TILs transduced with a retroviral vector. 9 This occurred almost concurrently with the first patient successfully treated with gene therapy using T cells transduced with the enzyme adenosine deaminase in children with severe combined immunodeficiency 166 and the first use of allogeneic T cells transduced with retroviral vectors encoding a suicide gene. 10 There is now increasing long-term confidence that overt transformation risks from using gamma-retroviral and lentiviral vectors in T cells are low. 167,168
Gene addition into differentiated T cells appears safer than in stem cells, 167,169 perhaps because polyclonal T cells are well differentiated and lack expression of genes associated with stemness and embryonic development and perhaps their diversity prevents outgrowth of potentially malignant T cells. 170 In contrast, for hematopoietic stem cell modification, there have been concerns relating to gamma-retroviral-mediated genotoxicity due to insertional mutagenesis in patients with inherited genetic conditions, 171 –174 and although these have been largely mitigated by switching to sin-lentiviral configurations, safety issues continue to be carefully investigated as wider applications develop. 175
In T cells, clonal expansion due to integration near the Casitas B-lineage lymphoma oncogene and disruption of the TET2 tumor-suppressor gene has been reported in patients treated with CAR T cells. 138,176 Interestingly, TET2 gene disruption resulted in the promotion of CAR T cell expansion and antitumor activity, indicating that viral vectors are able to influence gene expression and host cell behavior. Although there was clonal dominance in these subjects, there was no evidence of malignant transformation.
However, transformation of engineered T cells has been reported in a clinical trial of Piggybac transposon-modified CAR T cells where 2 cases of T cell lymphoma arose in a study of 10 patients treated. The mechanism of transformation is still uncertain, but excessive transposase activity, insertional mutagenesis, high transgene copy number, and the manufacturing process may have played a role. 177 Other transposon systems are based on the synthetic Sleeping Beauty transposon and have been used for T cell gene therapy, and so far proven safe. 68,178
Compared with technologies that achieve transgene integration by random gene insertion, precise genomic targeting should reduce risks of insertional mutagenesis or other undesirable influences on regional genes. However, off-target DNA cleavage activity may lead to point mutations, indels, or other changes at unintended sites at low frequency, whereas chromosomal aberrations such as deletions, translocation, and chromosome loss could also occur. A recent report of recombination-induced karyotype changes in a patient treated with TALEN-edited CAR19 T cells highlighted the importance of investigating such events when they arise. 179
Moreover, as a consequence of the DNA damage response, there is a risk of selection of p53-inactive clones. 180 To understand whether unintended off-target events are clinically relevant, Cromer et al. performed ultradeep sequencing of >500 tumor suppressors and oncogenes in hematopoietic stem cells electroporated with high-fidelity Cas9 protein and guide RNA; no evidence of mutations in tumor suppressors or oncogenes was found. 181 Conversely, large abnormalities were found in primary human T cells transfected with CRISPR/Cas9 and guide RNAs targeted to TCR and PD1. 182
The clinical relevance of Cas9-induced chromosomal loss was confirmed in preclinical CAR T cells, but reduced by clinical editing protocols that used a different order of operations in T cell manufacture. 183 Using this platform, early results from clinical trials of engineered TCR (NY-ESO-1 specific) T cells with TCR and PD-1 knockouts did not cause chromosome loss (NCT03399448). Moreover, the potential risk of immunogenicity did not appear to have affected the persistence of CRISPR/Cas9-edited T cells. 43
The safety of PD-1-edited T cells was also recently tested in a study for metastatic nonsmall lung cancer (NCT02793856), with an off-target event rate of 0.05%. 119 High safety and a first demonstration of CRISPR/Cas9-edited T cell efficacy have also been shown with edited anti-CD19 CAR T cells specifically targeted to PD-1 locus in B NHL, 184 with universal anti-CD19 CAR T cells edited in the TRAC and CD52 loci of pediatric B-ALL patients, 44 and with TRAC and TCRβ constant chain-edited neoantigen-specific TCR (neoTCR) in patients with refractory solid cancers. 185 Safety has also been demonstrated for anti-CD123 allogeneic CAR T cells in patients with AML. 186 GVHD has been generally absent or limited in severity and readily managed with immunosuppression.
Evaluation of long-term safety of genome editing is essential to ensure its application in the clinic, and the risk/benefit balance must drive the design of new therapies. It is anticipated that reducing the time of Cas9 activity by ribonucleoprotein complexes, 187 high-fidelity nucleases, 188,189 usage of nickase (n) Cas9 variants, 190 base editors, 47 and prime editors 55 have the potential to increase gene editing specificity and safety.
CONCLUSION
Advancements in genome editing technology and the identification of major targets and pathways involved in intratumoral T cell dysfunction have enabled a wide range of next-generation T cell therapeutic products. This new therapeutic pillar holds immense potential for cancer patients. Although further research and careful consideration of target specificity and safety concerns are necessary to fully exploit these promising approaches, gene ablation, gene overexpression, and the use of IFPs all offer new avenues to enhance T cell function and overcome inhibitory tumor environments.
Additional cellular platforms, including engineered NK,
119
Invariant natural killer-T cells,
191
gamma delta T cells,
192
and macrophages,
193
are also being investigated as part of an armamentarium of cancer immunotherapy using engineered cells. The proper clinical exploitation of engineered T cells will require efforts to increase sustainability and harmonization of manufacturing, use, and clinical monitoring. International coalitions and consortia, such as the GO-CART Coalition (
Footnotes
ACKNOWLEDGMENTS
AUTHOR DISCLOSURE
C.B. is inventor on patent applications and has been granted patents related to T cell engineering that have been, in part, licensed to industry. C.B. has been member of Advisory Board and Consultant for Intellia Therapeutics, Novartis, GSK, Allogene, Kite/Gilead, Miltenyi, Kiadis, Evir, Janssen, Alia and received research support from Intellia Therapeutics.
A.G.C. is a scientific co-founder of Affini-T Therapeutics, is an inventor on patents related to TCRs, and has received funding from Affini-T, Amazon and Lonza.
S.G. is an inventor on patents related to CAR-T cell therapy, filed by the University of Pennsylvania and licensed to Novartis and Tmunity, and has received commercial research funding from Gilead.
C.F.M. is an inventor on patents related to CAR T and regulatory T cell therapy, filled by the Tettamanti Research Center (under license to Coimmune), and Yale University, San Raffaele Hospital and the Telethon Foundation.
M.H. is inventor on patent applications and has been granted patents related to CAR technology that have been, in part, licensed to industry. Co-founder and equity owner T-CURX GmbH, Würzburg, Germany. Funding: BMS. Speaker honoraria: Janssen, BMS, Novartis.
W.Q. is inventor in patents filed in relation to genome edited T cells; Funding- Servier, Cellectis, Miltenyi; Consultancy Wugen, Virocell, Skylark.
FUNDING INFORMATION
C.B. is funded by AIRC (Ig 18458 and Ig 24965), AIRC 5xMille, Rif. 22737, Italian Ministry of Research and University (PRIN 2015NZWsec; PRIN 2017WC8499), Italian Ministry of Health (Research project on CAR T cells for hematological malignancies and solid tumors and RF-2019-12370243).
A.G.C. has received funding from the U.S. NIH (P01CA18029-41). S.G. has received funding from the Spanish Ministry of Science and Innovation under a Ramon y Cajal grant (RYC2018-024442-I). C.M. is supported by the Swiss National Science Foundation (PR00P3_201621), the Comprehensive Cancer Center Zurich, the San Salvatore Foundation, and the Helmut Horten Foundation.
M.H.: BMS. Speaker honoraria: Janssen, BMS, Novartis. W.Q. is supported by National Institute of Health Research, MRC, Wellcome Trust.