
Abstract
β-Thalassemia and sickle cell disease are autosomal recessive disorders of red blood cells due to mutations in the adult β-globin gene, with a worldwide diffusion. The severe forms of hemoglobinopathies are fatal if untreated, and allogeneic bone marrow transplantation can be offered to a limited proportion of patients. The unmet clinical need and the disease incidence have promoted the development of new genetic therapies based on the engineering of autologous hematopoietic stem cells. Here, the steps of ex vivo gene therapy development are reviewed along with results from clinical trials and recent new approaches employing cutting edge gene editing tools.
INTRODUCTION
Hemoglobin (HB),

The β-like genes are spatially ordered as they are temporally activated to form the embryonic, fetal (HbF), and adult Hb (HbA). The silencing of the γ fetal genes during adult life is the result of diminished interaction with transcriptional activators, engaged on the promoter of adult β and δ genes, and of silencing by repressor complexes, including BCL11A and lymphoma/leukemia related factor (LRF) factors, and possibly others. 2,3
The binding sites for transcription factors in the γ promoters are targets of mutations, which, together with deletions in the β-locus, are responsible for the benign condition of hereditary persistence of fetal hemoglobin (HPFH), which, if coinherited, ameliorate the pathology since 30% increase of HbF can prevent α-chains precipitation in β-thalassemia and sickle hemoglobin (HbS) polymerization in sickle cell disease (SCD). Most of the gene or base editing approaches are aimed at reproducing such conditions for therapeutic purpose. 4
Mutations in the adult β-globin gene cause β-thalassemia and SCD. Despite the common genetic cause, the pathophysiology is different and phenotypic diversity is also contributed by both genetic and non-genetic modifiers (Fig. 1C).
In β-thalassemia, more than 400 different mutations in the cluster, including deletions or points mutations in the coding and regulatory sequences, cause a broad spectrum of symptoms. Phenotypes are assigned based on the clinical presentation with dependence from transfusion as the main cutoff between the transfusion-dependent (TD) and the non–transfusion-dependent (NTD) patients, although stigmata of the disease can be present in both categories with different severity.
Mutations in hemoglobin subunit beta (HBB) gene result in reduced production of HbA, with a severity index of a relative reduction or a complete absence of β-globin chain synthesis (β+ and β0). The severity of anemia, transfusions requirement, and clinical morbidity are closely related to the level of α-globin and β-globin chains imbalance. The excess of unstable α-globin tetramers generates cytotoxic reactive oxygen species (ROS) and intracellular precipitates impairing maturation and viability of erythroid precursors, leading to ineffective erythropoiesis and hemolysis.
The permanent stimulus to produce RBC creates a chronic stress environment in the bone marrow, impairing not only the erythroid compartment but also primitive hematopoietic and stromal populations. 5,6 Recent phenotype classification introduces TD β-thalassemia (β-thalassemia major) or NTD β-thalassemia (β-thalassemia intermedia) with no transfusions or occasional transfusions in specific circumstances. Ineffective erythropoiesis leads to marrow expansion and bone deformity, contributing to β-thalassemia stigmata such as the craniofacial protrusions.
Compensatory extramedullary hematopoiesis can be activated in the spleen and liver (hepatosplenomegaly) and other tissues. Iron overload is a hallmark of the disease, even in TD patients, with potential significant consequences on several organs, contributing to a multisystem disorder.
Uncertain epidemiology data estimate 40,000 affected individuals born yearly, with more than 90% of patients living in a geographic area from Africa to Middle East, the Mediterranean basin, and Southeast Asia, because of the protective role against malaria associated to the thalassemia carrier state. Population movements extended β-thalassemia to Europe and Americas. Due to the complex clinical condition and the need of long-life clinical care, difference in survival rate is mainly dictated by the access to cures, and patients in resource limited countries are unable to achieve their fourth decade of life. 7
SCD, defined as the first “molecular disease,” is caused by abnormal HbS production in which the βGlu (6)A3→Val substitution changes a negatively charged side chain in a hydrophobic one, allowing deoxygenated HbS to polymerize, altering RBC architecture and flexibility to the sickle cell shape. The culprit of SCD is the deoxygenated HbS polymer, which damages the RBC, resulting in hemolytic anemia and vaso-occlusion by blocking small blood vessels.
Ischemic tissues damage exacerbates in severe pain, acute chest syndrome, and cerebral strokes that can be fatal. Genetic variants associated with HPFH in SCD may provide a milder phenotype, since HbF can inhibit HbS polymerization being excluded from the HbS polymer. The critical issue to achieve phenotype amelioration is the relative proportion of HbS and HbF content in each erythrocyte. Thus, 30% HbF homogenously distributed in RBC can alleviate SCD symptoms in compound heterozygous patients. Lessons from disease natural history set this threshold as mandatory in gene therapy approaches.
SCD is the most common monogenic disorder with a prevalence in malaria-endemic regions, sub-Saharan Africa, the Mediterranean basin, the Middle East, and India, because of the protective function of sickle cell trait against severe malaria. 8 The original distribution has changed due to slave trading and population migration, with ∼100,000 estimated patients in the United States.
Although difficult to predict, newborn estimates indicate 300,000 babies per year born with SCD, the vast majority being in Nigeria, Congo, and India. Life expectancy and mortality are strictly dependent on country income, and in lower-income countries is reduced by about 30 years and childhood mortality peaks 90% in children younger than 5 years born in Africa.
TREATMENTS
Allogenic stem cells transplant (HSCT) is offered as a curative treatment for β-thalassemia and SCD mainly to pediatric patients with matched related donors. Despite the increase availability of human leukocyte antigen-matched donors and the general improvement of pretransplant and transplant settings, HSCT still showed morbidity and mortality due to acute and chronic severe graft versus host disease and graft failure. Thus, the limitations of HSCT favored the development of gene therapy (Fig. 2) and of new drugs for the treatment of hemoglobinopathies.
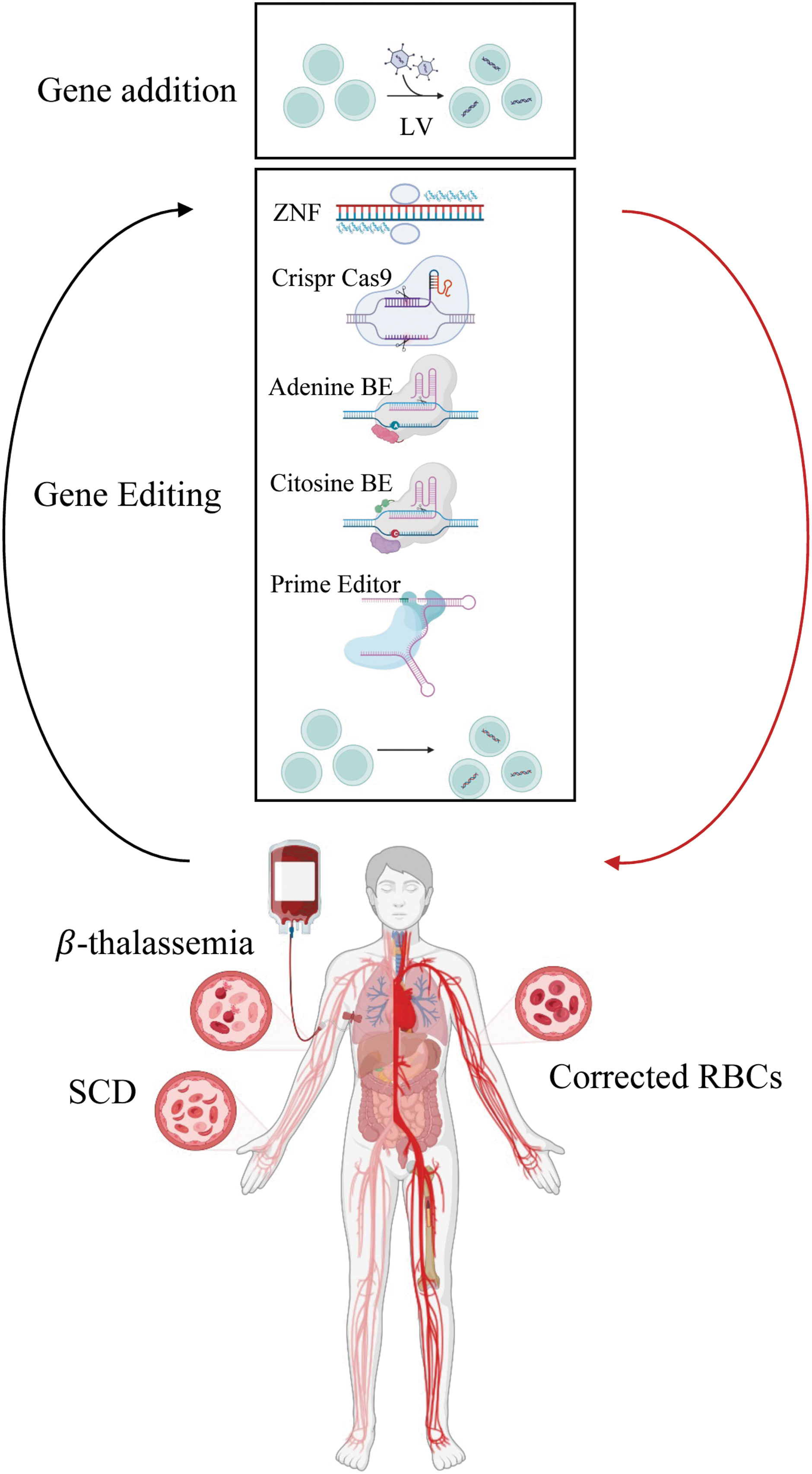
Gene addition and gene editing strategies to correct β-thalassemia and SCD. Created with BioRender.com
Novel drugs for β-thalassemia
Alternative treatments are now approved or under evaluation for patients unable to access the transplant. Emerging drug therapy for β-thalassemia aims mainly at restoring the α/b chains ratio, ameliorating the ineffective erythropoiesis, and correcting the iron overload.
Drugs targeting ineffective erythropoiesis or iron dysregulation such as pyruvate kinase activator, Janus Kinase 2 inhibitor, hepcidin stimulator, or mimetics and ferroportin inhibitors 9 improved anemia and iron-related factors in mice models but inconsistency was found in clinical trials. 10 –12 Among them, the pyruvate kinase activator Mitapivat, already approved for PK deficiency, improves anemia and iron overload by increasing ATP levels. Ongoing phase 2 trial (NCT03692052) showed increase in Hb level with decrease hemolysis marker levels, and mild-moderate adverse events that in one case led to a discontinuation. 13
Luspatercept is a recombinant fusion protein binding members of TGF-β superfamily ligands trapping TGF-β-like molecules and inhibiting SMAD2/3 signaling to increase the nuclear localization of GATA-1 and TIF-1, finally promoting late-stage erythropoiesis. The multicenter phase 2 (NCT01749540 and BEYOND NCT03342404) and phase 3 BELIEVE (NCT02604433) trials showed a rapid and sustained increase of >1.0 g/dL in Hb concentration in both non transfusion dependent thalassemia (NTDT) and transfusion-dependent β-thalassemia (TDT) patients, an amelioration of ineffective erythropoiesis, and a reduction in transfusion requirement up to transfusion independence in 11% of TDT patients.
However, the impact on iron overload, measured as serum ferritin levels, was less clear in liver iron concentration, likely due to short time of observation. 14 –16 Luspatercept, as Reblozyl®, was approved in 2019 and 2020 for TDT patients by Food and Drug Administration (FDA) and European Commission (European Medicines Agency [EMA]), respectively, and more recently also for NTDT patients by EMA. Although a general clinical benefit was observed, β0/β0 patients showed a lower response leaving open the need for different approaches. 14
Novel drugs for SCD
Clinical observation in patients with increased HbF levels reported a milder phenotype with a better survival of erythroid cells and provided the rationale for the use of HbF-inducing drugs. Among these, hydroxyurea, a potent inhibitor of ribonucleotide reductase, already used in the treatment of myeloproliferative disorders and known to induce HbF expression ameliorating clinical symptoms, was approved by the FDA in the late 1990s for the treatment of SCD. 17 Unfortunately, until now, there are no molecules showing response in β-thalassemia as reported for SCD. 18 Recently, in addition to hydroxyurea, FDA approved three new drugs for the treatment of SCD.
An alternative mechanism was evoked, based on the role of
Crizanlizumab was approved by the FDA in 2019: it is an intravenously administered humanized monoclonal antibody against P-selectin, which is upregulated in SCD cells. Blocking the interaction between P-selectin and P-selectin glycoprotein 1, Crizanlizumab can prevent neutrophils activation and adhesion of erythrocytes, leukocytes, platelets, and endothelial cells, reducing vaso-occlusion.
The main phase 2 clinical trial (SUSTAIN, NCT01895361) compared two agent doses with placebo and reported evidence of clinical improvements in adults and adolescents patients (>16 years), showing a decrease in the number and proximity of VOC episodes, only in a high-dose group, irrespective of Hydroxyurea treatment. 23,24 Phase 2/3 trials are now recruiting to study pharmacokinetics and pharmacodynamic parameters (SOLACE-adults, NCT03264989, and -kids, NCT03474965) and to evaluate efficacy and safety in younger patients (>12 years, NCT03814746 STAND).
Voxelotur was initially approved in 2019 by the FDA and in 2022 by the EMA to treat SCD patients ≥12 years. Based on the results of the HOPE clinical trial, the approval has been recently extended to pediatric patients (4–11 years). 25 It is an Hb allosteric modifier that directly binds the Hb molecules, increasing oxygen affinity and disrupting the sickling and unsickling cycle. In vitro studies demonstrated the improvement in RBCs deformability and half-life, the reduction of hemolysis and blood viscosity. 26 –28
The phase 2 HOPE and HOPE-kids trials reported increased Hb levels and an amelioration in hemolysis markers with moderate adverse effects. 29 Although VOCs were reduced, no statistically significant difference was reached as compared with the placebo group. Despite positive results, safety concerns are emerging to a potential risk of reducing ability to deliver oxygen to tissues. 30
GENE ADDITION STRATEGIES
Autologous transplantation with genetically corrected HSC has been pursued with the goal to offer a universal, one-shot treatment overcoming the major limitations related to allogeneic HSCT. Several vectors have been developed over the years to increase their efficacy and safety. In this review, we mainly focus on lentiviral vectors (LV) developed to restore β-globin expression or to induce the reactivation of γ-globin.
LV to express β-globin
For decades, investigators designed LV carrying β-globin genes with different portions of LCR elements to improve gene transfer efficiency and guarantee a sustained and stable transgene expression. The first study described TNS9 vector, carrying β-globin gene with an intron 2 internal deletion and under the control of a 625 bp endogenous promoter and the addition of a 3.3 kb LCR with HS2, HS3, and HS4. Thalassemic mice treated with TNS9 transduced hematopoietic stem and progenitor cells (HSPCs) showed the amelioration of disease features, indicating potential clinical benefits in patients. 31
Later, therapeutic potential in β-thalassemia was demonstrated both in mice and in patients' cells using GLOBE LV, encoding for a functional transgene with minimal LCR containing HS2 and HS3 elements. 32 Importantly, correction of patients CD34+ cells led to normal HbA expression with reduced apoptosis during RBC in vitro differentiation. 33 Non-clinical toxicity and biodistribution studies in thalassemic mice and in immunodeficient recipients transplanted with human transduced cells proved the safety of GLOBE LV. 34
The introduction of a β-globin transgene modified with the glutamine 87 (T87Q) amino acid variation, enabling the anti-sickling effect of γ globin chain, in an LV containing an LCR composed by HS2, HS3, HS4, generated a unique vector to treat SCD and β-thalassemia, and ameliorate the disease in SCD and β-thalassemia murine models. 35,36
The addition of the cHS4 insulator produced the Lentiglobin HPV569 LV, which was used in the first clinical trial for β-thalassemia. 37 The removal of cHS4 in the optimized Lentiglobin BB305 LV by Bluebirdbio (BBB) increased vector titer and gene transfer efficiency, making it more suitable for further clinical application. 36
To enhance the anti-sickling effect, a novel vector (Lenti-βAS3) was cloned carrying additional anti-sickling mutations: a substitution of glutamic acid with alanine (E22A), present in γ and δ-globin and of glycine with aspartic acid (G16D), present in the natural nonpathogenic Baltimore variant. The SCD mice transplanted with Lenti-βAS3 transduced HSC cells showed high and long-term expression of human Hb, leading to improved RBC and hematocrit values, amelioration in tissue damage, and reduction of reticulocytes, although in the absence of complete anemia rescue, mimicking the effect of HPFH in some SCD patients. 38
An improved version was obtained by cloning βAS3 transgene in the GLOBE LV framework, increasing vector titer and transduction efficiency. Variants of this vector design included different size and type of HS from LCR in the effort to increase vector titer and transgene expression 39 and ameliorate pathology in SCD mice. 40
In a different approach, a modified β-globin LV was developed to express a G16D variant of γ-globin under the control of minimal β-globin promoter and the HS2, HS3, and HS4 LCR elements, showing in vivo correction of SCD phenotype with high proportion of HbF-expressing cells. 41
Long-term safety and stability studies in pigtails macaques showed stable and sustained γ-globin expression with a polyclonal integration profile up to 3 years follow-up, and no perturbation of gene expression was observed supporting its use in a clinical trial (NCT02186418). 42
LV to reactivate γ-globin
Since the naturally occurring HPFH condition improves clinical outcome in both β-thalassemia and SCD patients, an alternative LV-based approach aims at inducing the γ-globin reactivation by silencing of the repressor BCL11A. A GLOBE-derived LV encoding for micro-RNA short harpin RNA (shRNAmiR) able to silence BCL11A was developed.
Preclinical data in human SCD cells showed HbF induction with a normal biodistribution in xenotransplanted immunodeficient mice. Absence of toxicity was observed in non-clinical studies, 43 and it supported the clinical translation to a first trial. Recently, a refined version included shRNAmiR for ZNF410 repressor, with an additional 10% increase in HbF content in erythroid differentiated cells and amelioration in SCD mice phenotype. 44
CLINICAL TRANSLATION OF GENE ADDITION
The first reported clinical trial treated a TD βE/β0 patient with Lentiglobin vector, who achieved transfusion independence with up to 10 g/dL Hb, with 1/3 contributed by the transgene. Clonality and vector integration analysis demonstrated the benign expansion of a dominant myeloid cell clone in which HMAG2 gene was trans-activated from vector integration, whose proportion decreased at later time points. 37,45 Following this pioneer trial, phase 1–2 multicenter trials using the Lentiglobin derived vector BB305, sponsored by BBB, opened in 2013 for TDT (NCT01745120, NCT02151526, Table 1).
Clinical trial of gene addition
BM, bone marrow; G-CSF, granulocyte-colony stimulating factor; mPB, mobilized peripheral blood; Plx, plerixafor; SCD, sickle cell disease.
Results reported showed a significant reduction in transfusion requirements up to transfusion independence in non-β0/β0 patients (12 out of 13) but less effective results in more severe β0/β0 patients (3/8). 45,46 Patients with more than 2 years of follow-up are now enrolled in long-term FU study (NCT02633943). To improve clinical outcome, a manufacturing process optimization was adopted in a later phase 3 clinical trial (NCT02906202, referred as Northstar2), increasing both vector copy number (VCN) and transduction efficiency, and reaching transfusion independence in 20 out of 23 patients. 47
A new phase 3 trial (NCT03207009) is now ongoing. Initial results showed a higher percentage of transfusion independence (89.5%, 34/38 treated Pts) as compared with phase 1/2 trials (68.2%, 15/22), with an overall 18% (11/63) of patients experiencing adverse events likely related to drug products; in particular, VOC are registered in 7 out of 63 subjects.
A preliminary multivariate statistical analysis indicated transduction efficiency and VCN in drug products as the major key factors responsible for better clinical outcome. 48 Based on these results, the BBB gene therapy product Zynteglo was approved in 2019 by the EMA but withdrawn from Europe by the sponsor in 2022. In the same year, the product was approved by the FDA.
Similar findings resulted from the phase 1 clinical trial based on the use of TNS9.3.55 vector showing a long-term stable but modest engraftment of transduced HSC and reduction in transfusion requirements in two out of four patients mainly due to low transduction rate in drug product (VCN range: 0.09–0.15), suggesting that reduced intensity conditioning is sufficient to achieve a stable CD34 engraftment but high VCN is needed to have a clinical improvement with transfusion independence. 49
In 2015, the first clinical trial treating both adults and pediatric TDT patients was based on the use of GLOBE vector, with intra-bone administration of transduced CD34+ cells. Reduction of transfusion requirements was achieved in all patients but one, with transfusion independence in three out of four pediatric patients. 50 Recent results showed robust and persistent engraftment in seven out of nine patients, with a reduction of transfusion requirement in both frequency and volume in adult patients up to transfusion independency in four out of six pediatric patients. 51
Among clinical trials on SCD patients, results from phase 1 to 2 multicenter trials using the BB305 vector (NCT02151526, NCT02140554) showed a sustained improvement in the SCD phenotype. 52 Due to low VCN and low amount of transgene expression, improved transduction protocol was performed to ameliorate clinical outcome.
Patients treated with an optimized manufacturing process (n = 35) showed reduction in HbS expression and a vast proportion (>90%) of RBC carrying the anti-sickling Hb, leading to resolution of VOC and improvement in clinical outcome. 53 –55 Recently, two serious adverse events of myelodysplastic syndrome progress to acute myeloblastic leukemia were reported. Investigational analysis indicated that association between gene therapy and malignancies development was unlikely, as described in a later section. 56
Another study treating a small cohort of SCD patients with an LV encoding for a G16D variant of γ-globin gene (ARU-1801, NCT02186418), with a mild conditioning, showed an improvement of clinical outcome with a reduction in VOC number.
57
In 2022, ARU-1801 has been discontinued due to company-wide cost optimization and pipeline reprioritization (press release ARUVANT, June 28, 2022,
More recently, phase1 and 2 trials in SCD (NCT03282656) based on HbF induction through BCL11A shRNAmiR vector silencing started in the United States. Results at 30 months median follow-up demonstrated stable and robust HbF expression with a significant amelioration of clinical manifestation of the pathology in 9 out of 10 treated patients. 58
Overall, results from clinical trials using LV showed an optimal risk/benefit ratio, with no insertional mutagenesis and adverse events mostly related to myeloablative conditioning or to the underlying pathology. However, the inclusion in globin LV of a surrogate version of the LCR is limiting the expression of physiological levels of transgene from a safe VCN per cell. In this respect, gene editing strategies are endowed with an intrinsic advantage.
GENE EDITING STRATEGIES
HbF induction by gene editing represents a promising approach to cure patients with hemoglobinopathies. In β-thalassemia, reactivation of HbF decreases α-chains precipitation and increases total Hb levels, reducing disease severity. SCD patients expressing higher levels of HbF have milder symptoms, because HbF works as an anti-sickling agent, preventing HBS polymerization. These individuals have a condition known as HPFH. The HPFH-related mutations occurred in specific regions comprising the HBB gene locus, the γ-globin promoter, the BCL11A gene, and the HBS1L-MYB region. 59
Gene editing approaches are based on the use of nucleases that generate DNA double-strand break (DSB) into the genome. The most used nucleases are transcription activator-like effector nucleases (TALENs), zinc finger nucleases (ZFNs), and clustered regularly-interspaced short palindromic repeats and cas (CRISPR associated) nucleases (CRISPR/Cas), with the last two being applied in clinical gene therapy.
Specifically, TALENs and ZNFs are engineered restriction enzymes 60 –62 composed of sequence-specific DNA-binding modules linked to a non-specific DNA cleavage domain of the FokI endonuclease. CRISPR/Cas nuclease was discovered in bacteria in 1993 and was used as a genomic editing tool from 2012. The CRISPR/Cas9 system is composed by a Cas9 nuclease driven on the genomic target sequence by a single-guide RNA.
It is flanked by a DNA motif called protospacer adjacent motif (PAM) that is inserted on the non-target strand of the genomic DNA, downstream of the target sequence. The Cas9 nuclease recognizes this interaction and cleaves the DNA.
The cell attempts to repair DSB by using different repair mechanisms: non-homologous end joining repair (NHEJ) and homology-directed repair (HDR).
Non-homologous end joining repair
The most investigated approach to treat hemoglobinopathies is NHEJ. This repair pathway, particularly active during G0 and G1 phases of the cell cycle, creates insertions or deletions at the cut site. This is an error-prone mechanism and usually produces frameshift mutations, generating premature stop codons and/or a non-functional protein. In hemoglobinopathies, a specific +58 nt of the erythroid enhancer of BCL11A gene, including a GATA1 binding site, is the most promising target site for gene editing. 63 Deletion of this region using CRISPR/Cas9 induces an increase in expression and in HbF-expressing cells. 64
Indeed, preliminary results of clinical benefit by targeting BCL11A with CRISPR/Cas9 system in both β-thalassemia and SCD have been recently reported. 65 Disruption of the other sites (+55 and +62 nt) led to a less HbF induction. 66 ZFNs have been also applied to disrupt the erythroid enhancer in BCL11A promoter to upregulate the expression of γ-globin in patients with β-thalassemia major. 67 An updated version of ZFN has been applied in a phase 1/2 clinical trial.
Other important targetable sites include the binding sequences for LRF and BCL11A upstream the γ-globin promoter. CRISPR/Cas9 technology has been used in CD34+ cells to recreate HPFH mutations in these binding sites, inducing HbF and correcting the sickling phenotype. 68 –72
In addition, it has been shown recently that also TALEN technology achieves efficient generation of DSBs targeting, via NHEJ, leading to different deletions in HBG1 and HBG2 promoters to mimic HPFH phenotype. 73
Homology-directed repair
The other gene editing arm exploits the HDR pathway, which is active only in S-G2 phases of the cell cycle, thus limiting its application in quiescent cells. HDR needs an exogenous double-stranded DNA template flanked by homologous sequences complementary to the region around the DSB.
The HDR pathway has been applied by using CRISPR/Cas9 system combined with recombinant adeno-associated virus serotype 6 (rAAV6) vector to deliver the donor template into HSPCs. In addition, the globin genes have been edited to correct the β-thalassemia 74 or the SCD mutations. 75 –78 These preclinical studies supported the translation into the clinical practice of the CRISPR-based methodology to target HSPCs by HDR at the globin locus.
Alternative approaches exploited single-stranded oligodeoxynucleotides (ssODNs) to correct β-thalassemia or SCD point mutations with inefficient results. The application of this approach has been investigated by using different nucleases. Using ZFNs 79 low efficiency and efficacy of gene editing in long-term HSPCs damped its possible therapeutic application. Other groups focusing on the modification of the single nucleotide mutation E6V in SCD cells have obtained similar results. 80,81
Recently, Pattabhi et al. compared the ssODNs with rAAV6 as delivery platform of exogenous template in SCD cells. This experiment confirms a higher gene correction by using ssODNs compared with rAAV6. 82 Different groups obtained controversial data. 75,77 Similarly, gene editing with ssODN obtained similar results in β-thalassemia. 83
Alternative strategies: base and prime editing
Nowadays, base editors (BEs) and prime editors are the cutting-edge technologies for gene editing, avoiding DSB. Two different BEs have been engineered: cytosine base editors (CBE) and adenine base editor (ABE).
CBE is a fused protein composed by an impaired Cas9 (dCas9 or Cas9 nickase) with cytidine deaminase enzyme that directly converts cytosine to uridine, transforming C · G to T·A. 84 ABE acts by turning A · T to G · C by fusing impaired Cas9 with an adenosine deaminase. 85 CBE can change sequence at the +58 nt of the BCL11A erythroid enhancer or in the HBG1/2 promoter or correct the mutated codons in hemoglobinopathies.
CBE editing of BCL11A induced therapeutically relevant HbF levels in erythroid progeny of both β-thalassemic and SCD CD34+ cells. Moreover, full correction of the β-thalassemic phenotype is feasible by combining BCL11A editing with correction of the disease-causing mutation. 86
BEs have been employed to target the HBG1/2 promoters to reproduce HPFH and induce changes in transcription factors binding. SCD CD34+ cells, efficiently edited in vitro (>80%), engrafted in immunodeficient mice, maintained the base-edited variant (>90%) with 65% erythroid cells expressing HbF. 87 Generation of a KLF1 activator binding site and/or disruption of the LRF repressor binding site led to therapeutically relevant HbF levels in both β-thalassemia and SCD HSPCs. 88
Recently, a new variant (ABE8e) efficiently introduced multiplex editing in the BCL11A enhancer and the HBG1/2 promoter in β-thalassemia CD34+ HSPCs, with durable editing in cells repopulating immunodeficient mice. 89 Finally, in a comparative testing of five different strategies, gene editing by ABE was the most efficient approach to edit HBG1 − 175A>G SNP in CD34+ cells generating a functional GATA1-TAL1 motif. 90
ABE can convert mutation of HbE to a non-pathogenic or normal variant with 90% of efficiency in CD34+ cells, with limited off-target effects and persisting in vivo editing following xenotransplantation assay in immunodeficient mice. 91 In β-thalassemia major, IVS1–110 mutation was efficiently edited by ABE with >80% gene correction and rescue of inefficient erythropoiesis both in vitro and in vivo. 92
BEs cannot be used to create a transversion mutation necessary to correct the SCD mutation 20A<T (Glu6Val), but ABEs can be applied to replace the A · T with a G · C generating the benign anti-sickling Hb-Makassar variant. High-frequency conversion was achieved in HSPCs from SCD patients and in a murine SCD model, with improvement of RBCs sickling and associated pathologies. 93
The new technology of prime editing can introduce insertion/deletion/substitution with Ca9-nickase fused to reverse transcriptase. Correction of the SCD allele with prime editing in human erythroblasts in vivo differentiated from HSPCs with minimal off-target supported the prediction of a therapeutic benefit. 94
CLINICAL TRANSLATION OF GENE EDITING
Gene editing trials for hemoglobinopathies are present at different stages and listed in Table 2, with disclosed results reported here. The most advanced program of gene editing in hemoglobinopathies exploits CRISPR/Cas9 disruption of BCL11A erythroid enhancer to reactivate endogenous HbF expression and is sponsored by CRISPR Therapeutics and Vertex Pharmaceuticals (NCT03655678, NCT03745287).
Clinical trial of gene editing
CRISPR/Cas9, clustered regularly-interspaced short palindromic repeats and cas (CRISPR associated) nucleases; ZFN, zinc finger nuclease.
Recent results showed clinical efficacy with transfusion independence in 42 out of 44 patients affected by TDT, and resolution of severe vaso-occlusive crisis in most of the 31 treated SCD patients. 65,95 In both trials, the proportion of edited cells at early time points in vivo was >60%. These findings are promising, and further follow-up is required to confirm therapeutic editing in long-term engrafting HSC.
Some cases of delayed neutrophil engraftment and thrombocytopenia deserve better investigation of the procedure on specific progenitors. A similar strategy was applied in Bioray sponsored phase 1/2 trial for TDT (NCT04211480), with six patients successfully achieving transfusion independence. 96,97
In Sangamo (NCT03432364) and Sanofi trials (NCT03653247), ZFNs were used to disrupt the BCL11A enhancer in TDT and SCD, respectively. Lack of efficacy was observed in five TDT patients, 98 whereas no recurrence of vaso-occlusion events was reported in four SCD patients. 99
Recently, Novartis trial (NCT04443907) reported the use of CRISPR-Cas9 disruption of the HBG1 and HBG2 gene promoters for HbF induction in SCD. Three patients received OTQ923 product and showed an improvement in hematological levels and some reduction of symptoms. 100
Correction of SCD mutation by HDR was developed by Graphite Bio by CRISPR-Cas9 and rAAV6-based HSPC gene editing, with the first patient treated in 2022 (NCT04819841). As of January 2023, the trial was voluntarily paused due to a serious adverse event likely related to study treatment (
In the context of base editing, advantage is funded on the precise intervention at single base level with limited off-target activity and reduced cell response to DNA engineering. However, basic research studies are highlighting unpredicted findings, such as bystander editing or activation of the IFN pathway in human HSPCs alerting for intensifying non-clinical studies. Recently, a phase1/2 clinical trial in SCD patients started with the administration of autologous HSPCs edited by ABE to mimic HPFH variants (NCT05456880), but results are not yet available.
FACTORS INFLUENCING GENE THERAPY EFFICIENCY AND OUTCOME
The clinical outcome of gene therapy for hemoglobinopathies depends on several factors, including the patient's clinical condition, the dose and quality of the engineered engrafting HSPCs, and the status of the recipient bone marrow (BM) microenvironment. 101 In the autologous setting, the additive effect of an impaired HSC function and a suboptimal supporting activity by the BM niche components can hamper the engraftment of genetically modified HSCs.
Although many studies explored the BM microenvironment regulation on induced stress signals or in malignancies, 102 HSC-niche interactions were under-investigated in hematological inherited disorders. Recently, alterations in BM microenvironment and HSC function were shown in both β-thalassemia and SCD. 5,6,103 In hemoglobinopathies, HSCs have decreased frequency, higher cycling activity, increased DNA damage, inflammation, and accumulation of ROS, associated with a reduced hematopoietic supportive ability of osteolineage and mesenchymal stromal cells.
Moreover, despite iron chelation therapy in TDT populations, there is still evidence that high iron and ferritin levels affect the mesenchymal stem cell support of HSCs. 6 Stromal injury may influence the quality of HSCs mobilized for ex vivo gene therapy/editing and the success of engraftment. In recent trials for hemoglobinopathies, some cases of absence of clinical benefit, despite the occurrence of early hematopoietic engraftment, have been reported. 46,47,50
Low levels of genetically modified HSPCs in patients with a lack of clinical benefit might be related to an impaired HSC function, as well as to a defective supporting activity by niche components, although the root causes have not been clarified yet.
Further, recent cases of myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) in SCD gene therapy trials (NCT04293185) 104 raise the need for a better characterization of the BM microenvironment in hemoglobinopathies toward the potential targeting of niche defects to ameliorate therapeutic outcomes. 105 Retrospective studies suggest that SCD patients have a high risk of development of hematological malignancies. 106 –108
Other studies indicate the presence of preceding clonal hematopoiesis (CH) evolving to MDS/AML when allogeneic transplant engraftment failed 109 and point out the need to quantify the burden of CH in SCD. So far, whole genome sequencing and whole exosome sequencing studies missed to identify somatic clones carrying CH, due to their low of frequency. 110 –112
Therefore, improving our comprehension of the status of the BM niche, the HSC biology, and their functional crosstalk in hemoglobinopathies is mandatory to optimize the long-term clinical outcomes.
The reduced number of HSPCs available for collection and the impaired engraftment potential in patients treated by gene therapy suggests potential limitations for clinical benefit, resulting in variability of gene therapy/editing efficiency and in vivo reconstitution. It is reasonable to expect that the status of the BM microenvironment influences the quality of HSPCs harvested for genetic engineering, as well as their engraftment and reconstitution capacity once transplanted.
Mobilization of HSPCs from BM to peripheral blood with granulocyte-colony stimulating factor (G-CSF) and/or plerixafor is commonly used to increase CD34+ cells harvest in gene therapy trials, although G-CSF is avoided in SCD because of severe adverse events due to the activation of neutrophils, endothelial cells, and platelets that affect the adhesion of RBCs to endothelial cells. Several factors can affect the yield of HSPCs from leukapheresis in SCD patients, including lower baseline CD34+ counts in the BM, higher baseline WBC and platelet counts, older age, and clinical severity. 113
One of the most relevant factors limiting efficacy of gene editing is the DNA damage response and p53 activation evoked by Cas9-mediated DSBs, leading to functional HSC impairment, and reduced in vivo engraftment. In HDR gene editing, this effect is amplified using rAAV6 as donor template. Mitigation strategies, including a different platform for template delivery, and inhibition of P53 have been successfully tested. 114
GENOMIC PERTURBATION BY GENE THERAPY
Undesired effects of LV
Genotoxicity represents a potential risk in gene therapy approaches using integrating vectors, due to their random insertions in the genome possibly activating oncogenes or repressing tumor suppressor genes. The use of self-inactivating LV reduced the safety concerns associated with first-generation gamma-retroviral vectors, with a recent meta-analysis survey highlighting the safety profile of LV-based ex vivo gene therapy. 115
Limited vector associated trans-activation of neighboring genes is expected in hemoglobinopathies due to the erythroid-specific activity of transgene promoter and enhancer elements. 34 In addition, analysis of potential induction of read-through transcription originated from provirus by aberrant splicing sites showed a lower probability to interfere with expression of neighboring genes. 116,117
In the first patient treated for β-thalassemia with Lentiglobin, a benign hematopoietic clone expanded due to the activation of tumor-associated transcription factor HMGA2 but no malignant expansion was observed in NHP with clonal expansion of cells expressing 3′ UTR-truncated form of HMGA2 more than 7 years post-transplant. 37,118
More recently, two AML cases were reported in SCD patients in clinical trials NCT02140554 and NCT042293185. Although the mechanism has not been fully understood, molecular analysis did not correlate AML to LV insertional mutagenesis but to SCD predisposing factors, as mutations in oncogenes or chromosomal alteration already present at baseline. 56,104
So far, at 15 years from the first patient treated, LV-mediated gene addition in hemoglobinopathies has been proven safe.
Undesired effects of gene editing
Off-target genome editing, including undesired point mutations, deletions, insertions, or inversions, can be a byproduct that requires rigorous monitoring both in non-clinical studies and in clinical trials. Targeting the β-globin locus nucleases generates off-target mutations at the δ-globin gene due to high sequence similarity. 79 In addition, rearrangements occurred in ZNFs edited CD34+ cells with intergenic deletion between targeted β-globin and δ-globin sites followed by inversion of the intergenic fragment. 119
To mitigate these phenomena, high-fidelity versions of Cas9 resulted in a 20-fold reduction in off-target compared with a wild-type Cas9, 120 and a modified version of gRNAs with <20 nucleotides allowed a 5,000-fold reduction in off-target. 121
Even if BEs lack DSBs, they can generate limited off-target DNA and RNA mutations, 122 such as bystander cytosine deamination that can be reduced by restricting the editing frame. 123 Several ABE and CBE have been generated with alternative PAM sites, or even PAM-less, to improve the precision of base editing technology. 124
Various methods for off-target effects detection have been developed so far, which are reviewed in Guo et al. 125 However, highly sensitive, unbiased, and precise assays are still a challenge for gene editing approaches. It has been shown that Cas nucleases could also generate long deletions and large insertions in human cells, including HSPCs. 114,126 –129
Gene editing chromosomal translocation events between on and off-target sites can occur, and they could persist in vivo, 130 –132 and also events of chromothripsis caused by rearrangements of targeted chromosomes. 133,134
CONCLUSIONS
Among hemoglobinopathies, β-thalassemia e SCD have provided the ground for the development of innovative advanced medical therapy and the sole diseases where the outcome from clinical trials of gene therapy via gene edition and via gene editing can be compared.
At more than 15 years from the first patient treated with LV-based gene therapy the analysis of risk-benefit and future challenges gives the opportunity for a more general consideration about sustainability and design of an exportable model of advanced therapies in a large patient population and in lower-income countries.
Although the recent advent of gene editing holds the promise of exact and potentially safer genetic engineering, the technical, economic, and ethical challenges bound to the ex vivo HSC manufacturing and transplantation remain. In addition, while the monitoring of genotoxicity by LV integration analysis for several years during patients' follow-up has become a consolidated practice, the field of gene editing lacks validated and sensitive methods to test and predict unwanted consequences at genomic level.
In addition, the clonal composition of the graft and absence of clonal dominance are relevant parameters for the prediction of a safely reconstituted hematopoiesis that needs specifically developed assays for gene edited cells. Thus, although initial results are encouraging, the jury is still out.
Footnotes
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
No funding was received for this article.