
Abstract
Oncolytic viruses (OVs) are appealing anti-tumor agents. But it is limited in its effectiveness. In this study, we used combination therapy with immune checkpoint inhibitor to enhance the antitumor efficacy of OVs. Using reverse genetics technology, we rescued an oncolytic influenza virus with the name delNS1-GM-CSF from the virus. After identifying the hemagglutination and 50% tissue culture infectivedose (TCID50) of delNS1-GM-CSF, it was purified, and the viral morphology was observed under electron microscopy. Reverse transcription quantitative-polymerase chain reaction (RT-qPCR) was used to identify the level of GM-CSF expression in delNS1-GM-CSF, and the GM-CSF expression level was determined after infection with delNS1-GM-CSF by enzyme linked immunosorbent assay (ELISA). To study the tumor-killing effect of delNS1-GM-CSF, we utilized the hepatocellular carcinoma (HCC) tumor-bearing mouse model. To examine signaling pathways, we performed transcriptome sequencing on mouse tumor tissue and applied western blotting to confirm the results. Changes in T-cell infiltration in HCC tumors following treatment were analyzed using flow cytometry and immunohistochemistry. DelNS1-GM-CSF can target and kill HCCs without damaging normal hepatocytes. DelNS1-GM-CSF combined with programmed cell death 1 blockade therapy enhanced anti-tumor effects and significantly improved mouse survival. Further, we found that combination therapy had an antitumor impact via the janus kinase-signal transducer and activator of transcription (JAK2-STAT3) pathway as well as activated CD4+ and CD8+T cells. Interestingly, combined therapy also showed promising efficacy in distant tumors. DelNS1-GM-CSF is well targeted. Mechanistic investigation revealed that it functions through the JAK2-STAT3 pathway. Combination immunotherapies expected to be a novel strategy for HCC immunotherapy.
Introduction
Hepatocellular carcinoma (HCC) is a global health problem with increasing incidence and mortality. 1,2 Of the five deadliest cancers, it is the only one that increases in incidence every year, 3 and is expected to affect more than one million people worldwide each year by 2025. 4 Unfortunately, current treatment for HCC remains unsatisfactory. Although surgical treatment is still the first choice for HCC, 4 the indication for surgery is extremely restrictive, with an overall 5-year survival rate of 60–70%. 5 The majority of patients had a late diagnosis of liver cancer and were found to be in mid-to-late stages.
At the same time, the presence of multiple tumors, vascular invasion, and many complications prevent the patient from undergoing surgical treatment. 6 It is estimated to eventually result in more than half of patients receiving systemic therapy, 7 but patient survival is reported to be less than 1 year. 8,9
HCC development and progression is associated with evasion of immune responses via multiple mechanisms, including positive regulation of immune checkpoints, namely programmed cell death 1 (PD-1) and its ligand (PD-L1). 10,11 The overexpression of PD-L1 by tumor cells enhances the negative feedback of PD-1/PD-L1 interaction on the immune response, hindering the immune response to epitopes associated with cancer. 12
In the first-line setting, two PD-(L) 1 inhibitors, namely, durvalumab and telezolizumab, were recently shown to be noninferior to sorafenib for OS in the phase III HIMALAYA trial and RATIONALE-301 (telezolizumab: hazards ratio [HR] 0.86; 95.67% confidence interval [CI] 0.73–1.03, duvalumab: HR 0.85, 95.003% CI 0.712–1.019). 13 In subsequent clinical studies, however, the Phase III trials of nivolumab and pembrolizumab failed to reach their primary endpoints. 14,15
This also exposes two problems with PD-1 therapy: most patients are resistant to PD-1 blockade and susceptible to immune tolerance due to the formation of an immunosuppressive microenvironment, 16 –19 and the rate of PD-1 response to treatment is relatively low. 20 Single PD-1 treatment is unsatisfactory and must be combined with alternative treatments to improve therapeutic outcomes. 21 It is important to note that the rate of adverse effects can be high with combination therapy. 22 Therefore, exploring safer and more effective PD-1 combination therapies is crucial for improving the prognosis of HCC patients.
Oncolytic viruses (OVs) hold great potential for cancer therapy. 23,24 OVs can selectively infect tumor cells, replicate indefinitely, lyse, destroy tumor cells, and release progeny virus particles without damaging normal tissue, 25 which can further expose tumor antigens to antigen-presenting cells (APCs). 26 In the early development of analytic virotherapy, naturally occurring OVs were used without modification of biological ones, and their specificity against tumors was limited. 27,28
Treatment outcomes for these viruses have not been satisfactory in terms of safety and efficacy. 28 But these studies have indicated that OVs can act as vectors to deliver foreign genes into tumors, thus modifying the tumor microenvironment. 29 As a result, research directions in the therapy of OVs have begun to focus on the application of reverse genetics techniques to modify wild-type viruses.
Modified OVs carrying genes encoding immunostimulatory molecules can enhance antitumor efficacy by combining oncolytic and immunotherapy. Several studies have evaluated novel OVs in various cancer models, such as OVs carrying genes encoding inflammatory cytokines that can alter the tumor microenvironment to augment the anti-tumor response. 30 In 2015, Talimogene laherparepvec (T-VEC) was approved by the Food and Drug Administration for use in the treatment of metastatic melanoma.
The gene encoding granulocyte-macrophage colony stimulating factor (GM-CSF) was inserted into herpes simplex virus. The inserted GM-CSF was allowed to modulate the immune microenvironment to kill the tumor cells better. 31 GM-CSF enhances antibody-dependent cell mediated cytotoxicity by promoting macrophage activity and numbers, to reverse immunosuppression in the tumor microenvironment, and to increase the activity of cytotoxic T cells to augment antitumor effects. 32
Due to the limited variety of OVs, there is a need to study viral vectors to improve their targeting and safety. It was discovered that the influenza A virus A/PR/8/34 (PR8), which is a single-stranded, segmented, negative strand RNA virus, can be used as an oncolytic virus vector. 33 More importantly, armed recombinant optimized OVs have the potential to alter the tumor immune microenvironment. 34
Using reverse genetics, we inserted the exogenous GM-CSF fragment into the NS fragment of influenza A virus PR8 and named it delNS1-GM-CSF. 35 It not only stimulated the body to produce an effective immune response but also expressed GM-CSF proteins that modify the tumor microenvironment and play an anti-tumor role. Our hypothesis was that delNS1-GM-CSF, a recombinant oncolytic influenza virus, would synergize with anti-PD-1 antibodies to maximize the therapeutic effect. For this reason, we chose to combine delNS1-GM-CSF with an anti-PD-1 antibody to explore the efficacy and mechanism of action of combination therapy on HCC.
Materials and Methods
Cells, viruses, and animals
The canine kidney cell MDCK was purchased from the Shanghai Institute of Life Sciences of the Chinese Academy of Sciences. Mouse hepatoma H22 and HepG2 were purchased from the ATCC (American Type Culture Collection) Cell Bank. Influenza virus A/PR/8/34 (PR8) and recombinant oncolytic influenza virus delNS1-GM-CSF were stored in our laboratory (The construction of delNS1-GM-CSF has been described in detail in our previous article 35 ).
The SPF chicken embryos were purchased from Boehringer Ingelheim (Beijing). Balb/c nude mice (6–8 weeks) and female Balb/c mice (6–8 weeks) were purchased from Spfbio (Beijing). All animal experiments were performed in accordance with the guidelines of the Experimental Animal Ethics Committee at the Fifth Medical Center of the PLA General Hospital.
Hemagglutination test
The delNS1-GM-CSF and PR8 were inoculated into 9-day-old SPF chicken embryos in 200 μL per embryo. Inoculated chick embryos were incubated in an incubator at 37°C for 72 h and then transferred to a refrigerator at 20°C for 1 h. The chick embryo allantoic fluid was collected for the determination of hemagglutination titer.
50% Tissue culture infectivedose
Cell plates were dispensed with MDCK cells, and cell counting was performed. The original upper medium was discarded and washed with phosphate-buffered saline (PBS). The virus was serially diluted 10–1 times and inoculated into the cell plate, the cell state was observed, and the results were judged after 5 days.
Concentrate and purify the virus
The delNS1-GM-CSF and PR8 were diluted 1:1,000 times and injected into 9-to-11-day-old chicken embryos. After 72 h, all positive fluid was collected and centrifuged at 4,000 r/min for 25 min; the sediments were discarded, the supernatant was centrifuged at 32,000 r/min for 2 h, and the precipitate was resuspended in PBS in 30% and 60% sucrose gradients by centrifugation, The virus band was then centrifuged at 32,000 rpm for 4 h before being resuspended in PBS. Purified delNS1-GM-CSF and PR8 were obtained by 2 h centering at 32,000 r/min.
Transmission electron microscope
The delNS1-GM-CSF was diluted 1:500-fold and injected into 9-to-11-day-old chicken embryos at 0.2 mL per egg. The allantoic fluid from the positive chicken embryo was collected, concentrated, and purified after 72 h in culture. Negative stain transmission electron microscopy (Phenom, Holland) was used to observe the morphology and size distribution of the delNS1-GM-CSF.
Enzyme linked immunosorbent assay
The GM-CSF antibody content was manipulated according to the instructions of the kit (HGM-CSF-100; Alpha Diagnostic International). The standard material, GM-CSF antibody, PBS control, and PR8 were added at a concentration of 50 μL/hole, respectively. OD values were detected with a microplate reader at a wavelength of 450 nm, and an absorbance more than 2.1 times greater than that of the control group was determined to be a positive result.
Reverse transcription quantitative-polymerase chain reaction
The RNA of delNS1-GM-CSF was extracted by the Trizol method and reverse transcribed into cDNA, then detected by real-time fluorescence quantitative PCR (probe method). The quantitative primers and the corresponding probe information are shown below. The M1 gene was amplified with the universal primers of the influenza A virus. The primers used to strengthen the M1 gene were influenza M1 5′-aagaccatctctg3′. 5′-caaacgtctacgcagtcc-3′, mouse GAPDH 5′-aggtcggtgaacggattgg-3′; 5′-tgtagaccatgtaggtca-3′, and the expression level of the delNS1-GM-CSF was analyzed by 2-ΔΔCq method using the expression level of internal reference gene GAPDH as a standard. qPCR was performed with an ABI 7500 polymerase chain reaction system in three wells of a 96-well reaction plate.
Establish a nude mouse model of HCC
Six- to eight-week-old Balb/c nude mice were injected intraperitoneally with H22 mouse hepatoma cells, and ascites were formed after 5–7 days. Ascites were harvested, centrifuged at 3,000 r/min for 5 min, discarded, and resuspended in PBS to a cell count of 5 × 106/mL, which was acceptable as supernatant. The H22 cells subcutaneously were injected into the right groin of the mice, and the cells were injected randomly into the right groin of the mice based on tumor volume and body mass.
Once the subcutaneous tumor reached 80–120 mm3, recombinant oncolytic influenza virus (107/100 μL) was injected into the tumor for 5 consecutive days. The PBS group received the same volume of PBS concurrently.
In the unilateral tumor model, 5 × 106 H22 cells were subcutaneously injected into the right groin skin of each mouse. In the bilateral tumor model, 5 × 106 H22 cells were subcutaneously injected into the proximal side of each mouse and 3 × 106 H22 cells were subcutaneously injected into the distal side of each mouse. When tumor size on the delivery side increased to 80–120 mm3, the mice were randomly divided into five groups: PBS group, PR8 group, delNS1-GMCSF group, antiPD-1 group, and delNS1-GMCSF+AntiPD-1 group; each group consisted of six animals that were inoculated with PBS solution, PR8 virus suspension, delNS1-GM-CSF virus suspension, PBS solution, and delNS1-GM-CSF virus suspension for 5 consecutive days.
The dose of PBS was 100 μL, and the quantity of virus was 107 PFU/100 μL. The Anti-PD-1 group and the delNS1-GM-CSF+ Anti-PD-1 combination group received 0.2 mg of anti-PD-1 antibody (BE0273; BioXcell) every other day starting at day 7, and the remaining groups received the same amount of PBS solution in the tail vein. Tumor volumes were measured every other day, and mouse vital signs were recorded. On the 7th day after the end of administration, mouse tissue samples were taken for subsequent experiments.
Hematoxylin and eosin staining distal side
Tissue samples from the heart, liver, spleen, lung, kidney, brain, and tumor were fixed in 4% formalin and then embedded in paraffin wax. Sections were cut into 5 μm thick sections, deparaffinized and hydrated, stained with hematoxylin and eosin (HE), dehydrated through a gradient, cleared in xylene, and sealed with neutral glue.
Flow cytometry
Mouse spleen tissues were harvested, minced, and filtered. To isolate mouse lymphocytes, a 6 mL mouse lymphocyte separation solution (7211011; Dakewe) was slowly added to the filtrate along the wall of the centrifuge tube. One microliter PE/Cyanine7 anti-CD3 antibody (100220; Biolegend), FITC anti-CD4 antibody (100406; Biolevend), and PerCP/Cyanine5.5 anti-CD8a antibody (100733; Biolevend) were added to each tube of the sample (Biolelend) APC/Cyanine7 anti-CD45 (103115; Bioleeng); Brilliant Violet 510 anti-CD69 (104532; Bioledeng) antibody, and 100 μL FACE wash solution were added, blown, and mixed well.
The mixed solution was incubated at 4°C for 30 min in an environment protected from light, excess antibody was washed away with an fluorescence activated cell sorting wash solution, centrifuged at 8,000 r/min for 1 min and the supernatant was discarded. A 200 μL/tube of 4% paraformaldehyde was added to the pellet, held out of the light for 20 min, and analyzed by flow cytometry.
Immunohistochemistry
Tumor tissue was freshly isolated and fixed with 4% paraformaldehyde, embedded in dehydration, dewaxed, hydrated, cut into 5 μm-thick sections, and sealed with 5% serum bovine serum albumin for 2 h at room temperature, and then added along with anti-Ki67 from the mouse (huaxingbio, Product number, Clone, 1:300, p < 0.05).), mouse anti-CD4 (1:1,000, 25228; Cell Signaling) and mouse anti-CD8 (1:500, 9891; Cell Signaling), incubated overnight at 4°C, washed with PBST the next day, and incubated with goat anti-rabbit secondary antibody (1:500; Abcam) at room temperature (1:2,000; ab205718), then incubated for 1 h, washed with DAB solution for color development, counterstained again with hematoxylin, and observed under a microscope.
Transcriptome sequencing
After the mice were sacrificed, the tumor tissues of each group were obliterated and sent to CapitalBio Technology (Beijing) for transcriptome sequencing analysis.
Bioinformatics analysis
Differential analysis of gene expression data between the PBS group and delNS1-GM-CSF combined with the PD-1 blockade group was performed using the “DESeq2” package. Gene ontology (GO) enrichment analysis and KEGG pathway analysis of differential genes were analyzed with the “clusterProfiler” package and “clusterProfiler” package.
Western blotting
HepG2 cells were infected with the oncolytic virus at multiplicity of infection of 0.1 and 1, respectively. The supernatant was collected after 48 h of incubation and then centrifuged at 10,000 r/min for 2 min. Cells were seeded in 1 × RIPA buffer and 1 × protease inhibitors for 15 min, followed by centrifugation at 10,000 r/min for 2 min. The separated protein samples were electrophoresed on a 12% polyacrylamide-sodium dodecyl sulfate gel and transferred onto a nitrocellulose membrane.
The cells were blocked in a TBS buffer containing 5% skim milk at room temperature for 60 min. Use of anti-janus kinase (JAK2) primary antibodies (1:1,000, ab108596; Abcam), signal transducer and activator of transcription (STAT3) (1:500, PTM-5090, 12680; Abcam), p-JAK2 (1:1,000, ab32101, clone; Abcam), p-STAT3 (1:1,000, ab76315; Abcam), and β-action (1:1,000, 4967S; Cell Signaling) primary antibody were incubated overnight at 4°C, washed with TBST, and incubated with sheep anti-rabbit secondary antibody (1:10,000, HX2030; Huaxingbio) for 1 h at room temperature. Exposures were carried out using Odyssey Imager. β-action served as an internal control.
Statistic analysis
Statistical analysis was performed using GraphPad Prism 8.0 software (Los Angeles, California, USA) and R 4.2.2. The data were analyzed by paired t-test using GraphPad Prism 8.0 software. The log-rank test assessed the significance of the Kaplan-Meier survival curves (SPSS software, version 16.0; SPSS, Inc., Chicago, IL, USA). Differences were deemed to be statistically significant when p < 0.05.
Results
Construction and identification of the delNS1-GM-CSF
The open reading frame sequence of GM-CSF genome was inserted into the NS plasmid sequence of PR8 main frame. A schematic diagram of the structure of the recombinant oncolytic virus plasmid is shown in Fig. 1A. The recombinant plasmid was subsequently cloned into the phw2000 vector. The enzyme-cleaved fragments were electrophoresed on an agarose gel, as shown in Fig. 1B, and the size of the fragments was as expected.

Construction and identification of pfluΔns/GM-CSF.
These results indicated the successful construction of the recombinant pfluΔns/GM-CSF plasmid. Transmission electron microscopy after purification of the delNS1-GM-CSF by gradient analysis revealed that the delNS1-GM-CSF was spherical, the nucleocapsid surface was coated with a capsule, and the surface of the envelope was covered with cilia (Fig. 1C). Viral particles were between 80 and 120 nm in size (Fig. 1D).
This analysis indicated that the morphological structure and particle size of the delNS1-GM-CSF were consistent with typical features of influenza viruses. DelNS1-GM-CSF was continuously amplified in chicken embryo. The hemagglutination titer on SPF chicken embryo is 28–29 (Fig. 1E). The titer of delNS1-GM-CSF virus was measured on MDCK cells, reaching 107–8 log10TCID50/mL (Fig. 1F).
DelNS1-GM-CSF targeted killing of HCC
After viral inoculation in the H22 HCC hormonal Balb/c nude mouse model, we took nude mice for examination of each organ. We found that delNS1-GM-CSF was present and replicated in significant amounts only in tumors. DelNS1-GM-CSF was not detected in the heart, liver, spleen, kidney, and brain; the concentration of delNS1-GM-CSF in tumors was significantly higher after administration than before, and the replication of delNS1-GM-CSF was time-dependent (Fig. 2A).
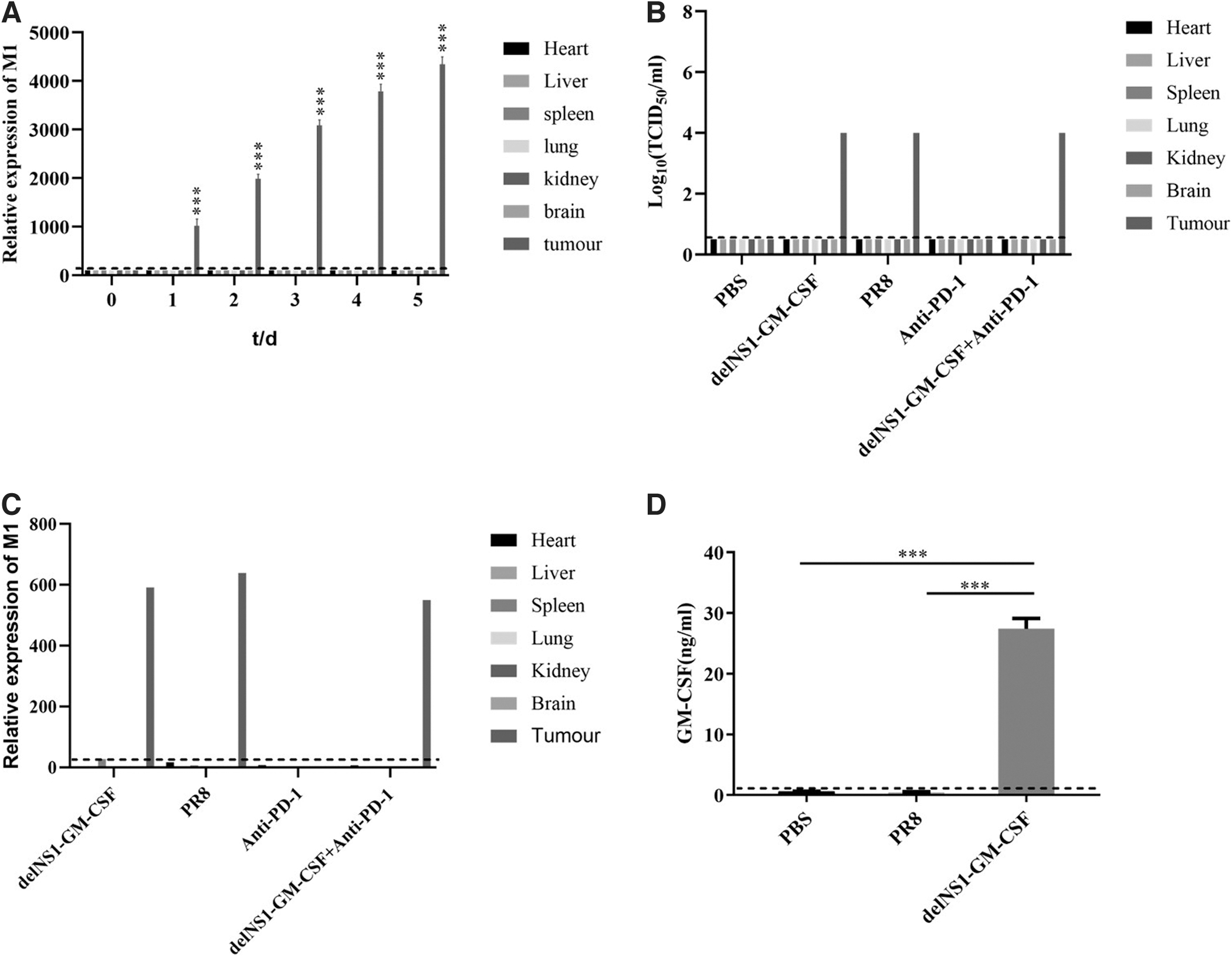
HCC targeting of delNS1-GM-CSF and GM-CSF protein expression.
The 50% tissue culture infectivedose (TCID50) assay was performed on the tissues of each group, and the results showed that the delNS1-GM-CSF had the same virulence as the backbone of the original influenza virus. The insertion of the exogenous gene did not affect the virulence of the actual influenza virus (Fig. 2B). Subsequently, we assessed the bio distribution and replicative capacity of the virus by RT-qPCR, and we observed a large amount of virus replication in the tumor site of delNS1-GM-CSF.
Nevertheless, we did not detect the presence of delNS1-GM-CSF in the mouse's heart, liver, spleen, lung, kidney, and brain (Fig. 2C). These results indicated that the delNS1-GM-CSF could target enrichment, which is only enriched in the tumor but not in the other important organs. Further, to further test whether GM-CSF fragments inserted by delNS1-GM-CSF could lead to changes in peripheral serum GM-CSF protein levels, serum samples were collected from different groups 2 days after treatment of H22 tumor-bearing mice.
The results showed that the expression of GM-CSF in the serum of the delNS1-GM-CSF group was higher than that of the other two groups (p < 0.01), which indicated that the foreign gene GM-CSF in delNS1-GM-CSF was successfully expressed.
The combination of delNS1-GM-CSF with PD-1 blockade has an excellent therapeutic effect on HCC
The subcutaneous H22 tumor model was established in immunoreactive mice. We adopted the dosing regimen demonstrated in the figure (Fig. 3A) to assess whether delNS1-GM-CSF combined with PD-1 blockade synergistically enhanced the antitumor immune response. Tumor growth for each group was shown in Fig. 3B and D. In this study; we observed that administration of delNS1-GM-CSF in combination with PD-1 blockade significantly inhibited tumor growth, which was found to be more significant than both the delNS1-GM-CSF and PD-1 blockade groups (p < 0.05).

Enhanced antitumor activities against primary tumors.
Based on these findings, delNS1-GM-CSF and PD-1 antibody might synergistically treat HCC to enhance each other's antitumor effect. Most importantly, the delNS1-GM-CSF and PD-1 blockade significantly prolonged survival (Fig. 3C). All the mice in the PBS group died on day 27, and all the mice in the delNS1-GM-CSF group combined with PD-1 blockade group were in good condition on day 33, indicating that delNS1-GM-CSF combined with PD-1 blockade could significantly benefit mouse survival.
In addition, the delNS1-GM-CSF combined with the PD-1 blockade group had significantly smaller tumor volume and weight than the other groups (p < 0.01) (Fig. 3E, F). These results proved that delNS1-GM-CSF with PD-1 blockade has an excellent therapeutic effect on HCC.
Safety evaluation of the delNS1-GM-CSF
DelNS1-GM-CSF has been shown to have excellent targeting and no killing effect on normal hepatic cells. To further evaluate the safety of delNS1-GM-CSF in other vital organs, we killed mice and pathologic sections of the liver and lung were stained with HE staining. This analysis revealed that lung and liver tissue from delNS1-GM-CSF treated mice did not have significant pathological changes. Likewise, no significant pathologic changes were detected in the heart, liver, spleen, kidney, and brain (Supplementary Fig. S1). HE staining demonstrated that treatment with delNS1-GM-CSF did not damage other organs and was safe.
DelNS1 GM-CSF combined with PD-1 blockade exerted anti-tumor effect through JAK2-STAT3 pathway
To explore the mechanism of action of delNS1 GM-CSF in combination with PD-1 antibodies in treating HCC, we performed transcriptome sequencing on each set of tumor tissues. Subsequently, we analyzed the transcriptome data of the delNS1 GM-CSF combined with anti-PD-1 and the PBS treatment groups. Figure 4A and B show the sequence and volcano plots of the differential expression genes screened.

Analysis of the mechanism of delNS1-GM-CSF.
The differentially expressed genes were then analyzed for KEGG and GO enrichment. These differential genes were primarily enriched for immune-related functions and may exert anti-tumor effects via the JAK2-STAT3 signaling pathway (Fig. 4C). Protein expression in the JAK2-STAT3 signaling pathway before combined treatment at the cellular level is shown in Fig. 4D.
Figure 4E explained the protein of the JAK2-STAT3 signaling pathway proteins after combined treatment in tissue. As can be seen, the combination treatment inhibited the expression of JAK2, STAT3, p-JAK2, and p-STAT3 in the JAK2-STAT3 signaling pathway. These results suggested that delNS1-GM-CSF combined with PD1 antibody treatment inhibited cancer proliferation and metastasis by inhibiting the JAK2-STAT3 signaling pathway.
The combination of delNS1-GM-CSF and PD-1 blockade activated CD4+ and CD8+ T cells
In previous studies, OVs have been shown to exert antitumor effects primarily through activating CD4+ T cells and CD8+ T cells. Our transcriptome analysis supported this. Flow cytometry analysis of single-cell suspensions from the spleen of mice showed the percentage of CD4+ CD69+ T cells in spleen cells from tumor-bearing mice treated with delNS1-GM-CSF plus PD-1 blockade to be 18.9% (Fig. 5A), and the CD8+ CD69+ T cell rate to be 16.6% (Fig. 5B). In contrast, CD4+ CD69+ and CD8+ CD69+ T lymphocytes in the control group accounted for only 4.98% and 4.77% of lymphocytes, respectively.

Treatment with delNS1-GM-CSF plus Anti-PD-1 activated both CD4+ and CD8+T cells.
There was a significant increase in the number of splenic lymphocytes in the delNS1-GM-CSF combined with the anti-PD-1 group (p < 0.01). The results of this study indicated that delNS1-GM-CSF combined with PD-1 blockade treatment could activate T cells from tumor-bearing mice and enhance adaptive immunity in mice. In addition, we examined lymphocytic infiltration into tumor tissues.
We verified by HE and IHC staining that tumor tissues from the delNS1-GM-CSF combined with the anti-PD-1 group exhibited extensive necrosis compared with the other groups. The tumor tissues in the delNS1-GM-CSF combined with the anti-PD-1 group showed a significant increase in CD4+ and CD8+ T cells, and the degree of infiltration was considerably higher than in other treatment groups (Fig. 5C).
The staining levels of Ki67 in the PBS control and monotherapy groups were remarkably higher than those in the delNS1-GM-CSF combined with the anti-PD-1 group. This observation also meant combining delNS1-GM-CSF with anti-PD-1 treatment inhibited HCC cell proliferation. These results suggested that treatment with delNS1-GM-CSF combined with PD-1 blockade could effectively activate T cells and inhibit tumor cell proliferation by stimulating T cells to exert immunotherapy effects.
The combination of delNS1-GM-CSF and PD-1 blockade enhances anti-tumor activity against untreated distant tumors
Using a mouse model of H22 cells bearing subcutaneous tumors, we evaluated whether delNS1-GM-CSF in combination with anti-PD-1 treatment could stimulate a systemic antitumor response. Both sides of Balb/c mice were inoculated with H22 cells, and when the tumor on the administered side reached 80–120 mm3, tumor-bearing mice were treated in different groups as described in the Materials and Methods section. The combination of delNS1-GM-CSF and anti-PD-1 treatment group inhibited the growth of distant H22 tumors that were not treated more efficiently than the other groups (p < 0.05) (Fig. 6A, B).
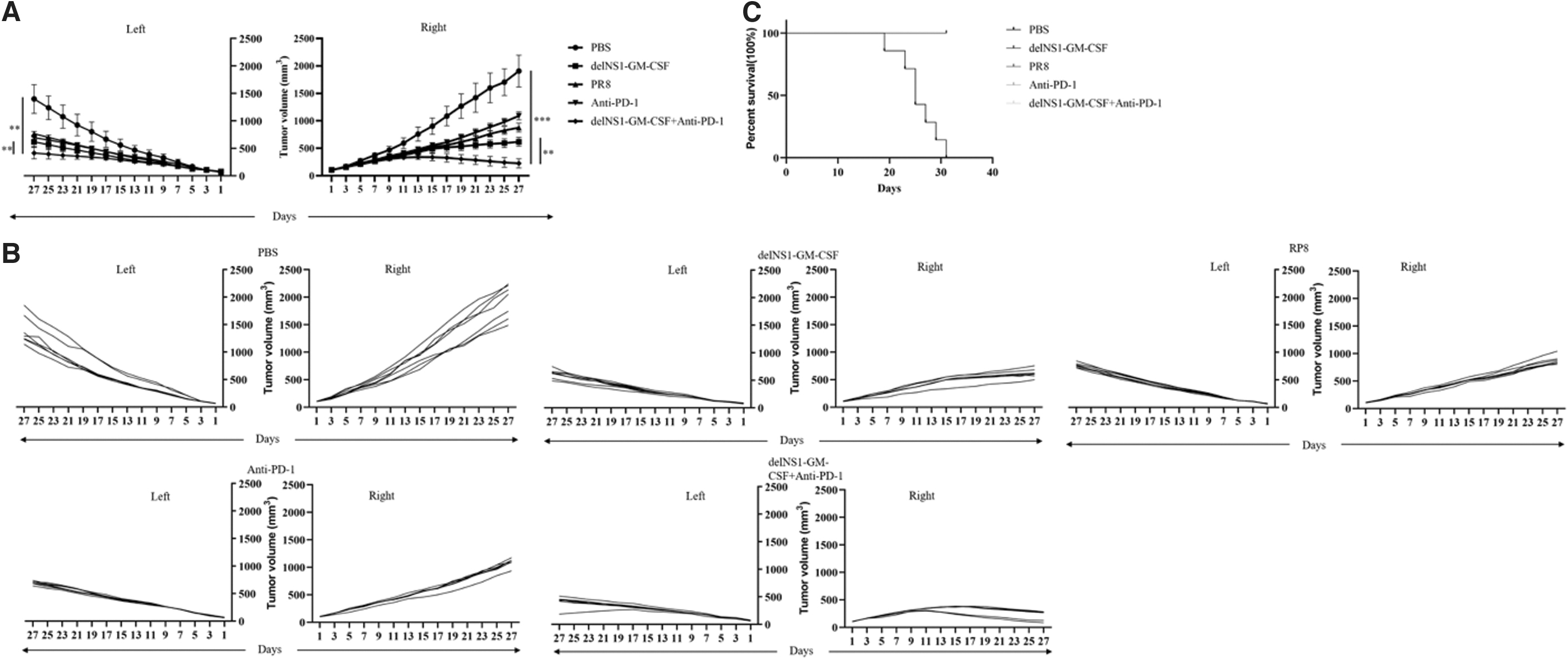
delNS1-GM-CSF plus Anti-PD-1 enhanced antitumor activity against untreated distant tumors.

Besides that, mice receiving delNS1-GM-CSF plus PD-1 blockade had significantly longer survival than those receiving PBS (Fig. 6C). All mice in the PBS group died on day 19, and all of the mice died on day 31. At the end of the study, mice in the delNS1-GM-CSF combination with the anti-PD-1 group remained healthy. In this study, we examined T cell infiltration in untreated tumor tissue. By HE and IHC staining of the tumor tissue (Fig. 6D), we observed that the amount of tumor necrosis on the untreated side was significantly higher in the delNS1-GM-CSF plus anti-PD-1 group compared with the other groups.
There was also a significant infiltration of CD4+ and CD8+T cells in the untreated tumor tissue from the delNS1-GM-CSF plus anti-PD-1 group by staining with IHC, the expression levels of the Ki67 in the delNS1-GM-CSF plus anti-PD-1 group were significantly less than those in the other treatment groups. The results of this study suggested that the combination of delNS1-GM-CSF and PD-1 blockade could activate a systemic immune response that substantially killed the contralateral untreated tumor and inhibited tumor cell proliferation. PD-1 blockade in combination with delNS1-GM-CSF had more excellent systemic antitumor activity than either PD-1 blockade or delNS1-GM-CSF alone.
Discussion
In the present study, we reported that delNS1-GM-CSF could successfully express GM-CSF. PT-qPCR, TCID50, and HE staining of various tissues confirmed the excellent safety profile of delNS1-GM-CSF, which infected only tumor cells, had no effect on normal tissues, and was well-targeted. Moreover, the antitumor effect of delNS1-GM-CSF was more substantial than that of A/PR/8/34 (PR8). One possible explanation for this is that GM-CSF expressed by the foreign gene in delNS1-GM-CSF played an essential role in the anti-tumor effects. GM-CSF was shown to recruit APCs to the tumor microenvironment, enhance dendritic cell function, and promote cytotoxic T cell responses to tumor associated antigens. 36 –38
Prior studies have shown that direct intratumoral injection of a single GM-CSF, which has been shown to have limited preclinical and clinical activity, 39 –41 produces more significant tumor suppression and survival of mice in GM-CSF-armed vector viruses compared with the same virus alone. 42 This was consistent with the results of our study. Our study found that GM-CSF may enhance local expression and immune effects. Local injection of oncolytic tumors activated T cells that induce a distant immune response. 43
Dmitriy et al. 44 also found that topical treatment with OVs caused inflammatory immune infiltration in distant tumors, making them susceptible to systemic therapy with immunomodulatory antibodies. However, delNS1-GM-CSF alone may lower the limit of antitumor immunity. 45
To facilitate the antitumor effect of recombinant OVs, we used combination therapy with immune checkpoint inhibitor (ICI), in line with previous studies by Shi et al. 45 The DelNS1-GM-CSF and PD-1 blockade significantly activated T lymphocytes, had significant anti-tumor effects, inhibited tumor proliferation, and elicited systemic immunity compared with a single virus or antibody.
T cells were critical immune cells that play a role in anti-tumor immunity in the tumor immune microenvironment. T cells can be activated to kill tumor-associated antigens. The major problem is that the majority of patients with cancer remain unable to activate neoantigen-specific T cells spontaneously and are resistant to immune checkpoint blockade.
This mechanism could promote tumor immune escape and avoid T cell recognition of tumor neoantigens and tumor destruction. 46,47 This may be primarily due to the poor presentation of tumor antigens and tumor immunosuppressive microenvironment. Neoantigens on the surface of tumors were particularly relevant. We found a possible synergistic effect of ICIs, oncolytic viral replication, and GM-CSF stimulation to activate specific T-cell responses.
DelNS1-GM-CSF modifies the tumor microenvironment by attracting T cells, which attract dendritic cells that process tumor cell antigens and present them to T cells. T cells were then programmed to recognize and kill tumor cells. 43 In fact, during intratumoral administration of the single-agent delNS1-GM-CSF, we demonstrated an apparent increase in CD4+ and CD8+ T cells and, more importantly, an increase in intratumoral permeability. CD4+ T cells have direct anti-tumor activity, mainly by killing tumors directly via MHC II molecules, 48 whereas CD8+T cells have long-lasting effects against tumor cells. 49
DelNS1-GM-CSF created an immunogenic tumor microenvironment that could enhance the effectiveness of the anti-PD-1 therapy. T cells induced by delNS1-GM-CSF may generate a systemic response in distant tumor metastases following PD-1 antibody blockade. These T cells expressed PD-1, and the tumor cells expressed PD-L1, which may limit the antitumor activity of the single agent delNS1-GM-CSF. We thus maximized the benefit of delNS1-GM-CSF in combination with PD-1 blockade. This resulted in combinations being used beyond either therapy.
Tumor tissue transcriptome analysis was performed, and differentially expressed genes were enriched primarily in the JAK2-STAT3 pathway. Combined treatment with delNS1-GM-CSF and PD-1 blockade has been shown to have anti-angiogenesis and anti-metastatic effects in tumor tissue, mainly via the JAK2-STAT3 pathway.
The JAK-STAT signaling pathway has been commonly expressed in various tumors and directly or indirectly affects cellular proliferation, apoptosis, invasion, metastasis, and angiogenesis. 49,50 Multiple cancers, tumor invasion, and metastasis were significantly associated with the Overactivation of STAT. JAK2 recruited STAT3 to participate in uncontrolled cellular proliferation, angiogenesis, and inhibition of apoptosis to transfer carcinogenic signals. 51
This study used Western blot to detect decreased STAT3 and JAK2 phosphorylation in HepG2 cells and tumors. The phosphorylation of STAT3 and JAK2 was significantly higher in the different groups than in the delNS1-GM-CSF combined with anti-PD1 group. By inhibiting STAT3 activity in hematopoietic cells, the innate immune surveillance system can be activated, thereby inhibiting tumor growth and metastasis. 52
In addition, the level of phosphorylated STAT3 was closely related to T cell response function. 53 It was the leading cause of additional activation of T-cell infiltration in the tumor after combination therapy. STAT3 inhibition reduced the effect of fatty acid oxidation on CD8+ T cells. 54 Similar to the earlier study by Song et al., we also found that delNS1-GM-CSF, in combination with anti-PD1 therapy, inhibited the expression and phosphorylation of STAT3. 55 Most importantly, however, the JAK/STAT3 pathway regulates the immune microenvironment in a PD-1independent manner. 56 This is the reason why delNS1-GM-CSF, in combination with PD1, may exert a synergistic therapeutic effect.
This study aimed at describing the efficacy of treatment with delNS1-GM-CSF and PD-1 blockade and demonstrating the killing effect of the combination on tumor tissue in mice. However, limitations remain in the present study. First, the analysis was performed in mice, and further prospective studies in clinical trials are required to validate its clinical effectiveness. Clinical trials are needed to test the safety and efficacy of delNS1-GM-CSF in patients with HCC. Further, intramuscular drug administration in clinical practice is expensive and challenging, and we need a new method of drug delivery to avoid the above problems.
In summary, delNS1-GM-CSF interacts synergistically with PD-1 antibodies to activate a specific T-cell response and a systemic immune response, inhibiting tumor cell proliferation and angiogenesis and promoting tumor cell apoptosis. It offers effective immunotherapy for liver cancer treatment.
Footnotes
Authors' Contributions
Study concept and design: Ho. Y., F. S., P. Y.; Data extraction: Ho. Y., Ha. Y., C. T., C. L.; Data analysis and interpretation: Ho. Y., F. S., Y. X.; Drafting of the manuscript: Ho. Y., F. S., Y. X.; Critical revision of the manuscript for important intellectual content: F. S., Y. X; Statistical analysis: Y. K., L. H.
Author Disclosure
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.