
Abstract
Inherited kidney diseases are among the leading causes of chronic kidney disease, reducing the quality of life and resulting in substantial socioeconomic impact. The advent of early genetic testing and the growing understanding of the molecular basis and pathophysiology of these disorders have opened avenues for novel treatment strategies. Viral vector-based gene therapies have evolved from experimental treatments for rare diseases to potent platforms that carry the intrinsic potential to provide a cure with a single application. Several gene therapy products have reached the market, and the numbers are only expected to increase. Still, none target inherited kidney diseases. Gene transfer to the kidney has lagged when compared to other tissue-directed therapies such as hepatic, neuromuscular, and ocular tissues. Systemic delivery of genetic information to tackle kidney disease is challenging. The pharma industry is taking steps to take on kidney disease and to translate the current research into the therapeutic arena. In this review, we provide an overview of the current viral vector-based approaches and their potential. We discuss advances in platforms and injection routes that have been explored to enhance gene delivery toward kidney cells in animal models, and how these can fuel the development of viable gene therapy products for humans.
Introduction
Societal and economic impact of chronic kidney diseases
Chronic kidney disease (CKD) affects more than 9.1% of the population worldwide. 1 Unlike other important noncommunicable diseases, CKD-related mortality has increased by more than 40% in the last decades. 1,2 As such, by 2040, CKD is estimated to become the fifth leading cause of death. 1,3 On top, CKD poses an important economic cost. In 2020, treating Medicare beneficiaries with CKD cost $85.4 billion, and treating people with end-stage renal disease cost an additional $50.8 billion. 4 There is no cure for end-stage kidney disease, leaving patients with only the invasive and high-burden treatment options of renal replacement therapy (RRT) comprising dialysis and kidney transplantation. While the advancements in RRT have increased life-expectancy, it has not necessarily improved quality of life, due to its high burden. Current treatments for CKD are symptomatic and supportive and do not target the underlying cause of the disease, which stresses the unmet need in the management of CKD.
Gene therapy as a treatment approach for CKD
Both genetic kidney disorders and systemic conditions that affect the kidneys can lead to CKD. Recently, it has been reported that as much as ∼30% of adult and ∼50% of pediatric cases of nondiabetic CKD are caused by monogenic diseases with more than 600 genes implicated. 5 Monogenic kidney diseases, where changes in a single gene result in abnormalities in the structure or function of the kidney and potentially cause systemic effects with examples, such as polycystic kidney disease (PKD), cystinuria, congenital nephrotic syndrome, and Alport syndrome, are targetable by gene therapeutic approaches. In line, systemic CKD is also amenable to gene therapy, even though it typically is not caused by a single genetic mutation but rather results from the combination of genetic predisposition (multiple genes and genetic variants) in interaction with environmental factors (such as diabetes mellitus, hypertension, autoimmune diseases, infectious diseases, and certain medication side effects). 6
General principles of gene therapy
In its simplest form, gene therapy has the overarching aim to complement, remove, or change genetic information in the patient's cells to treat, or ultimately cure, the disease.
Over the past decades, gene therapy has recorded serious adverse events but also remarkable advances, especially for a wide range of inherited diseases that lacked therapy, or where treatment is complex and/or unsatisfactory. 7,8
Molecular gene therapy approaches include (Fig. 1A): (i) gene replacement, which involves replacement of a nonfunctional gene by a functional copy; (ii) gene addition, where a new gene function is added which, in turn, alters/rescues the cellular function; (iii) gene silencing, which involves suppression of a gene with toxic effects, for example, by siRNA/miRNA-based knockdown; and (iv) gene editing, where the genetic information in the genome of the patient is modified at a specific genomic position, for example, by CRISPR/Cas.
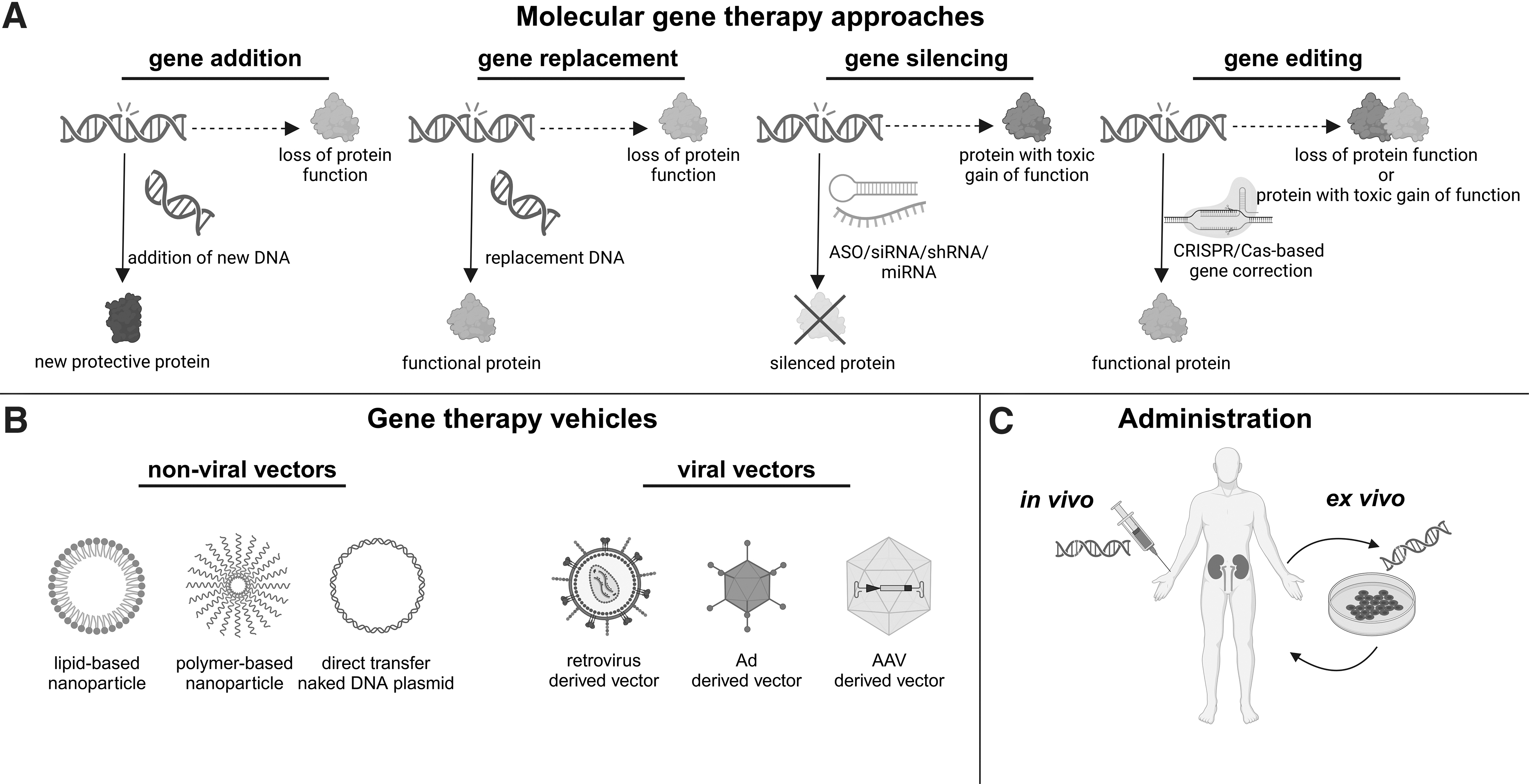
General principles of gene therapy: overview of molecular gene therapeutic approaches
Transfer of the required genetic information to the specific cells and tissues in the patient is facilitated by viral or nonviral vector approaches (Fig. 1B). Viral vectors employ the potential of their parental virus to transfer their genetic information to host cells (Table 1). Nonviral vector approaches comprise physical methods including needle or ballistic RNA/DNA injection and chemical carriers like lipid-based and polymer-based nanoparticles (recently reviewed in Refs. 9,10 ).
Characteristics of viral vectors and their parental viruses
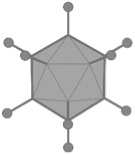
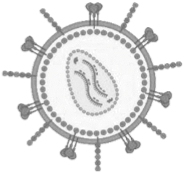
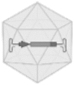
AAV, adeno-associated virus; Ad, adenovirus; AdV, adenoviral vector; dsDNA, double-stranded deoxyribonucleic acid; HAd5, human adenovirus type 5; HIV, human immunodeficiency virus; LV, lentiviral vector; MMLV, Moloney murine leukemia virus; rAAV, recombinant adeno-associated viral vector; RV, retroviral vector; ssDNA, single-stranded deoxyribonucleic acid; ssRNA, single-stranded ribonucleic acid.
In ex vivo gene and stem cell therapies, patient (stem)cells are cultured ex vivo to be genetically modified, and subsequently reinjected to reconstitute the tissue (Fig. 1C). In the past decades, this approach has been shown to be particularly successful for inheritable blood-based immune disorders that could be treated/cured following gene addition approaches (reviewed in Refs. 11,12 ). Yet, blood is an exception to the rule. Indeed, most other tissues do not readily allow for this approach and necessitate genetic material to be delivered directly to the patient through an in vivo approach. Mostly, this implies gene transfer technology to accurately target a specific tissue or subset of cells: some tissues are simple in architecture and complexity, consisting of a few to several different cell types and are easy to target, whereas other tissues are difficult to target, more complex, and, therefore, more challenging.
Given the breadth of molecular gene therapy, an overview of all methods in this field is beyond the scope of this article. In this review, we aim to introduce the reader to the current state of viral vector technology to target diseases affecting the kidney and to provide considerations and perspectives on the potential of viral vector-based gene therapy, including the first steps toward clinical implementation (for a more detailed review see Davis and Park 13 ).
Tackling Genetic Kidney Disorders: Vehicles and Routes to Reach the Kidney
Although there are several approved gene and cell therapies for inherited disorders that affect other organs, none target the kidney directly (also reviewed in Peek and Wilson 14 ). The kidney is a challenging target. Currently, an effective delivery method to the kidney is lacking, first and foremost due to its complex tissue architecture.
For efficient in vivo delivery and long-term effects, viral vectors are currently the preferred and most potent platform, but nonviral platforms, such as lipid nanoparticles (LNPs), are catching up. 15,16 Nanoparticle-based RNA delivery has several advantages, such as cost-effectiveness, flexibility, tuneability, and low immunogenicity (reviewed in Bondue et al. 17 ).
Theoretically, the kidney is accessible via diverse routes of delivery, including (i) systemic (intravenous) delivery, (ii) targeted delivery via the renal artery or intraparenchymal administration, or (iii) through retrograde introduction via the renal vein or ureter. Depending on the specific cells to be targeted, a particular route may be favored. Inherited kidney diseases generally either affect the glomerular complex (more specifically, the podocytes and/or mesangial cells), or the tubular epithelial cells (proximal tubule, loop of Henle, distal tubule, and collecting duct). There are 51 main cell types in the mammalian kidney with at least 16 different highly specialized epithelial cell types, while the number of specialized endothelial cells, immune cells, and interstitial cell types is even larger, and their intricate, concerted interplay is critical for kidney function. 18,19 Also, depending on the size of the vehicle carrying the genetic information, and the route of delivery, access to specific kidney cell types is restricted either due to the glomerular filtration barrier and/or elimination via the liver. 20
Viral Vector-Based Approaches to Tackle Kidney Diseases
Studies conducted in murine models have revealed promising advances in gene therapy for kidney disease, providing encouraging evidence that this form of therapy holds promise as a potential solution for treating kidney diseases in the future. 21
Viral vectors carry the intrinsic potential of their parental viruses to transfer genetic information in the targeted cell (Table 1). However, viral vectors are replication-defective, and the viral sequences are largely replaced by genetic information to be provided to patient's cells. 16 The most common viral vector platforms that have been applied to transfer genetic information in animal models to various kidney cell types are adenovirus (Ad) or adeno-associated virus (AAV)-based, considered best to transfer genetic information in vivo. A major challenge for in vivo application is specific on-target delivery of the genetic payload without eliciting an immune response. Still, even though retrovirus-based vectors (lentiviral vector [LV] or γ-retroviral vector [RV]) are primarily used for ex vivo applications, they may be of interest for in vivo kidney therapy. Here, we discuss the literature on viral vector-based approaches to tackle diseases affecting the kidney.
Adenoviral vectors and their potential in kidney gene transfer
General characteristics
Adenovirus-based vectors (AdV) are derived from nonenveloped viruses with a double-stranded (ds)DNA genome. There are at least 57 serotypes of human adenovirus (Ad1-Ad57) that form 7 species (A-G). 22 Most AdV are Ad5-derived and can carry a large genetic payload (8–36 kb). AdV transduce a wide range of dividing and nondividing cells and do not integrate in the host cell genome (episomal), explaining their transient nature.
With adenoviruses being ubiquitous and very immunogenic, people of all ages carry neutralizing antibodies and Ad-specific CD4+ T cells due to childhood infections. 23,24 In 1999, high-dose systemic AdV administration in a gene therapy trial to treat ornithine transcarbamylase deficiency elicited a strong innate immune response, multiple organ failure and eventually the death of a patient. 25,26 Because of the strong humoral and T cell responses to the AdV particle and T helper 1 type response to the vector-expressed transgene, AdV are employed extensively as vaccine vectors, as exemplified in the recent COVID-19 vaccination campaign. 27,28
Applications for targeting the kidney
AdV gene transfer to the kidney in animal models showed that direct targeting of the kidney is challenging (Fig. 2). Systemic injection of AdV in the rat resulted in liver and spleen transduction, but was ineffective to target the kidney. 29,30 Blocking the portal vein before systemic AdV infusion (superior mesenteric artery) effectively transduced the glomeruli. 31 Direct intraparenchymal injection showed transduction of the tubular epithelial cells, although restricted to the site of injection. 13,32 Likewise, retrograde infusion via the ureter or the bladder, and anterograde administration via the renal artery resulted only in modest tubular epithelial cell transduction, with higher AdV doses resulting in a profound immune response in rats. 32,33 AdV platforms based on alternate Ad species (porcine, bovine) showed elevated kidney transduction relative to human Ad in a mouse model. 34

Viral vector-based approaches and injection routes to target the kidney in a murine model. The respective approaches and vector platforms that are currently studied and are discussed in the text are shown, and the different injection routes explored in animal models are indicated. 1. Systemic routes; 2. Retrograde routes; 3. Local-direct routes. AdV, adenoviral vector; LV, lentiviral vector; rAAV, recombinat adeno-associated vector.
In all, the profound immune responses following AdV administration have reinforced substantial safety concerns for their use in gene therapeutic approaches and limiting their application to treat inherited disease. Accordingly, they are proven as a vaccine platform, with approvals for Ebola vaccines, and groundbreaking COVID-19 vaccines over the past year and have become the promise of new therapeutic cancer treatments. 28,35
Retroviral vectors and application in kidney therapy
General characteristics
Retroviruses carry two identical single-stranded (ss)RNA genome strands in an enveloped capsid structure. The hallmark of these viruses and their derived vectors is the reverse transcription of the ssRNA genome into dsDNA, which is subsequently stably integrated into the genome, thereby permanently linking its fate with that of the host cell, ensuring long-term and stable expression. 36 Although insertional mutagenesis remains a concern, the vector genome has been engineered to address this liability. 37
Retroviral vectors are designed to be replication defective and either based on Moloney Murine Leukemia Virus, referred to as retroviral vector (RV), or the complex retrovirus human immunodeficiency virus, referred to as lentiviral vector (LV). RV only transduce dividing cells, whereas LV can pass the nuclear membrane and transduce nondividing cells as well. 38,39 Still, LV transduction in vivo is only effective in cycling cells. 40 RV or LV can carry a genetic payload up to 9–10 kb. Rather than the canonical envelope protein, they are decorated with alternative glycoproteins to improve safety (replication-competent retrovirus) and broaden the range of target cells/tissues, which are referred to as pseudotyping. 41
Applications for targeting the kidney
Systemic administration of LV in mice resulted in massive transduction of the liver and the spleen, with minimal to no activity in the kidney and other peripheral organs. 42 In line with AdV, alternative administration routes have been explored. Injection of LV into the kidney via the retrograde infusion in the ureter displayed moderate reporter gene expression in the renal outer medulla and corticomedullary junction, while injection in the parenchyma extended expression to the renal cortex. 43 Vector delivery via the renal artery and vein was markedly less efficient. Despite the sparse transduction, therapeutic efficacy was reported in a model for tubular damage associated with ischemia-reperfusion injury. 44,45
Diverse groups explored alternate approaches to increase LV kidney transduction, such as iterative infusions, combining entry routes, or high-pressure injections over longer time periods, with limited improvement. 46,47 Recently, a minimally invasive, ultrasound-guided method was reported for LV injection in the mouse renal parenchyma showing reporter expression in proximal and distal tubular epithelial cells that sustained beyond 2 months. 48
RV/LV stably integrate into the genome ensuring the genetic information is passed on to all progeny of the initial target cell, underscoring why ex vivo cell therapy approaches are effective. 37 This approach has been applied in animal models to demonstrate proof of principle to treat systemic kidney diseases, such as the attenuation of renal cell carcinoma by CAR-T cells secreting PD-L1 antibodies in a humanized mouse model, or by the gene replacement approach in autologous hematopoietic stem cells (HSCs) to treat cystinosis and Fabry disease. 49 –52
RV/LV pseudotyping can enhance in vivo gene delivery to the kidney. Taking advantage of the natural tropism of other membrane-enveloped viruses allows for modulating RV/LV, altering the interaction with the host immune system and broadening the target cell range. 41,53 Also, envelope proteins can be engineered to target specific cell types, for example, by fusing a ligand or single-chain antibody that complements specific receptor proteins on the envisioned target cell/tissue. 54 A recent study demonstrated that high transduction of renal tubular epithelial cells in a mouse model for systemic injection of a LV pseudotyped with the Zika virus envelope glycoprotein. 55 Notably, a recent study reported on ex vivo kidney gene therapy in a rat model, perfusing a donor kidney ex vivo with LV to efficiently silence MHC antigens and thus improve graft survival after transplantation. 56
Recombinant adeno-associated vectors to target the kidney
General characteristics
Currently, recombinant adeno-associated virus (AAV)-based vectors (rAAV) are the ruling platform for in vivo gene delivery. AAV is a nonenveloped virus and is considered nonpathogenic. For the completion of its life cycle, the AAV depends on a helper virus, such as adenovirus, herpes simplex virus, or cytomegalovirus among others. 57 The viral genome consists of ssDNA flanked by inverted terminal repeat regions packaged in an icosahedral protein capsid of 25 nm. Next to natural occurring and ancestral AAV capsid serotypes, rational design, directed evolution, or in silico discovery generated a broad portfolio of capsids with specific tropism and properties. 58 rAAV vectors are indistinguishable from natural AAV viruses as to capsid sequence and structure, but are devoid of the viral protein coding sequences, maximizing the genetic payload to 4.8 kb. 59
Careful design is indispensable to accommodate all therapeutic genetic information within the limited boundaries along with regulatory elements to support gene expression. Owing to their small size and their high titers, rAAV may be better suited to transduce organs with complex architectures compared to the other viral vector platforms.
Applications for targeting the kidney
Direct injection into the renal artery or the parenchyma did not result in transduction of kidney cells in early studies with AAV1, AAV3, AAV4, or AAV5 serotypes. 60 rAAV2 showed transduction of intercalated cells of the collecting duct and proximal tubular epithelial cells only at doses >1010 vector genomes combined with clamping of the renal hilum, illustrating the importance of both the dose and the route of administration. 61,62 The use of more recent serotypes (AAV6.2, AAV8, AAV9) at higher doses resulted in promising transduction efficiencies following retrograde infusion in the ureter, transparenchymal renal pelvis injection, or subcapsular infusion. 63 –66
Systemic rAAV infusion initially resulted only in poor or undetectable kidney cell transduction in different animal models. 67 A recent study showed efficient rAAV9 delivery of therapeutic genetic information by systemic and renal vein injection, improving kidney dysfunction in a mouse model for acrodysostosis. 68 However, a comparative study of six AAV serotypes could not show rAAV9 transduction in the mouse kidney following systemic infusion via the retro-orbital venous plexus, whereas rAAV8 and the ancestral Anc80 serotype transduced kidney interstitial cells. 69
A recent study has shown that in vitro transduction of human podocytes by rAAV-LK03 and rAAV2/9 is highly efficient. 70 In addition, they demonstrated that the systemic delivery of rAAV2/9 (tail vein injection) in mice demonstrated successful AAV-mediated gene rescue in a monogenic disease with podocyte as a target for a gene therapeutic approach. Efficient delivery and expression of rAAV9-sphingosine phosphate lyase (rAAV-SPL) by injection into the superficial temporal vein in a mouse model demonstrated a dramatically prolonged survival and prevented nephrosis for Sphingosine-1-phosphate lyase insufficiency syndrome. 71 Furthermore, in a recent study on primary hyperoxaluria type 1, systemic administration of rAAV8-CRISPR/Cas9 targeting glycolate oxidase in Agxt1− /− mice showed reduced oxalate production and kidney damage with no signs of toxicity. 72
Employing systemic infusion to other tissues are also targeted. Synthetic engineering of novel capsids to introduce new properties and characteristics has been a constant pursuit. Particular mutations can uncouple interactions thereby de-target rAAV from distinct tissues or cells, for example, the liver, while others add another layer of specificity by triggering an interaction with a cell-specific receptor, for example, nanobody-VP1 or insulin-mimetic peptide. 73,74 Selective transduction and gene expression can be achieved by the combined manipulation of the rAAV capsid and the use of cell-specific promoters or enhancers, such as renal nephron segment-specific gene expression that was achieved by combining a rAAV9 capsid with the segment-specific promoters and administration by retrograde urethral infusion. 75 Additional complexity has been added by the recent documentation of rAAV capsid/promoter interactions that impact cell-specific gene expression independent of capsid permissiveness across species, experimental manipulations, and engineered capsids. 76,77
Clinical Trials for Molecular Therapies Targeting Diseases Affecting the Kidney
Gene therapy is becoming an integral part of the medical armamentarium, as exemplified by the EMA marketing authorization for 16 gene therapeutic products for orphan disorders. 78 –81 To this day, no gene therapeutic drugs specifically targeting the kidney are available in the clinic. Currently, several clinical gene therapy trials are registered for four kidney diseases: autosomal dominant polycystic kidney disease (ADPKD), primary hyperoxaluria, cystinosis, and Fabry disease. For completeness and to underscore the potential, we here also included the ongoing nonviral vector-based trials. The approaches used, however, do not directly target the kidney but employ methods that were successful for other systemic disorders: either they target the liver using rAAV or LNPs, or they use LVs to modify HSCs (Table 2).
Overview of ongoing clinical trials employing gene transfer for kidney diseases
This table was built based on kidney diseases mentioned in the review papers of Rubin and Barry and Bondue et al.
17,21
Clinical trials were selected on
ADPKD, autosomal dominant polycystic kidney disease; ASO, antisense oligonucleotides; cDNA, complementary DNA; co, codon-optimized; Hs, Homo sapiens; HSC, hematopoietic stem cell; IV, intravenous; LV, lentiviral vector; NA, not applicable; NV, nonviral vector; rAAV, recombinant adeno-associated viral vector; SC, sub-cutaneous.
Targeting extra-renal organ systems to prevent CKD: siRNA treatment in metabolic disease for prevention of CKD
In primary hyperoxaluria (1, 2, or 3), mutations in the AGXT, GRHPR, and HOGA1 genes result in a disease-causing oxalate overproduction in the liver, which in turn causes recurrent kidney stones, urinary tract infections, and progressive CKD. Lumasiran (Oxlumo; Alnylam Pharmaceuticals) provides an siRNA to silence the HAO1 mRNA via RNA interference reducing the levels of glycolate oxidase enzyme in the liver. 82 Another RNA interference therapy is DCR-PHXC (Nedosiran), a siRNA that targets the LDHA gene encoding lactate dehydrogenase, an enzyme involved in the conversion of glyoxylate to oxalate in the liver, and thus prevents excessive oxalate production. 83,84
Systemic LV-based HSC treatment in metabolic disease for alleviation of CKD
The monogenic lysosomal storage disorders cystinosis and Fabry disease are systemic disorders mainly affecting the kidney. Thus, treatment or cure aims at expression of the defective gene both in the kidney and all the extra-renal organs. Cystinosis is an autosomal recessive lysosomal disorder, caused by defective mutations in, or lack of, the CTNS gene, resulting in cystine accumulation in all body cells but primarily affecting the kidney. By introduction into the patient's HSCs of a functional CTNS complementary DNA (cDNA) via LV and subsequent re-administration of these gene-modified cells, the Cherqui group together with AVROBIO aimed to target the root cause of cystinosis. 50,85 Recently, the AVROBIO cystinosis gene therapy program was acquired by Novartis (Switzerland). 86
Fabry disease is another X-linked recessive lysosomal storage disease caused by mutations in the GLA gene, resulting in alpha-galactosidase A deficiency and accumulation of globotriaosylceramide in endothelial, parenchymal, and vascular smooth muscle tissues. Clinical gene therapy trials targeting different organs are ongoing. 87 In line with the cystinosis trial, an ex vivo gene therapeutic approach with autologous HSC transduced with LV encoding GLA cDNA was set up by AVROBIO (but discontinued early 2022) and Ozmosis Research, Inc. (Canada). 88 Additionally, three in vivo approaches using intravenous rAAV are in clinical trials. Freeline Therapeutics (United Kingdom) uses rAAVS3 (but was discontinued early 2023), whereas 4D Molecular Therapeutics uses a cardiotropic rAAV4D-C102 capsid (and is currently under FDA clinical hold). 89 –91 Sangamo Therapeutics uses rAAV2/6 to deliver the GLA coding sequence in its investigational Fabry disease gene therapy and is gearing up for a Phase III clinical trial in 2024.
Direct targeting of the kidney
ADPKD is caused by mutations in the PKD1 or PKD2 genes and is characterized by excessive kidney cyst cell proliferation that ultimately leads to ESRD in ∼50% of ADPKD patients by the age of 60. RGLS4326 (Regulus Therapeutics) is an oligonucleotide designed to inhibit miR-17 and designed to preferentially target the kidney. 92 Preclinical studies have shown that RGLS4326 directly regulates PKD1 and PKD2 expression, reduces cyst growth in human in vitro ADPKD models, and attenuates of cyst proliferation and improvement of kidney function in mouse models of ADPKD. 93
There are no registered clinical trials directly targeting the kidney using viral vectors. Pioneering kidney-focused gene therapy companies are coming round with targeted programs to bring these disease-modifying therapies to the patients. PurespringTX (United Kingdom) and NinevahTX (Spain/United Kingdom) employ a rAAV-based platform to treat CKD by directly targeting the podocytes exploring new rAAV capsids with improved tropism for kidney cells. Nephrogen aims to employ gene-editing technologies (CRISPR-Cas) in both viral and nonviral vector-based platforms (rAAV and nanoparticles) for local and systemic delivery in vivo but are also exploring ex vivo kidney perfusion and subsequent kidney re-transplantation.
Considerations and Perspectives for the Clinic on Kidney-Targeted Gene Transfer
The progress in gene transfer to the kidney has been limited compared to other tissues due to its complex anatomical architecture and a lack of understanding of the biological molecular cues required for efficient gene transfer to specific kidney cells. Viral and nonviral approaches should be considered as complementary and mutually reinforcing gene transfer platforms. Currently, no universal approach can be identified, and depending on the target cells, the disease considered, and the payload to be delivered, a gene transfer platform and route of administration will be selected.
The importance of the injection route
Although systemic administration via peripheral vein injection seems a straightforward method of delivery, it is known that most vector particles are retained in the liver. Different injection routes have been studied, primarily in mouse models, including infusion via the renal artery, retrograde infusion via the renal vein or ureter, and direct intraparenchymal injection into either the cortex or medulla of the kidney (reviewed in Rubin and Barry 21 ). It will be key to assess the transduction potential of different viral vector capsids/pseudotypes for the different injection routes, especially when strategies are envisioned to reach specific subsets of cells in the kidney, such as specific glomerular and tubular cells. Together, studies underscore that route of administration, vector titer (dose), injection method, and frequency are to be carefully weighted.
The kidney filtration barrier
Viral vector particle dimensions are quite large, limiting gene transfer to the kidney due to the glomerular filtration barrier, formed by podocyte slit diaphragms, transcellular epithelial fenestrations (60–80 nm), together with the dense, negatively charged, hydrated glycocalyx mesh at the endothelial surface. 94 Still, the fact that filtration function is frequently compromised in kidney disease due to loss of cells reduced endothelial fenestration and glycocalyx and decreased filtration slit frequency, may thus permit more efficient passage. Also, when considering degenerative kidney disease, it is key to treat the cells before they are lost, or before the point of no repair. 95 Mimicking these conditions in research setting with better animal or organoid models will be essential to assess the full potential of gene transfer platforms.
Safety, immune response, and durability of gene therapy treatment
Gene therapy is generally presented as a single-dose, once-in-a-lifetime, curative treatment; however, the durability of these treatments remains to be proven. Even if the effect of gene therapy would not be long lasting or only slow down disease progression, it could be applied to prolong the kidney lifetime, thereby improving quality of life for the patient, and possibly avoiding or postponing renal transplantation therapy.
Though the advent of rAAV-enabled gene transfer with less toxicity, setbacks related to vector toxicity and immunogenicity represent major challenges to the field. 96 Safety and efficacy of rAAV vectors, particularly for indications that require high doses is a major concern. 97 In line with other drugs, this approach also provokes immune responses (antibodies against the viral vector and the encoded product) reducing effectivity. 98 In addition, the fact that an immune response is mounted annihilates the potential of re-administration. Therefore, new approaches must be explored to mitigate immune response and to improve transduction at lower vector doses, and ultimately to allow re-dosing. If re-dosing were possible, multiple lower doses may achieve comparable therapeutic benefit, avoiding the toxicity and adverse events associated with the high-dose treatments. In addition, therapeutic benefit could be restored in case therapies would decline over time. 96
Correction does not always result in cure
Addition of a healthy copy or restoration of a gene of interest is expected to restore their function and thus to revert the disease phenotype, or at least some aspects thereof; however, this will not be the case for all diseases. We currently lack insight into the underlying molecular pathways and compensations occurring in diseased cells and tissues. Indeed, our knowledge on reversal of pathophysiology and restoration of normal physiology on the course of the disease is still evolving and is unique for each disorder. Our insight and understanding of disease will improve considerably by taking on these genetic approaches, which in turn will provide new therapy options to treat disorders where no other treatment is available.
Better humanized models to assess gene therapy potential
Viruses and their derived vectors display a particular host cell specificity. Hence, animal models do not confidently predict the tropism and biology of rAAV or other viral vectors in primates and humans. Preclinical model systems that accurately recapitulate human physiology are needed to evaluate a particular gene transfer platform. The use of ex vivo perfused kidneys may be one option. 99 Alternatively, human 3D kidney organoids, body-on-chip models, and micro-physiological systems can be employed as a measure for clinical translation potential. 94
Conclusion
CKD affects a significant portion of the population, and CKD-associated mortality rates have been steadily increasing in recent decades. With treatment of CKD being symptomatic and supportive, there is a pressing need to explore innovative translational and molecular renal therapies that target the root cause of the disease. While gene therapy has demonstrated remarkable success for various diseases without existing cures over the past few decades, only a limited number of gene therapy approaches targeting kidney disease have progressed to clinical trials. It is important to recognize that further innovation is required and obstacles to be overcome. Delivery of the genetic cargo directly to the kidney remains the main hurdle. Gene therapy, though still in its early stages, is growing into a valuable clinical approach that holds tremendous potential to profoundly transform the lives of the many patients affected by CKD, opening new avenues for treatment and potential cure.
Figures
Created with Biorender.
Footnotes
Authors' Contributions
Writing—original draft preparation, L.M. and R.G.; Writing—review and editing, L.M., K.V. and R.G.; Critical reading—L.M., K.V. and R.G.
All authors have read and agreed to the published version of the manuscript.
Author Disclosure
No competing financial interests exist.
Funding Information
Louise Medaer and Rik Gijsbers perform research that is funded by Cystinosis Ireland and Cystinosis Foundation UK (co-funded Research Funding Award 2021; CI-CFUK 2021-02).