
Abstract
Fabry disease (FD) is a multisystemic lysosomal storage disorder caused by the loss of α-galactosidase A (α-Gal) function. The current standard of care, enzyme replacement therapies, while effective in reducing kidney pathology when treated early, do not fully ameliorate cardiac issues, neuropathic manifestations, and risk of cerebrovascular events. Adeno-associated virus (AAV)-based gene therapies (AAV-GT) can provide superior efficacy across multiple tissues owing to continuous, endogenous production of the therapeutic enzyme and lower treatment burden. We set out to develop a robust AAV-GT to achieve optimal efficacy with the lowest feasible dose to minimize any safety risks that are associated with high-dose AAV-GTs. In this proof-of-concept study, we evaluated the effectiveness of an rAAV9 vector expressing human GLA transgene under a strong ubiquitous promoter, combined with woodchuck hepatitis virus posttranscriptional regulatory element (rAAV9-hGLA). We tested our GT at three different doses, 5e10 vg/kg, 2.5e11 vg/kg, and 6.25e12 vg/kg in the G3Stg/GLAko Fabry mouse model that has tissue Gb3 substrate levels comparable with patients with FD and develops several early FD pathologies. After intravenous injections of rAAV9-hGLA at 11 weeks of age, we observed dose-dependent increases in α-Gal activity in the key target tissues, reaching as high as 393-fold of WT in the kidneys and 6156-fold in the heart at the highest dose. Complete or near-complete substrate clearance was observed in animals treated with the two higher dose levels tested in all tissues except for the brain. We also found dose-dependent improvements in several pathological biomarkers, as well as prevention of structural and functional organ pathology. Taken together, these results indicate that an AAV-GT under a strong ubiquitous promoter has the potential to address the unmet therapeutic needs in patients with FD at relatively low doses.
INTRODUCTION
Fabry disease (FD) is an X-linked, multisystemic lysosomal storage disorder caused by mutations in the GLA gene resulting in significant reduction or complete loss of alpha-galactosidase A (α-Gal A) enzyme activity. The deficiency of α-Gal leads to accumulation of globotriaosylceramide (Gb3) causing cellular dysfunction and resulting in multiple-organ pathology affecting kidney, heart, gastrointestinal (GI), and peripheral nervous systems (PNS). The introduction of enzyme replacement therapy (ERT), Fabrazyme® and Replagal®, in 2001 led to a significant improvement of complication rates and early death. Despite the life-altering impact of ERT on amelioration of clinical symptoms and slowing disease progression, ERT has limited efficacy in halting disease progression as well as mitigating symptoms related to PNS, GI, and central nervous system (CNS) pathology. 1 A new ERT, Elfabrio®, was recently approved and is associated with a longer pharmacokinetic half-life but so far has not demonstrated superior efficacy. 2 An orally bioavailable chaperone therapy Galafold® was approved in 2018 for patients with amenable α-Gal mutations, but real-world data on efficacy for specific pathologies are still emerging.
Cell and adeno-associated virus (AAV)-based gene therapies (GTs) have shown promise in mouse models and in early clinical trials in patients with FD. 3 –13 Isaralgagene civaparvovec (ST-920) uses a liver tropic vector to produce circulating α-Gal A. While circulating levels of liver-expressed α-Gal A have been relatively stable for more than 2 years, plasma lyso-Gb3 increased upon stopping ERT in some patients. 7 4D-310 uses a cardiotropic AAV vector to deliver human GLA transgene directly into cardiomyocytes with preliminary results indicating improvements in left ventricular contractility in all 6 patients treated and in exercise capacity for 3 patients. 10 GT can become a transformative treatment eliminating the burden of bi-weekly ERT infusions and more fully addressing therapeutic needs. Nevertheless, additional research is still needed to better understand limitations of current GTs and to develop next generation therapies with better efficacy and safety.
One of the primary limitations of the AAV-GT is requirement of very high doses to achieve sufficient transgene expression and optimal efficacy. Herein, we report a proof-of-concept study conducted to demonstrate that low doses of a well-designed GT can be efficient in tissue substrate clearance as well as in mitigating tissue structural pathology, improving disease biomarkers, and functional disease manifestations. We chose to conduct the study in the G3Stg/GLAko mouse model that was developed by crossing lactosylceramide 4-alpha-galactosyltransferase (A4GALT) transgenic mouse line with α-Gal A knockout (KO) mice. Unlike commonly used mostly asymptomatic α-Gal A KO mice, G3Stg/GLAko mice accumulate higher levels of tissue Gb3 approaching that found in patients with FD. 14,15 Higher substrate accumulation in this model leads to the development of several pathologies characteristic of early FD. In this study, we demonstrate that our GT construct can achieve supraphysiological levels of tissue α-Gal activity, normalize substrate burden, and ameliorate pathologies in this symptomatic FD model.
MATERIALS AND METHODS
rAAV9-hGLA vector
rAAV9-hGLA was designed and produced using an approach and method described earlier. 16 Briefly, the vector genome (VG) for rAAV9-hGLA contains a 5′ inverted terminal repeat (ITR), a chicken β-actin (CB) promoter, human α-Gal gene (hGLA) gene sequence, a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE), a bovine growth hormone polyadenylation sequence, and a 3′ ITR (Supplementary Fig. S1). The null vector control genome comprises of a noncoding DNA sequence flanked by ITR sequences. ITR plasmid containing transgene expression cassette was constructed by standard restriction cloning techniques. rAAV9-hGLA vector was produced using standard triple-transfection methods. HEK293 cells were co-transfected with three plasmids: the ITR plasmid containing the transgene expression cassette, a RepCap expression cassette, and an adenoviral helper gene expression cassette. After 72–96 h of transfection, the cells were lysed, and cell lysate was cleared with benzonase treatment. The clarified lysate was loaded onto a POROS Captureselect AAVX column connected to an AKTA purification system, and eluate fractions containing AAV particles were collected according to manufacturer’s recommendation. The AAV vectors were formulated in phosphate-buffered saline containing 0.001% PF-68 (Thermo). The AAV vectors were subjected to standard characterization, including a quantitative polymerase chain reaction (qPCR) for titration, silver staining for purity, an amebocyte lysate assay (Endosafe) for endotoxin measurement, and an in vitro transduction assay to determine biological activity. Characterized vectors were aliquoted and frozen at −80°C.
rAAV9-hGLA study design
The G3Stg mouse model was licensed from JCRB Laboratory Animal Resource Bank at National Institutes of Biomedical Innovation, Health and Nutrition, Saito-Asagi 7-6-8, Ibaraki, Osaka 567-0085, Japan. The G3Stg/GLAko colony was established through breeding and maintained at Taconic Biosciences (Germantown, NY, USA). For the study, male G3Stg/GLAko mice were transferred to Melior Discovery (Exton, PA, USA) and acclimated for 5 days. C57BL/6 were acquired from Charles River Laboratories (Wilmington, MA, USA) to serve as wild-type (WT) control. Mice were housed on a 12 h light/dark cycle in a ventilated cage rack system. Animals were assigned randomly to treatment groups. Groups were balanced based on pre-dose hot plate sensitivity data and body weight. Approximately 11–13 male G3Stg/GLAko mice were assigned to each group. Mice were treated with a single intravenous injection of rAAV9-hGLA at 5 × 1010 vg/kg (low dose), 2.5 × 1011 vg/kg (mid dose), and 6.25 × 1012 vg/kg (high dose) or null vector control at 6.25 × 1012 vg/kg at 11 weeks of age. Age-matched WT mice were treated with vehicle as a control group. The study protocol was reviewed and approved by the Melior Discovery Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the guidelines of the U.S. National Research Council on Animal Care and with the Melior Standard Operation Procedures (SOP). SOPs were reviewed and approved by Takeda prior to the initiation of the study.
Necropsy and sample collection
Mice were bled at 2, 5, 10, and 14 weeks after dosing via tail vein bleed or retro orbital eye bleed. Blood was processed to obtain serum by sample centrifugation at room temperature. Serum was collected, frozen, and stored at −80°C. Urine was collected at 14, 18, 22, and 28 weeks, by putting mice in a metabolic chamber overnight then stored at −80°C. Eighteen weeks after vector administration, mice were euthanized using carbon dioxide inhalation. Terminal blood was collected via cardiac puncture, serum was separated, and samples were stored at −80°C. Then mice were perfused with 10 mL of phosphate-buffered saline (PBS). Kidney, heart, liver, duodenum, colon, whole spinal column with dorsal root ganglia (DRG), left hind paw, and brain were harvested. Samples intended for biochemical and molecular analysis were frozen. Histological samples were fixed with 10% neutral buffered formalin (NBF). For each animal liver was split into two samples; one for histology and one for biochemical and molecular assays. The left kidney was used for histology and the right for biochemical/molecular analysis. Whole brain was collected and used only in biochemical/molecular assays. Colon, duodenum, and heart samples were split between biochemical and histological analysis and 5–7 samples per group were analyzed for these tissues. For VG distribution and mRNA, 5–7 samples per group were analyzed from all tissues. Terminal serum and tissue samples for one mouse in the null vector and two mice in the low-dose groups were not collected. These mice had to be sacrificed per protocol before study termination because of the neurological pathology progression characteristic for this model.
Tissue homogenization
Frozen tissues were pulverized using Covaris tissue pulverizer (Covaris LLC, Woburn, MA, USA) and homogenized in lysis buffer containing 10 mM 4-(2-hydroxyethyl)−1-piperazineethanesulfonic acid, 0.5% Triton-X 100, and 1× Halt Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA, USA), in a ratio of 250 mg of tissue per mL of lysis buffer. Homogenization was performed using homogenizing beads and processed with Precellys homogenizer (Bertin Corporation, Rockville, MD, USA) for 3 cycles of 6500 rpm at 20 s each cycle. Tissue homogenate was then centrifuged at maximum speed, at 4°C for 10 min before removing supernatant to run α-Gal activity and protein quantification, Gb3 and lysoGb3 analyses, and bicinchoninic acid (BCA) assay.
α-Gal activity
α-Gal activity was determined using 4-methylumbelliferyl-D-galactopyranoside (4-MU-α-gal) fluorescent substrate. Briefly, 2 μL of a biological sample was incubated with 15 μL 4-MU-α-gal substrate solution (Research Products International Company, Mt. Prospect, IL, USA, catalog # M65400) and α-Gal B inhibitor (N-acetyl-D-galactosamine, Sigma-Aldrich Chemicals, St. Louis, MO, USA, catalog # A-2795) at 37°C for 60 min. The enzymatic reaction was stopped by an addition of 200 μL glycine carbonate stop solution, pH 10.7. The 4-methylumbelliferone (4-MU) product was measured at the excitation wavelength 360 nm and emission wavelength 465 nm by a fluorescence plate reader. The concentrations of 4-MU in testing samples were calculated from the 4-MU calibration curve in the same plate. Serum and tissue α-Gal activity was normalized to the volume of serum or tissue total protein concentration of the same prepared samples determined by BCA assay and expressed as nmol of 4-MU produced per hour per mL of serum of mg of total protein.
α-Gal protein concentration
Meso Scale Diagnostics LLC [Rockville, MD, USA]) ELISA method was used to quantify α-Gal protein in serum and tissues. All reaction was carried at room temperature unless stated. Briefly, a High Bind MSD (Meso Scale Diagnostics) black plate (MSD, Catalog No. L15XB) was coated with a polyclonal sheep antihuman α-Gal capture antibody (R&D Systems [Minneapolis, MN, USA], Catalog No. AF6146) in 0.2 M sodium carbonate-bicarbonate buffer (Thermo Fisher Scientific, Inc., Catalog No. 28382) overnight at 4°C. The coated plate was washed three times with wash buffer containing Dulbecco’s PBS (DPBS) and 0.05% Tween-20. The plate was blocked with blocking buffer (3% bovine serum albumin [BSA] in PBS) for 1 h before samples or purified α-Gal protein standard in diluent buffer (1% BSA in PBS) were added. Binding was carried for 1 h with low-speed shaking and washed three times with wash buffer. Then, polyclonal rabbit anti-human α-Gal (Novus Biologics [Littleton, CO, USA], Catalog No. H00002717-D01P) in diluent buffer was added and incubated for 1 h before washing three times with wash buffer and adding a detection antibody, sulfo-tagged goat antirabbit antibody (MSD, Catalog No. R32AB-1), for 1 h at room temperature. The plate was read with 1× read buffer (MSD, Catalog No. R92TC-1) in the MSD Sector Imager S600 system. Using a standard curve, final values of α-Gal concentration were calculated. Tissue α-Gal protein concentration was normalized by total protein concentration determined by BCA assay. None of antibodies used in this assay recognized mouse α-Gal protein.
BCA protein quantification
Tissue protein was quantified using Pierce™ BCA kit (Thermo Fisher Scientific, Inc., Catalog No. 23255) following manufacturer’s protocol. Protein concentration was used to normalize tissue α-Gal activity, protein level, Gb3, and lysoGb3 substrate levels.
Genomic DNA isolation and quantification
Approximately 50 mg of tissues aliquoted into Precellys CK14 lysing tubes were subjected to DNA extraction. The MagMAXTM DNA Multi-sample Ultra 2.0 kit combined with an automated Kingfisher flex magnetic particle processor (Thermo Fisher Scientific, Waltham, MA, USA) was used for genomic DNA extraction with modification. Briefly, 1 mL of tissue lysis buffer (containing 7 μL of β-mercaptoethanol) was added to the samples and homogenized in Precellys beads beater (5800 rpm; 15 s twice with a 30-s pause in between). The homogenization cycle was repeated twice to ensure complete homogenization of the tissue. One-hundred microliter of the lysate was transferred to a 96-well (deep well) plate; 20 μL of enhancer solution, and 40 μL of proteinase K were added and incubated at 65°C overnight. The next day, 5 μL RNaseA was added and mixed for 5 min at 900 rpm at room temperature to keep RNA contamination negligible. The bead mixtures (40 μL binding beads with 400 μL binding solution) were added to each well, and lysates were immediately processed for DNA extraction using the automated Kingfisher flex magnetic particle processor program (MagMAX_Ultra 2_Tissue_V). Genomic DNA concentrations and purity of extracted DNA were monitored by a Lunatic UV/Vis absorbance spectrometer (Unchained Labs, Brighton, MA, USA). The genomic DNA concentration was quantified using a QuickDrop spectrophotometer (Molecular Devices, San Jose, CA, USA). The genomic DNA was then added to the ddPCR reactive mix containing the respective primers and probes for target molecules or an un-transcribed region between ITRs and mouse Acta gene as a reference. Droplets were generated using the automated droplet generator (Bio-Rad Laboratories, Hercules, CA, USA). Following PCR amplification of the target sequence by end-point PCR in each droplet, the positive droplets were then quantified using the QX200 droplet reader (Bio-Rad Laboratories). After the assay completion, the threshold was manually set, and the viral genome copy number was normalized based on the reference copy number.
Gb3 and lyso-Gb3 analysis
Gb3 and globotriaosylsphingosine (lyso-Gb3) analyses were performed by NovaBioassays LLC (Woburn, MA, USA) using the method described in Sueoka et al, 2015. 17 Briefly, samples were extracted using chloroform:methanol (volume over volume [v/v] 2:1). Lyso-Gb3 and Gb3 Isoforms (C16:0, C18:0, C20:0, C22:1, C22:0, C22:0-OHCTH, C24:1 C24:0, C24:0-OH C26:0) were measured by high-performance liquid chromatography and liquid chromatography tandem mass spectrometry (LC/MS/MS) (Applied Biosystem, Foster City, CA, USA, API5000, Turbo Ion Spray Ionization, positive-ion mode). The total Gb3 concentration was calculated from the sum of all the Gb3 isoforms and normalized by the volume of serum or tissue total protein concentration.
Measurement of blood urea nitrogen, urine albumin, and creatinine levels
For measurements of blood urea nitrogen (BUN) in serum and albumin and creatinine levels in urine, samples were analyzed using a COBAS C 311 analyzer (Roche Diagnostics [Risch-Rotkreuz, Switzerland]). Serum BUN (Roche Diagnostics, Catalog No. 04460715 190) was determined by a couple of enzymatic reactions with urease dehydrogenase and glutamate dehydrogenase. For urine albumin, ALBT2 (Roche Diagnostics, Catalog No. 04469658 190), an immunoturbidimetric assay, using an anti-albumin antibody to form antigen/antibody complexes following agglutination was used, where turbidimetry was measured. Urine creatinine was analyzed in the CREJ2 (Roche Diagnostics, Catalog No. 04810716 190) assay, which is a kinetic colorimetric assay based on the Jaffé method. Urine albumin concentration was normalized to urine creatinine for final values.
Histological analysis
Duodenum, colon, heart, liver, kidney, spinal column, and left hind paw samples were collected 18 weeks after dosing and fixed in 10% neutral buffered formalin (NBF) for 48 h. Spinal column with DRG and the left hand paw samples containing bone tissues were subjected to decalcification with RapidCal Immuno Decalcifier (BBC Biochemical, Mt Vernon, WA, USA, Catalog No. 6089) for 24 h after the fixation and then washed with running distilled water for 30 min. Other samples were quickly washed with 1× PBS three times. All samples were processed to be embedded in paraffin and sectioned. Five-micron sections were used for immunohistochemistry (IHC) with Bond Polymer Refine Detection kit (Leica Biosystems, catalog # DS 9800) on the automated Leica Bond system (Leica Biosystems, Wetzlar, Germany). Immunostaining was performed using the following antibodies: α-Gal (Takeda Pharmaceuticals Inc., Cambridge, MA); Beta-Catenin (Abcam, catalog # ab32572,); Collagen I (Boster Bio, Pleasanton, CA, USA, catalog # RA2140-2); CD68 (Abcam, catalog # ab125212); Iba-1 (Wako Chemicals, Richmon, VA, USA, catalog # 019–19741); LAMP1 (Abcam, catalog # ab24170); myelin protein zero (MPZ) (Abcam, catalog # ab183868); p62 (Abcam, catalog # ab91526); PGP9.5 (Abcam, Catalog No. ab8189); and WT1 (Abcam, catalog # ab89901). A polymer antirabbit poly-HRP antibody from the Bond Polymer Refine Detection kit (Leica, Catalog No. DS9800) was used as a secondary antibody. Hematoxylin and eosin (H&E) staining was performed in an automated Leica ST5020-CV5030 Stainer Integrated Workstation. Stained slides were scanned with Leica AT2 Scanner. The whole digital slide images were viewed with Aperio ImageScope and Halo image system to select representative images for figures. Quantitative biomarker image analysis was performed using HALO® image analysis software. Positivity was calculated using the following formula: positivity (%) = positive area (pixels)/total analyzed area (pixels) × 100%.
Hot plate sensitivity analyses
A hot plate test was conducted on all mice at 10 weeks of age (baseline) and 5, 9, 13, and 17 weeks after dosing with AAV as is described in the following method. The hot plate (Columbus Instruments) was preheated to 55°C. An open-ended cylindrical plexiglass tube with a diameter of 30 cm was placed on top of the hot plate to prevent mice from escaping but leaving the animals’ paws exposed to the hot plate. Using a stopwatch, the time from placing the mouse on the hot plate to the time of the first pain response (paw lick, paw flick, or jump) was recorded. The mouse is immediately removed once this response is observed. If there was no response within 60 s, the test was terminated, and the mouse was removed from the hot plate to prevent heat-related injury.
Statistical analysis
The results are presented as the mean ± standard error of the mean. Statistical analysis was performed in GraphPad Prism v.9.2. Data were tested for Gaussian distribution, probability of normality, or lognormality. Untransformed data were analyzed when values in most group were normally distributed. If lognormality was predicted to be more likely, data were log transformed for statistical analysis. Groups were compared using one-way analysis of variance followed by post hoc pair-wise comparison with Dunnett’s multiple comparisons correction. Differences were considered significant when p ≤ 0.05. Nonparametric Spearman’s correlation analysis was performed to measure the significance and the direction of association between the α-Gal activity and substrate level.
RESULTS
Administration of rAAV9-hGLA increased plasma and tissue α-Gal A activity and reduced Gb3 and lyso-Gb3 in the TgG3S/GLAko mice in a dose-dependent manner
Dose levels for rAAV9-hGLA were selected in a 1-month dose range finding study (data not shown). A threshold dose that resulted in transduction of at least one of the tissues tested was used as the lowest dose in the current study. A single dose of either 5e10 vg/kg (low dose), 2.5e11 vg/kg (mid dose), or 6.25e12 vg/kg (high dose) of rAAV9-hGLA was administered by tail vein injection to 11-week-old male G3Stg/GLAko mice. Null vector was administered at 6.25e12 vg/kg. Age-matched WT animals were injected with the buffer. VG biodistribution analysis was performed in tissues collected 18 weeks after dosing. VG level was dose-dependent with the highest copy number in the liver detected in all animals in all dose groups. In the kidney and heart, VG was detected only in the high- and middle-dose groups. In other tissues, stable transduction was only observed in the high-dose group (Supplementary Fig. S2). Tissue level of hGLA mRNA was consistent with VG copy number (Supplementary Table S1).
Dose-dependent increase in serum α-Gal activity was detected at 2 weeks after dosing, and the level was maintained through the end of the study (Fig. 1A). At the terminal endpoint, serum α−Gal activity was 14,689-fold of WT; 4,976-fold of WT; and 161-fold of WT in the 6.25 × 1012 vg/kg, 2.5 × 1011 vg/kg, and 5.0 × 1010 vg/kg dose groups, respectively (Supplementary Table S2). Supraphysiological α-Gal activity was also detected in the terminal tissue samples, with the highest activity observed in the liver, followed by the heart, colon, duodenum, kidney, and brain (Fig. 1B). α-Gal activity was 364-fold of WT, 14-fold of WT, and 3.5-fold of WT in the kidney and 5,964-fold, 1,042-fold, and 111-fold in the heart in the high-, medium-, and low-dose groups respectively (Supplementary Table S2). The lowest level of α-Gal activity was observed in the brain, with only the highest dose of GT achieving activity levels within the range of the WT control (Fig. 1B). Serum and tissue activity were consistent with α-Gal protein concentration (Supplementary Fig. S3 and Fig. S4).

rAAV9-hGLA vector genome distribution, tissue and serum α-Gal activity, and protein level.
Supraphysiological α-Gal levels led to near complete or significant Gb3 clearance in the high- and medium-dose groups in most tissues evaluated, namely serum, heart, kidney, liver, colon, and duodenum (Fig. 2A). Increase in activity in the low-dose group was variable (Fig. 1B). Nevertheless, Gb3 clearance in this group was also complete or near complete in serum and liver. In the kidney, duodenum, and colon Gb3 reduction was partial but still statistically significant (Fig. 2A). A trend toward substrate reduction was also observed in the heart of the low-dose group but did not reach statistical significance (Fig. 2A). In the brain, statistically significant Gb3 reduction was only observed in the highest dose group. Modest brain pharmacologic efficacy was congruous with observed α-Gal activity (Fig. 1B). Consistent with the Gb3 reduction pattern, complete or significant reduction of lyso-Gb3 was observed in all dose groups and tissues except for the brain in the middle- and low-dose groups and heart in the low-dose group (Fig. 2B). The level of the residual tissue substrate inversely correlated with tissue α-Gal activity (Supplementary Table S3). Spearman’s correlation coefficient (r) between α-Gal activity and residual Gb3 was negative 0.78 with p < 0.0001 in the kidney and negative 0.82 with p < 0.0001 in the heart.

Tissue and serum Gb3 and lyso-Gb3 reduction in mice treated with rAAV9-hGLA.
AAV9-hGLA treatment improved kidney structure and function and reduced exploratory pathological biomarkers in G3Stg/GLAko mouse model
Kidney H&E staining in the control G3Stg/GLAko mice revealed tubular atrophy evident by the presence of tubular epithelial thinning, pyknotic nuclei, and lumen protein casts. Treatment with rAAV9-hGLA had protective effects on kidney structure, and normal kidney morphology was preserved in the high-dose group (Fig. 3A). Immunohistostaining revealed an increase in the lysosomal marker LAMP1 in the null vector treated G3Stg/GLAko mice kidney indicating substrate accumulation. The highest LAMP1 intensity was detected in the collecting ducts. Treatment with rAAV9-hGLA reduced LAMP1 IHC intensity (Fig. 3B), consistent with observed substrate reduction in kidney tissue homogenates (Fig. 2A). Analysis of additional exploratory biomarkers relevant to Fabry renal pathology was conducted to assess their translatability. Strong immunostaining for a well-established marker of fibrosis Collagen I 18 –20 was detected in the medulla of kidneys in the control G3Stg/GLAko mice indicating tubulointerstitial fibrosis. Collagen I was reduced in the rAAV9-hGLA treated animals achieving complete normalization in the high-dose group (Fig. 3C). WT1, an exploratory marker of early fibrosis, 21 was also upregulated in the control G3Stg/GLAko mice and normalized after treatment (Fig. 3D). To evaluate kidney function, we measured BUN and urine albumin levels. BUN levels were measured at the terminal time point and urine albumin was assessed every 4 weeks starting at 14 weeks of age. The level of BUN was significantly increased in the G3Stg/GLAko control mice. rAAV9-hGLA treatment reduced BUN in a dose-dependent manner (Fig. 4A). Urine albumin was also elevated in the control G3Stg/GLAko mice even at 14 weeks and continued to increase through the duration of the study. Low and mid dose of rAAV9-hGLA slowed down albuminuria progression while the level of urinary albumin in the high-dose group was not statistically different from WT control (Fig. 4B, Supplementary Table S4).

Improvement of kidney structural pathology and exploratory disease biomarkers in TgG3S/GLAko mice treated with rAAV9-hGLA.
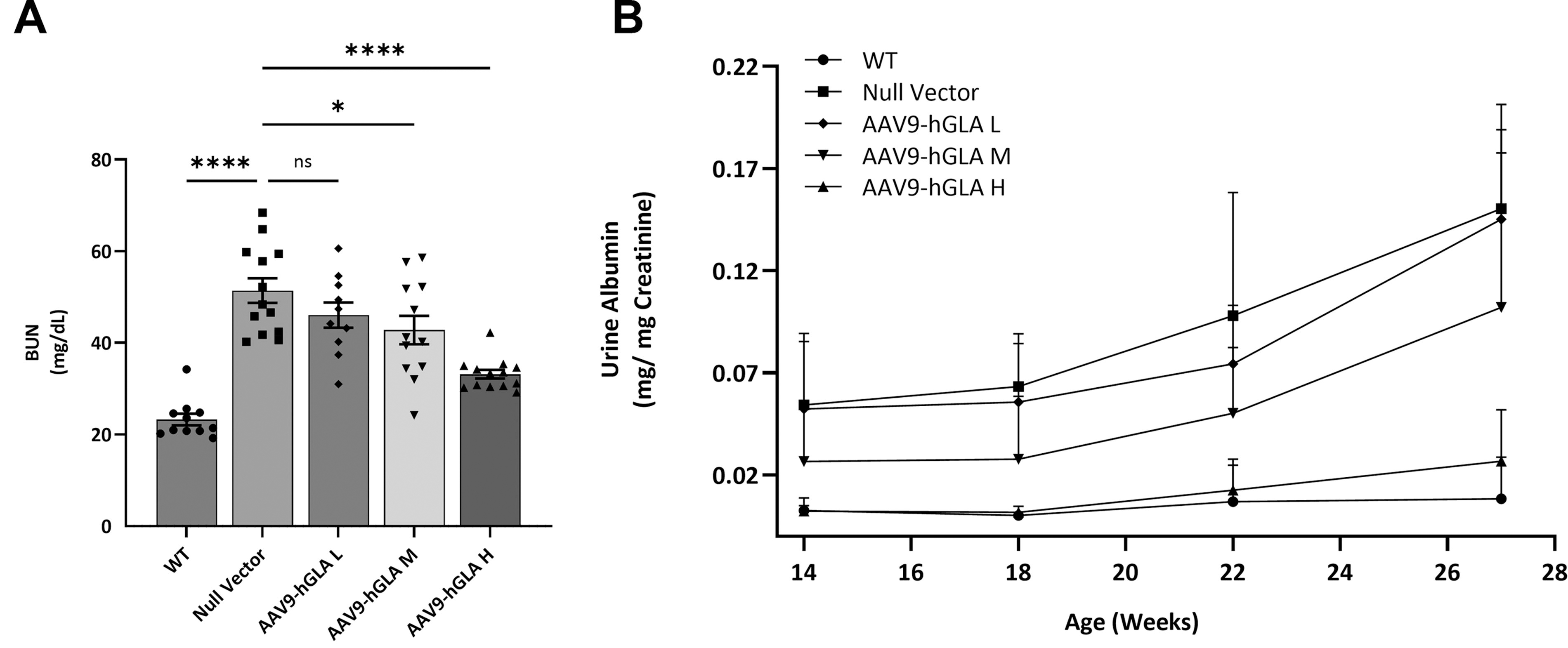
Improvement in kidney function in TgG3S/GLAko mice treated with rAAV9-hGLA.
AAV9-hGLA treatment reduced lysosomal burden in cardiomyocytes and normalized exploratory biomarkers of cardiac pathology
LAMP1 immunostaining in the heart of the control G3Stg/GLAko mice was detected across all cell types including cardiomyocytes. High dose of rAAV9-hGLA treatment normalized LAMP1 staining in the cardiomyocytes known to be highly affected in patients with FD 22 (Fig. 5A). Persistent LAMP1 staining in the interstitial cells was likely the result of the excess α-Gal uptake by phagocytic resident macrophages because near-complete substrate clearance was observed in the cardiac tissue homogenates in this group (Fig. 2A). In addition, we investigated Collagen I, identified in patients with FD as an early cardiac fibrosis biomarker, 23 as well as β-catenin, a novel, exploratory biomarker of cardiac pathology. 24 –26 Collagen I immunostaining was increased in the control G3Stg/GLAko mice and reduced after rAAV9-hGLA treatment, completely normalizing in the high-dose group (Fig. 5B). Immunostaining also revealed β-catenin accumulation in the disorganized intercalated discs of the G3Stg/GLAko control mice which is consistent with its possible role in the development of cardiac hypertrophy. GT prevented β-catenin accumulation and in the high rAAV9-hGLA dose group β-catenin cellular distribution was similar to WT control (Fig. 5C). Cardiac function of the G3Stg/GLAko mouse model was assessed by echocardiography and reported earlier. 27
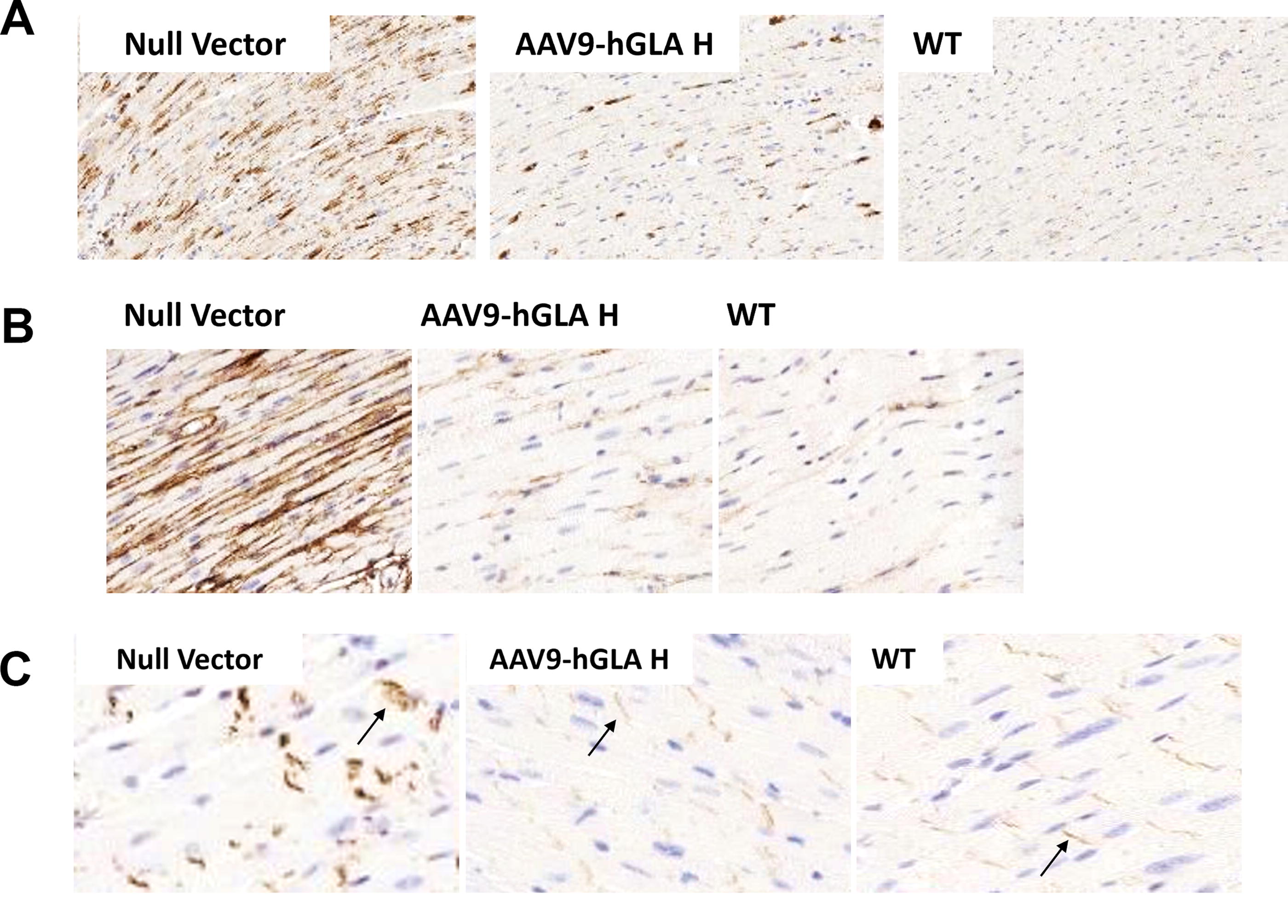
Efficacy of rAAV9-hGLA in the heart of the TgG3S/GLAko mice.
PNS pathology progression in the G3Stg/GLAko Fabry mouse model was prevented by rAAV9-hGLA treatment
In FD patients, Gb3 accumulation in the PNS leads to GI dysfunction and peripheral neuropathy. Myenteric plexus (MPG) and DRG) of the control G3Stg/GLAko mice had significant substrate accumulation as evident by the intensity of the LAMP1 IHC. Treatment with rAAV9-hGLA reduced LAMP1 immunostaining indicating Gb3 clearance and the staining in the MPG was normalized in the high-dose group. Substrate accumulation was also cleared in the GI smooth muscle layer (Fig. 6A).
Consistent with other reports, 28 we observed heterogeneous subpopulation-specific substrate accumulation in the DRG neurons of G3Stg/GLAko mice. Intensity of LAMP1 immunostaining was high in some DRG neurons and modest or undetectable in others. High-dose rAAV9-hGLA treatment resulted in normalization of LAMP1 in most highly and modestly affected neurons. A few neurons retained significant LAMP1 positivity potentially pointing to a treatment resistant subpopulation (Fig. 6B).
Contribution of immune cell infiltration 29 –31 to Fabry PNS pathology was investigated using microglia/macrophage marker Iba1 and macrophage/phagocyte marker CD68. Increased Iba-1 staining was detected mostly around DRG neuronal cell bodies of the control G3Stg/GLAko mice (Fig. 6C), whereas CD68-positive clusters were observed in the axonal bundle regions (Fig. 6D). Both Iba-1 and CD68 biomarkers were reduced with treatment and normalized in the high-dose group (Fig. 6C, D).
We also assessed p62 autophagy biomarker as defects in autophagy have been implicated in the pathology of multiple lysosomal storage diseases and neuropathy. 32 –35 p62 immunostaining was clearly detected in neuronal bodies and was essentially undetected in nerve fibers in the WT control mice. In the G3Stg/GLAko control mice, p62 immunostaining was increased in nerve fibers and decreased in neuronal cell bodies potentially indicating a defect in autophagic flow. Distribution of p62 was normalized in mice treated with rAAV9-hGLA and was comparable to the WT controls (Supplementary Fig. S5). p62 was also increased in the nerve fibers of the posterior/dorsal root containing afferent nerve fibers, which return sensory information to the CNS (Supplementary Fig. S6).
Fabry peripheral neuropathy is characterized by loss of small thinly myelinated nerves followed by unmyelinated fibers. 36,37 Immunostaining of the paw dermis with MPZ antibodies revealed significant loss of thinly myelinated fibers in the control G3Stg/GLAko mice. Treatment with rAAV9-hGLA preserved small fiber density. Increase in MPZ positivity was statistically significant even in the lowest dose group (Fig. 6E, Supplementary Fig. S6). Similar results were observed using the pan-neuronal marker PGP 9.5 (Supplementary Fig. S7).
Functional consequences of the pathological changes in the PNS were assessed in the hotplate test at 10 weeks (baseline) and 5, 9, 13, and 17 weeks after dosing. At baseline latency to respond to heat stimuli was not different between the groups. At 5 weeks after dosing, when mice were 16 weeks old, the increase in the hot plate latency test was already statistically significant in control G3Stg/GLAko and the low dose group mice. Heat sensitivity of the G3Stg/GLAko mice in the middle- and high-dose groups was not different from WT control through 13 weeks post-dose. At the final time point, 17 weeks post-dose, hot plate latency of the G3Stg/GLAko mice in the high-dose group remained in the normal range. The increase in average latency of the G3Stg/GLAko middle-dose group between 13- and 17-week post-dose time points was small and was still ∼50% lower than in the G3Stg/GLAko control mice (Fig. 6F, Supplementary Table S5). Overall, rate of heat sensitivity loss was decreased in the low and middle and prevented in the high-dose group.
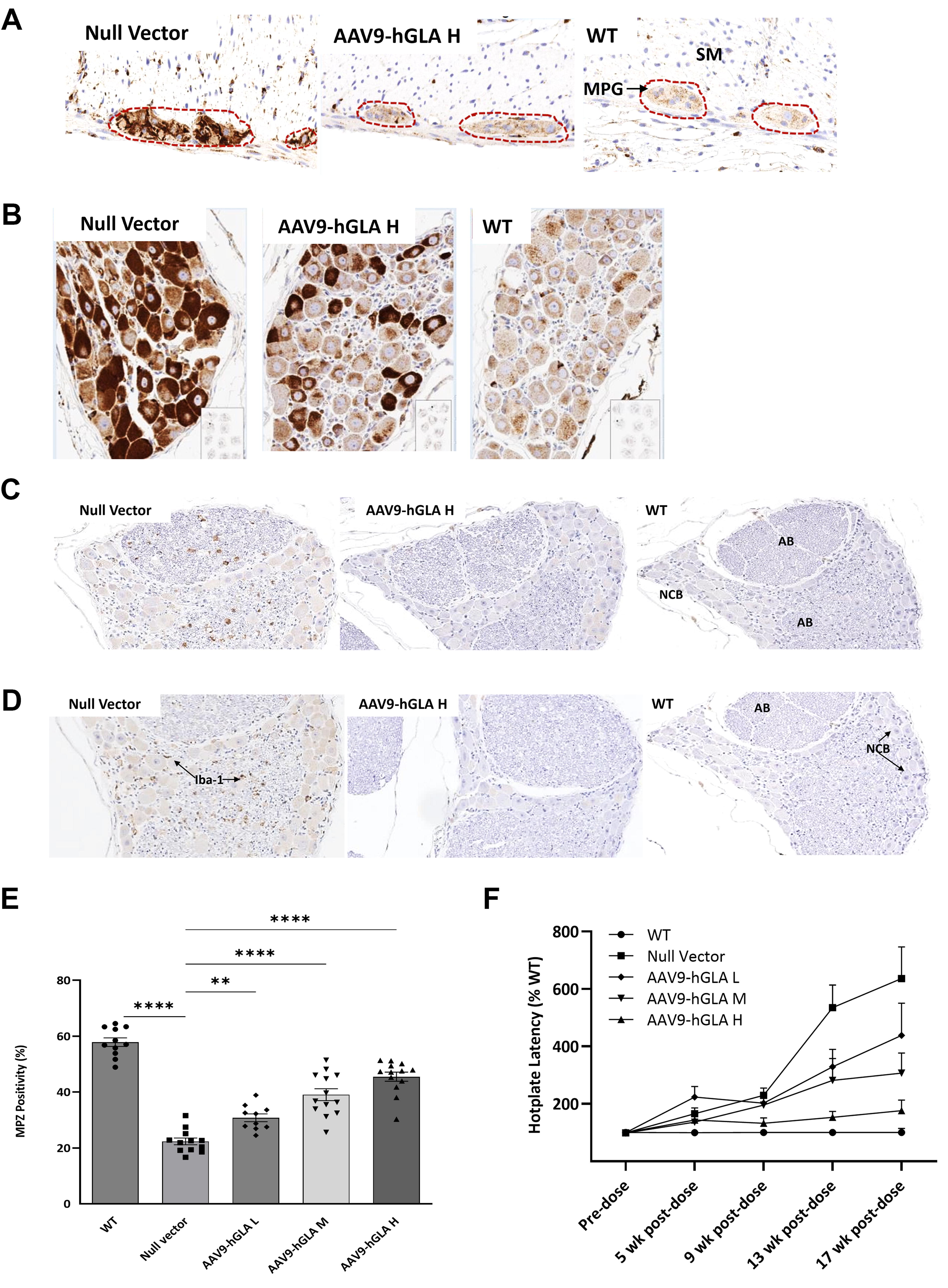
Efficacy of rAAV9-hGLA in mitigating PNS pathology in the TgG3S/GLAko mice.
DISCUSSION
Over the past two decades, multiple studies have demonstrated the efficacy of AAV GT in clearing tissue substrate in the Gla KO mouse model. 3,4,6,11 –13 Preclinical successes paved the way for clinical trial initiations. Two rAAV gene therapies for FD, 4D-310, and ST-920, are currently in Phase I/II. 7,10,38,39 Preliminary results indicate potential improvement in quality of life and GI symptoms in patients treated with ST-920 although small increase in serum lyso-Gb3 was observed in a few patients after ERT discontinuation. Although 4D-310 showed promising improvements in cardiac pathology, it was also associated with serious adverse events, leading to a clinical hold. 40 Further research and better preclinical models are needed to develop second-generation AAV GTs with potentially better safety and efficacy.
The G3Stg/GLAko mouse model accumulates higher levels of tissue substrate compared with α-Gal A KO mice, better reflecting substrate burden in FD patients. Recently, limited AAV GT efficacy in this model was reported by Hayashi et al., where a high 2e12 vg (∼1e14 vg/kg) dose of rAAV9-hGLA under ubiquitous promoter was administered to young, pre-symptomatic 6-week-old mice with modest substrate accumulation. 8 Even with early treatment initiation in this model, significant Gb3 reduction was only observed in the liver and heart.
In our study, we optimized rAAV9-hGLA GT for enhanced efficacy. We were able to achieve sustained dose–response in older G3Stg/GLAko mice by using a combination of a strong ubiquitous promoter and WPRE to boost transgene expression, with similar rAAV9 capsid. We assessed AA9-hGLA across a wide dose range, with the lowest dose (5e10 vg/kg) near the transduction threshold and the highest dose (6.25e12 vg/kg) still within a moderate range for an AAV-based GT study assessed in mouse models. Treatment initiation at 11 weeks was chosen based on significant substrate accumulation and pathology development observed in G3Stg/GLAko mice at this age. 9,14,18
Following rAAV9-hGLA administration, dose-dependent supraphysiological levels of α-Gal were detected in serum and tissues. Tissue substrate reduction at 18 weeks posttreatment was consistent with α-Gal activity and complete or near-complete clearance of Gb3 and lyso-Gb3 was achieved in the high-dose group across serum and all tissues examined (except for the brain). We found that despite higher level of Gb3 accumulation in the kidney, lower level of α-Gal in comparison to the heart was required for complete clearance. The lack of efficacy in the brain at low and medium doses was expected, as rAAV9 GT does not efficiently transduce the brain in adult mice at the low-dose levels used in this study. In addition, circulating α-Gal enzyme does not efficiently cross the blood–brain barrier. 1 As others have demonstrated, the translatability of AAV9 transduction efficacy in mouse brain to intravenous GT administration in humans and translatability of G3Stg/GLAko mouse model brain pathology to FD patients is limited. 14,16,36,41 Therefore, establishing rAAV9-hGLA brain efficacy was not one of the objectives for this study. After observing remarkable efficacy of rAAV9-hGLA in substrate reduction in tissues relevant to FD, we explored the treatment’s impact on structural and functional pathologies and biomarkers relevant to FD.
Similar to patients with FD, G3Stg/GLAko mice exhibit significant Gb3 accumulation in kidney distal tubules and collecting ducts, leading to tubular atrophy. 18,42 –44 AAV9-hGLA treatment dose-dependently reduced tubular atrophy and completely normalized morphology in the high-dose-treated mice. Positive effects were also observed on fibrotic markers. Specifically, rAAV9-hGLA treatment prevented an increase in Collagen I and re-expression of WT1. WT1 is an embryonic development transcription factor and is only active in a few selected cell types in adults. 45 Observed reactivation of WT1 in tubular epithelial cells provides in vivo support for the hypothesis that the mechanism of renal fibrosis initiation in FD could be similar to the one described for other renal diseases and is prompted by tissue damage because of substrate accumulation. WT1 tubular reexpression stimulated by Gb3/lyso-Gb3 tissue damage can then induce epithelial-to-mesenchymal transition—a well-established driving force of fibrosis. 20,21,46,47
rAAV9-hGLA treatment improved kidney function reducing BUN and albuminuria. It is noteworthy that normal or near normal kidney function was only observed in the highest dose group where kidney α-Gal exposure was ∼400-fold higher than WT. At the same time, some improvement in albuminuria was reported when ERT treatment of G3Stg/GLAko mice was initiated at 5 weeks before the onset of pathology. 14 This is consistent with the prevalent view that at some point in the disease course, renal pathology becomes irreversible. Efficacy of the high dose could be explained by very rapid substrate clearance prior to the mice encountering this intractable state. Additional studies with treatment initiation at different ages could help clarify the relationship of dose level versus treatment timing.
In contrast to cardiomyocytes in α-Gal A KO mice, significant level of Gb3 accumulation is detected in cardiomyocytes of the G3Stg/GLAko mouse model. This accumulation more accurately reflects Fabry cardiac pathology. 48 Efficient cardiac Gb3 clearance in G3Stg/GLAko mice by rAAV9-hGLA treatment has the potential to translate into better cardiac efficacy in patients with FD. In addition, our finding that Collagen Type I could be reduced by rAAV9-hGLA treatment may provide a potentially useful biomarker to monitor rAAV9-hGLA cardiac efficacy in patients. 23 Dysregulation of β-catenin localization in the heart of the G3Stg/GLAko mice presents an interesting hypothesis on potential early mechanisms in Fabry cardiac pathology since aberrant β-catenin signaling has been implicated in cardiac fibrosis, arrhythmias, and hypertrophy. 23 –25,49,50 But further studies in mouse models and patients are needed to elucidate its role in Fabry.
rAA9-hGLA efficacy in PNS substrate clearance was evident with histological examination of GI MPG and DRG. rAA9-hGLA treatment also preserved small fiber density, sensory function, and normalized pathological biomarkers. Peripheral neuropathy is one of the most debilitating manifestations of FD poorly addressed by current treatments. Several potential mechanisms of Fabry neuropathy triggered by substrate accumulation have been proposed. 51 Inflammation is one of the proposed mechanisms and DRG macrophage infiltration is implicated in the initiation of neuropathic pain. 31 Upregulation of CD68 monocyte/macrophage markers in the DRG of α-Gal A KO mouse model has been reported. 29 We also found increase in CD68 as well as Iba-1 immunostaining in the DRG of the control G3Stg/GLAko mice that was normalized by rAA9-hGLA treatment. Iba1-positive cells were detected around a subset of the neuronal bodies, consistent with the finding in the mechanical injury PNS pain model where increase in Iba1 was detected around NB200+ neurons. 30 Somatosensory NB200+ neurons are the most affected neuronal subtype by Gb3 accumulation in the α-Gal A KO mice; 28 therefore, it would be reasonable to expect higher Iba-1 around this neuronal subtype. Double staining will be needed to confirm this finding.
We also investigated the potential role of autophagy in FD-associated neuropathy. Autophagy is an important mechanism for maintaining neuronal health. In axons, autophagosomes collect damaged proteins and organelles and move it for degradation into neuronal body enriched with lysosomes. Defects in autophagosome formation and/or retrograde flow lead to axonal degeneration. 34 p62 is a shuttle that facilitates the retrograde flow. We found a significant increase in p62-positive clusters in nerve fibers of the control G3Stg/GLAko, whereas intensity of p62 immunostaining in cell bodies was reduced. An analogous staining pattern was observed in the posterior/dorsal root containing afferent nerve fibers of the DRG neurons which return sensory information to the CNS indicating that both afferent and efferent branches of pseudo-unipolar DRG axons were affected. Accumulation of p62 in nerve fibers is indicative of the defect in the retrograde autophagic flow implicated in neurodegeneration and neuropathic pain. 33,35 Our findings are consistent with the results of α-Gal A KO mice DRG transcriptional profiling where retrograde transport pathway was found to be affected. 32 To our knowledge, this is the first report on the potential involvement of retrograde flow defect in Fabry peripheral neuropathy. Further studies in mouse models and patient tissue samples will be needed to confirm this hypothesis. rAA9-hGLA treatment also preserved small fiber density in the paw of the G3Stg/GLAko mouse model and prevented progression of sensory deficiency.
CONCLUSIONS
We found that rAAV9-hGLA treatment prevented and in some cases resolved pathological molecular processes in G3Stg/GLAko mice known to contribute to FD manifestations. Pharmacologic efficacy was already evident at the lowest feasible threshold transduction dose of 5 × 1010 vg/kg. Along with substrate normalization, structural and functional pathology in multiple tissues was completely reversed or prevented by a relatively low dose of 6.25e12vg/kg. Altogether, our findings provide new insights into potential pathological pathways activated in FD that can be mitigated by GT.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Bhanu Dasari and Pramod Rompikuntal for producing and performing vector QC for reagents used in the study, Yan Huang for pathology discussions, and Madhusudan Natarajan, Galen Carey, and Vivian Choi for study and article discussions.
AUTHORS’ CONTRIBUTIONS
N.B.: Conceptualization, formal analysis, writing—original draft, and writing—review and editing. S.Y.: Conceptualization, methodology, validation, formal analysis, investigation, and writing—review and editing. W.R.: Conceptualization, methodology, formal analysis, data curation, and writing—review and editing. S.A. Investigation. A.R. Investigation. M.R-H. Investigation. S.S. Investigation. K.K. Investigation. E.Y.H.P.: Supervision and writing—review and editing. M.D.: Supervision, conceptualization, methodology, resources, investigation, project administration and writing—review and editing. R.I.: Supervision, conceptualization, validation, methodology, formal analysis, writing—original draft, writing—review and editing, and project administration.
DATA AVAILABILITY
The data supporting this study could be available upon reasonable request to the corresponding author upon Takeda approval. Please contact the corresponding author with any inquiries.
AUTHOR DISCLOSURE
All authors are current or former employees of Takeda and may hold stock or equity interests in Takeda Pharmaceuticals USA, Inc. Rizwana Islam, Mugdha Deshpande, and Eric Park are authors on patents and patent applications related to this work.
FUNDING INFORMATION
This study was internally sponsored and funded by Takeda Pharmaceuticals USA, Inc.
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.