
Abstract
Adeno-associated virus (AAV) gene therapy is making rapid strides owing to its wide range of therapeutic applications. However, development of serious immune responses to the capsid antigen or the therapeutic transgene product hinders its full clinical impact. Immune suppressive (IS) drug treatments have been used in various clinical trials to prevent the deleterious effects of cytotoxic T cells to the viral vector or transgene, although there is no consensus on the best treatment regimen, dosage, or schedule. Regulatory T cells (Tregs) are crucial for maintaining tolerance against self or nonself antigens. Of importance, Tregs also play an important role in dampening immune responses to AAV gene therapy, including tolerance induction to the transgene product. Approaches to harness the tolerogenic effect of Tregs include the use of selective IS drugs that expand existing Tregs, and skew activated conventional T cells into antigen-specific peripherally induced Tregs. In addition, Tregs can be expanded ex vivo and delivered as cellular therapy. Furthermore, receptor engineering can be used to increase the potency and specificity of Tregs allowing for suppression at lower doses and reducing the risk of disrupting protective immunity. Because immune-mediated toxicities to AAV vectors are a concern in the clinic, strategies that can enhance or preserve Treg function should be considered to improve both the safety and efficacy of AAV gene therapy.
INTRODUCTION
Adeno-associated virus (AAV) gene therapy is rapidly advancing to correct defective genes, resulting in potentially lifelong cures for previously untreatable diseases. Food and Drug Administration (FDA) and European Medicines Agency (EMA) approvals for Luxturna, Zolgensma, Hemgenix, Roctavian, Upstaza, and Elevidys highlight the widespread applications for this viral delivery system. 1,2 Clinical success critically depends on immune tolerance to both the viral vector and the transgene product. The expanded use of gene therapy is currently impeded by the development of innate and adaptive immune responses to the capsid elicited in response to the high vector dose required for many gene therapy applications. 3,4 In addition, pre-existing humoral and cellular immune responses to natural infection with AAV can be amplified upon gene therapy. Neutralizing antibodies (NAbs) to the viral vector and transgene product can render gene therapy ineffective, whereas cellular T cell responses can reduce efficacy and pose significant safety risk. Consequently, there is strong interest in immune modulatory strategies to limit immune responses.
Several cell types play a role in modulating immune responses and maintaining lasting tolerance to AAV gene therapy. These include forkhead box P3 (FoxP3) expressing regulatory T cells (Tregs), which play a major role in immune homeostasis and have the potential to dampen cellular immune responses to AAV gene therapy. In this review, we focus on CD4+CD25+FoxP3+ Tregs and will highlight the importance of Treg homeostasis in mediating sustained tolerance to AAV gene therapy. We propose strategies to enhance the Treg suppressive effect by promoting in vivo Treg induction, cellular therapy with expanded or genetically manipulated Tregs, or interleukin (IL)-2 delivery to selectively enhance Treg in vivo persistence and expansion.
REGULATORY T CELLS
Tregs are small, specialized T cell subset that are essential for maintaining immune homeostasis, preventing autoimmunity, and moderating inflammation induced by pathogens and environmental insults. The dominant population of Tregs are CD4+ T cells defined by constitutive expression of the IL-2 receptor (IL-2R) α chain or CD25, and the master transcription factor FoxP3. FoxP3+ Tregs can be either thymus derived (tTregs or natural Tregs) with specificity to self antigen or can be peripherally induced to express FoxP3 (pTregs) following T cell receptor (TCR) stimulation under immune modulatory conditions. Other T cell subsets capable of exerting suppression to various self and nonself antigens in several distinct tissue locations have also been identified. Among these are CD49b+LAG3+FoxP3− Tr1 cells, which produce high levels of the immune modulatory IL-10 cytokine, and latency-activated peptide (LAP) expressing CD4+CD25−LAP+FoxP3− Th3 cells, which produce both transforming growth factor (TGF)β and IL-10. 5,6
Mechanisms of suppression
Tregs do not produce the cytokine IL-2 but critically depend on exogenous IL-2 produced by other cells for their development, survival, and maintenance of FoxP3 expression. 7 This forms the basis for one of the mechanisms for limiting conventional T cell growth and function, by sequestering IL-2 from the environment. Tregs can also inhibit immune function by other mechanisms. This can involve suppression of antigen-presenting cells (APCs) directly through engagement of inhibitory receptors such as CTLA-4 with costimulatory receptors such as CD80/86, and stripping peptide- major histocompatibility complex (MHC) II or costimulatory receptors off dendritic cell (DC) surfaces by a process of transendocytosis. 8,9 Tregs can also function indirectly via the production of suppressive cytokines (including IL-10, IL-35, and TGF-β), and alter the local metabolic environment to limit conventional T cell activity. 10 This ability to alter the local milieu offers the potential for bystander suppression.
In bystander suppression, Tregs specific for one antigen can suppress immune responses against other antigens because of their proximity to the antigen-specific response. 11 –13 Finally, Tregs can also mediate long-term suppression through a process of conversion of activated endogenous conventional T cells into de novo Tregs (particularly in the presence of TGF-β). 14 This ability of Tregs to promote the generation of other regulatory cells is likely to provide more robust and durable tolerance. In addition to inhibition of inflammation, Tregs also promote the repair of damage by releasing tissue and stem cell factors. 15 –18
IMMUNOGENICITY TO AAV GENE THERAPY
Immunity to AAV gene therapy is multifactorial and involves activation of multiple arms of the immune system. Depending on the disorder, AAV vectors are administered systemically or locally into the eye, central nervous system, or into specific muscles. 19 –24 The route of administration and vector dose can considerably impact immunogenicity of gene therapy. Other factors include the AAV serotype, genomic CpG content, promoter (ubiquitous vs. tissue specific), transgene, as well as underlying inflammation caused by the disorder (as seen with neuromuscular disorders). 3,25 –29 Although recent studies have improved our understanding of innate and adaptive immune responses to AAV vector–mediated gene transfer, several questions remain unanswered.
Innate immune response
AAV particles have been shown to potently activate the innate immune system not only by activating pathogen-associated molecular patterns (PAMPs) but also through the response to the high capsid protein load associated with systemic AAV administration (milligram quantities that can aggravate the risk of complement activation). 30 PAMPs in the AAV capsid and viral genome are recognized via Toll-like receptors (TLRs), primarily TLR2 and TLR9, which can trigger an innate immune response and promote the activation of adaptive immunity through chemotaxis, inflammation, and interferon production that in turn activate CD8+ T cells. 31 –35 CD4+ T cell help is requisite for driving cytotoxic T cell responses to the viral capsid through the TLR9-Myd88 signaling pathway. 25,35
A very recent finding by Kumar et al. documents a previously unknown dependence for IL-1 receptor 1-MyD88 signaling to generate cytotoxic CD8+ T cells against both a model antigen, ovalbumin, and the coagulation factor VIII (FVIII) transgene in liver-directed AAV gene therapy, which is independent of TLR9 or type I interferons. 36 This complex response requires cooperation between IL-1–producing plasmacytoid DCs, Kupffer cells, and XCR1+ DCs for antigen priming and cross-presentation, which is crucially dependent on CD4+ T cell help. 36
Adaptive immune response
Innate immune sensing initiates the activation, clonal expansion, and differentiation of antigen-specific B and T cells that can eliminate the antigen through humoral or cytotoxic responses and generate immunological memory. The activation of APCs such as DCs is a critical step in linking innate to adaptive immunity. Whereas antigen presentation/cross-presentation can prime and activate MHC I–restricted CD8+ T cells that are capable of lysing vector-transduced cells, this is critically dependent on MHC II–restricted CD4+ T cell help. 35 CD4+ T helper cells are also critical for the formation of memory responses. 37
B cell response
Natural infection with AAV or vector gene transfer leads to capsid antigen presentation, B cell activation, and differentiation into neutralization antibody (NAb) producing plasma cells and memory B cells, which is greatly facilitated by CD4+ T cell help. In addition to the presence of NAbs as a major exclusion criterion for patients receiving AAV gene therapy, capsid opsonization by antibodies can also enhance complement activation, thus amplifying innate immune responses. 30 Various protocols are currently being explored (mainly in preclinical models) to allow AAV administration in seropositive subjects, or to enable vector readministration. 38 Transient elimination of NAb-producing B cells can also reduce complement activation in some cases. 30
T cell response
Despite the perceived low immunogenicity of AAV gene therapy, systemic and localized vector capsid and transgene-specific T cell responses have been detected in patients. 20,39 –42 These responses range from localized and limited to systemic inflammatory toxicities with severe outcomes, particularly in a subset of high-dose clinical recipients. 40 Semiquantitative assays such as enzyme-linked immunospot (ELISpot) for human peripheral blood mononuclear cells (PBMCs) are generally used to detect T cell responses to the vector capsid or the transgene, although PBMCs may not reflect T cell responses within target tissues. Cellular infiltrates in tissue biopsies are detected by immunohistochemistry when possible. 24,43 It is entirely likely that T cell responses to AAV gene therapy are underreported, as many clinical trials do not test for T cell responses, and for the purely technical reason that many tests require a minimum threshold of T cells to determine an appreciable immune response, and current ELISpot analyses may not be sensitive enough. Subjects at risk of developing transgene-specific responses because of gene mutations or other factors are also typically excluded from clinical trials (NCT03362502, NCT06270719).
AAV capsid-specific T cell responses are thought to occur as reactivation of a memory T cell response from previous exposure to AAV but may also occur de novo. 44 –46 Capsid-specific T cells can eliminate transduced cells, causing a complete loss in transgene expression in some cases. 44,45 Capsid-specific T cells are also found in the blood of patients treated with retinal gene therapy, but it is unclear whether they enter the eye and contribute to the elimination of transduced cells. 47 Efforts are being made to rationally engineer less immunogenic capsids by predicting and silencing immunodominant T cell epitopes. 48 Although this would considerably help in mitigating memory T cell reactivation against conserved immunodominant epitopes, it is unclear whether this would affect de novo responses against other subdominant epitopes.
Transgene-specific T cell responses often occur owing to a lack of central tolerance, which is more prominent in patients who have mutations in the portion of the therapeutic gene that is present in the transgene. For example, Duchenne muscular dystrophy (DMD) patients with deletions in exons 8–11 of the dystrophin gene overlapping with those expressed in mini/microdystrophin may be at risk of developing transgene-specific T cell responses following AAV gene therapy (NCT04626674, NCT04281485, 2020-002093-27). 3,49
TREGS IN LIVER GENE TRANSFER
AAV gene therapy into the liver has achieved measurable success, as shown by FDA approvals for hemophilia A and B. 50,51 The liver microenvironment is considered tolerogenic, supported by a combination of several factors, such as hepatocyte-restricted transgene expression, antigen presentation by nonconventional parenchymal and nonparenchymal liver cells including liver sinusoidal endothelial cells, stellate cells and Kupffer cells, and high levels of anti-inflammatory cytokines IL-10 and TGF-β that promote nonresponsiveness to a potentially immunogenic antigen. 52 –54 Early preclinical studies for hemophilia B showed that hepatic AAV gene therapy led to sustained systemic expression of the clotting factor IX transgene product at therapeutic levels. 55 This correlated with the induction of Tregs and a lack of cellular or adaptive responses directed toward the transgene. Several subsequent studies confirmed the critical role of antigen-specific Tregs in sustaining this liver-restricted tolerance.
Treg depletion in mice and in a nonhuman primate (NHP) model of hemophilia B resulted in the mounting of immune responses that quickly led to loss of transgene expression. 51,53,56,57 Patients receiving liver-directed AAV gene therapy for hemophilia A or B have so far not developed antidrug antibody responses (also called inhibitors) against the therapeutic clotting factor. In fact, a recent report for liver-directed gene therapy for hemophilia A in patients with active FVIII inhibitors showed a decline in inhibitor titer, which correlated with detectable FVIII activity in a subset of patients, and no inhibitor recurrence in patients with prior inhibitors (NCT04684940) 58 validating early preclinical AAV liver gene transfer studies in hemophilia A dogs with inhibitors. 59
However, the viral vector load can contribute to toxicity in the liver, and hepatotoxicities 60,61 have been observed, even leading to fatality of subjects in certain liver gene therapy clinical trials. 4,61 Elevated liver enzymes that may be associated with a high viral vector load 51,62,63 or reactivation of memory CD8+ T cell responses to the capsid have also been observed. 45 Furthermore, in hemophilia A gene therapy clinical trials, stable clotting factor expression adequate to eliminate or nearly eliminate bleeding in all patients has not been achieved. 64
Proof for the role of Tregs in mediating liver tolerance toward an ectopic antigen was demonstrated in preclinical studies in autoimmune murine models. Keeler et al. showed that hepatic AAV gene delivery of the myelin oligodendrocyte glycoprotein (MOG) neuroprotein can prevent and even reverse autoimmune disease symptoms in mice with experimental autoimmune encephalomyelitis, which is the preclinical model for multiple sclerosis. 65 AAV gene therapy caused the induction/expansion of antigen-specific CD4+FOXP3+ Tregs in the liver, which correlated with an absence of inflammatory lesions within the spinal cord of mice treated with AAV8.MOG. 65 The authors subsequently described a role for multiple CD8+FoxP3− Treg subsets in mediating this tolerogenic effect. 66
Similarly, a study by Akbarpour et al. showed that lentiviral insulin gene expression targeted to hepatocytes induced antigen-specific Tregs, which halted immune cell infiltration into the pancreatic islet and protected from type 1 diabetes. 67 The tolerizing effect of liver-directed gene therapy was used by Bartolo et al. who demonstrated that dual muscle–liver-targeted AAV gene therapy allows for long-term transgene expression in the muscle even in the face of pre-existing immunity to the transgene. 68 This was associated with an exhausted profile in transgene-specific CD8+ T cells and lack of interferon-γ production.
Therefore, it appears that targeting gene transfer to hepatocytes can favor the induction of Tregs toward the encoded transgene, making the liver an attractive target for achieving transgene tolerance. Efforts to dissect mechanisms of antigen-specific Treg induction showed that virally encoded antigen is presented in liver-draining lymph nodes, which is also the site for Treg induction. 69 Hepatic-induced Treg rapidly disseminate through the systemic circulation, where they regulate immune responses. 69 These studies therefore suggest that augmentation of Treg activation in the liver can provide new means to avoid destructive immune responses to gene transfer.
TREGS IN MUSCLE GENE TRANSFER
Muscle-directed gene therapy, either systemically injected to target skeletal and cardiac muscle, or locally injected into multiple sites in the skeletal muscle, can provide therapeutic benefit for degenerative disorders like the muscular dystrophies, X-linked myotubular myopathy, metabolic disorders such as Pompe or Danon disease. 4,19,70 –72 Ectopic expression of corrective transgenes in skeletal muscle cells can also provide lasting transgene expression for disorders such as lipoprotein lipase (LPL) and alpha-1 antitrypsin (AAT) deficiency. 24,73 A protective role for the accumulation of Tregs that secrete IL-10 and amphiregulin have been shown to modulate myeloid cell infiltration and inflammation in a mouse model of muscular dystrophy, thereby improving muscle repair. 16 This therapeutic effect can be enhanced by adoptive transfer of antigen-specific Tregs or by the in vivo expansion of polyclonal Tregs with low-dose IL-2/anti-IL-2 complexes. 74,75 These studies demonstrate the potential of Tregs and Treg-inducing agents to reduce muscle inflammation and immune cell infiltration.
Although delivering the therapeutic transgene into various muscles bodywide via intravenous or intramuscular gene transfer is considered more immunogenic, and anti-capsid and anti-transgene T cells have been detected in subjects, 76 evidence for the protective role of Tregs in driving sustained transgene expression has been demonstrated in AAV clinical trials for LPL deficiency and AAT deficiency. 24,73,77 In the AAT deficiency trial, all treatment recipients developed immune responses to the AAV1 capsid and one of the nine patients had a positive T cell response associated with a single AAT peptide polymorphism present in the subject but not in the wild-type AAT therapeutic product. 24,78 Despite the presence of anti-capsid CD4+ and CD8+ effector T cells, sustained presence of the transgene in the injected muscle was observed in all subjects.
A substantial portion of the muscle infiltrating lymphocytes were Tregs suggesting that they actively suppressed vector-induced effector T cell responses and thereby prevented loss of AAV-transduced muscle cells. 77 A further follow-up study conducted 5 years after gene transfer showed the persistence of Tregs and potentially exhausted AAV capsid-specific CD8+ T cells within the injected muscle. 79,80 Similar findings were observed for muscle-directed AAV gene therapy for LPL deficiency, where CD8+ T cells in muscle infiltrates lacked cytotoxic potential, correlating with the infiltration of FoxP3+ CD4+ T cells, indicating that both T cell exhaustion and active tolerance contributed to long-lived transgene expression. 73 Taken together, these studies suggest that in some cases, intramuscular AAV delivery can induce a regulatory T cell response and T cell exhaustion profile that can favor sustained transgene expression.
CHOICE OF IMMUNE SUPPRESSIVE DRUG
Immune suppressive (IS) drugs are commonly used in AAV clinical trials to manage immune-related adverse and severe adverse events (SAEs). However, there is no consensus as to which drug/drug combinations should be used for type of SAE, dose, or duration of treatment. In some cases, wrongly timed intense IS, in particular Treg-depleting drugs, can exacerbate the risk of transgene immunogenicity as shown in mice and NHP models. 56,81 First-line treatments to dampen T cell immune responses include steroid drugs such as methylprednisolone/prednisone or deflazacort, given until resolution of inflammation. These drugs control inflammation and are well tolerated if given transiently. Although they do not act selectively on T cells, they do affect multiple aspects of T cell responses by attenuating TCR and proliferation signals, prevent memory CD8+ T cell formation, cause impaired cytokine production and effector function, and even induce Treg formation. 82 –84
Corticosteroids have been combined with other treatments that are more T cell targeted such as tacrolimus, given either prophylactically or postgene therapy, as shown in clinical trials for hemophilia B, 85 Danon disease, and DMD (NCT03369444, NCT03641703, NCT03882437, NCT03769116). Tacrolimus is a calcineurin inhibitor, which inhibits TCR-induced T cell activation and IL-2 transcription by inhibiting the translocation of nuclear factor of activated T cells (NFAT). High-dose tacrolimus may hamper the function of tissue-protective Tregs, given the dependence of these cells on IL-2 and their need for nuclear NFAT to express FOXP3 efficiently. 86,87 At a low enough dose, tacrolimus can preserve Treg survival, as shown in an NHP study with AAV8 and AAV9 gene therapy for DMD, where continuous treatment with 0.06 mg/kg tacrolimus was able to preserve expression of a Lacz or microdystrophin transgene for 42 weeks. 88
Another common IS combined with steroid drugs and/or B cell depletion compounds is the macrolide mTOR inhibitor sirolimus or rapamycin, which is known to selectively inhibit survival signals in activated T effector cells, while sparing Tregs, because they are less dependent on mTOR signaling. 89 This selective inhibition skews the tissue to a Treg-rich environment, further enhanced by the conversion of activated conventional T cells into antigen-specific Tregs in the absence of mTOR signaling. Furthermore, in preclinical models of hepatic gene transfer, sirolimus can enhance AAV hepatic transduction because of autophagy. 90 –92 Sirolimus administered intravenously or encapsulated as tolerogenic nanoparticles provides a dose-dependent and long-term suppression of cellular responses against AAV. 93,94 Sirolimus can also inhibit germinal center B cells and antibody class switching. However, this can result in an accumulation of IgM expressing B cells with AAV neutralizing and complement activating properties. 95,96
In AAV gene therapy clinical trials, sirolimus is administered for the resolution of inflammation (NCT03362502). Furthermore, a combination of the B cell–depleting anti-CD20 chimeric antibody, rituximab and sirolimus with limiting corticosteroids is being evaluated to transiently inhibit humoral and cell-mediated immune responses in Danon disease (NCT03882437) and Tay Sachs disease. 97 Because this combination can also reduce NAbs to the vector capsid, it is also being considered to enable AAV redosing (NCT02240407) and reduce complement severity, thrombotic microangiopathy, thrombocytopenia, and atypical hemolytic uremic syndrome–like symptoms (NCT04519749). This is because capsid-specific antibodies to systemic high-dose AAV administration are shown to initiate the classical pathway of complement activation, which is further amplified by the alternative pathway. 30 Therefore, the right IS treatment regimen can aid tolerance to both the vector and transgene product.
ROLE FOR TREG CELLULAR THERAPY
Cellular therapy with Tregs has shown promise in several clinical trials for autoimmune and inflammatory disorders and in transplantation studies. Therapeutic Tregs can be either ex vivo expanded cells of polyclonal specificity or enriched for donor alloreactivity by coculture with donor APCs, as in the case of living donor transplants. 98 –103 These phase I/II studies have demonstrated that Treg therapy is feasible, can be administered safely, and is well tolerated with potential benefit. Many academic and industry-sponsored clinical trials for synthetically modified and unmodified Tregs are currently underway to further assess the benefits of Treg therapy. 15
The potential benefit for Treg therapy administered concomitantly with therapeutic gene delivery has been established in various preclinical models, although it has not been tested in patients to date. In early proof of concept studies, adoptive transfer of FoxP3 overexpressing T cells was shown to suppress inhibitory antibody development that neutralized functional FVIII activity in a hemophilia A model of nonviral FVIII plasmid-mediated gene transfer. 104 A potentially superior suppressive capacity of ex vivo expanded polyclonal Tregs in protein replacement therapy for hemophilia A and AAV gene therapy for hemophilia B was then demonstrated by Sarkar et al. 105 This was attributed to the activated status of the expanded Tregs, which resulted in upregulation of coinhibitory receptors such as CTLA-4, making them more potent mediators of suppression. 105 CTLA-4 also competes with CD28 for binding to the costimulatory molecules CD80/86 on APC, thereby inhibiting antigen presentation to conventional T cells owing to downregulation/trans-endocytosis of CD80/86. 8,106 This suboptimal antigen presentation by APC can facilitate the induction of de novo antigen-specific Tregs, 106,107 which can lead to lasting tolerance because of the emergence of an endogenous antigen-specific Treg population.
Role for engineered Treg therapy
Engineering approaches to generate antigen-specific Tregs are a feasible strategy to restore tolerance, especially to the therapeutic transgene in case of gene mutations that produce very little or no endogenous protein. Recent synthetic approaches to engineer chimeric receptors have enhanced Treg antigen-specific tolerance while increasing specific activity, stability, efficacy, and in vivo persistence. 10 Engineering Tregs to recognize a specific antigen can lead to an activated, “effector-like” phenotype with improved migratory capacity toward a target tissue. 108,109
Tregs can either be engineered to express antigen-specific transgenic TCRs, or antibody-based chimeric receptors such as chimeric antigen receptors (CARs). 10 Multiple preclinical studies in autoimmunity and transplantation tolerance establish the safety and potency of engineered Tregs, 110 and early clinical trials (NCT04817774, NCT0523419) are ongoing. Engineered TCR or antibody-based receptor expressing Tregs have also been shown to be suppressive in murine models of protein replacement therapy, by modulating antibody responses against the therapeutic protein product. 111 –114
CAR engineering has transformed the landscape for blood cancer therapy. Unlike TCRs, CARs combine the antigen recognition specificity of an antibody with T cell primary and costimulatory signaling domains, to allow for antigen-specific cell activation and proliferation independent of MHC presentation. 115 The impact of antibody affinity and antigenic density on CAR Treg signaling and its effect on suppressive function is still being understood. In line with recent findings that CAR signaling is stronger and distinct from endogenous TCR signaling, high-affinity FVIII-CAR Treg signaling led to overstimulation and subsequent destabilization into a proinflammatory phenotype. 112 Development of a TCR fusion construct by tethering the FVIII single chain variable fragment onto the TCR-CD3 complex was able to control Treg signaling upon antigen recognition and reduce proinflammatory cytokine production, which led to suppression of inhibitory antibody formation in a murine model of FVIII protein replacement therapy. 112
A caveat with engineering antibody-based chimeric receptors is that these are usually directed against cell surface antigens, and therefore cannot recognize intracellularly expressed antigens or enzymes. One way to circumvent this obstacle is leveraging a Treg's bystander suppression capacity, whereby antigen-stimulated Tregs can also suppress against third party antigens in their vicinity by mechanisms including coinhibitory receptor expression, IL-2 consumption, and localized production of anti-inflammatory cytokines. 116,117 Of importance, bystander suppression does not impact the ability to respond against infections or tumor immunity. 117 This was demonstrated in a preclinical model of intramuscular AAV gene transfer by Arjomandnejad et al. who designed a CAR construct with specificity to the intact AAV capsid. The construct also contained a FoxP3 gene that induced a Treg-like phenotype in transduced cells. These engineered AAV-CAR Tregs were able to suppress both capsid-specific CD8+ T cells as well as CD8+ T cells directed toward the AAV-delivered transgene product 118 (Fig. 1).
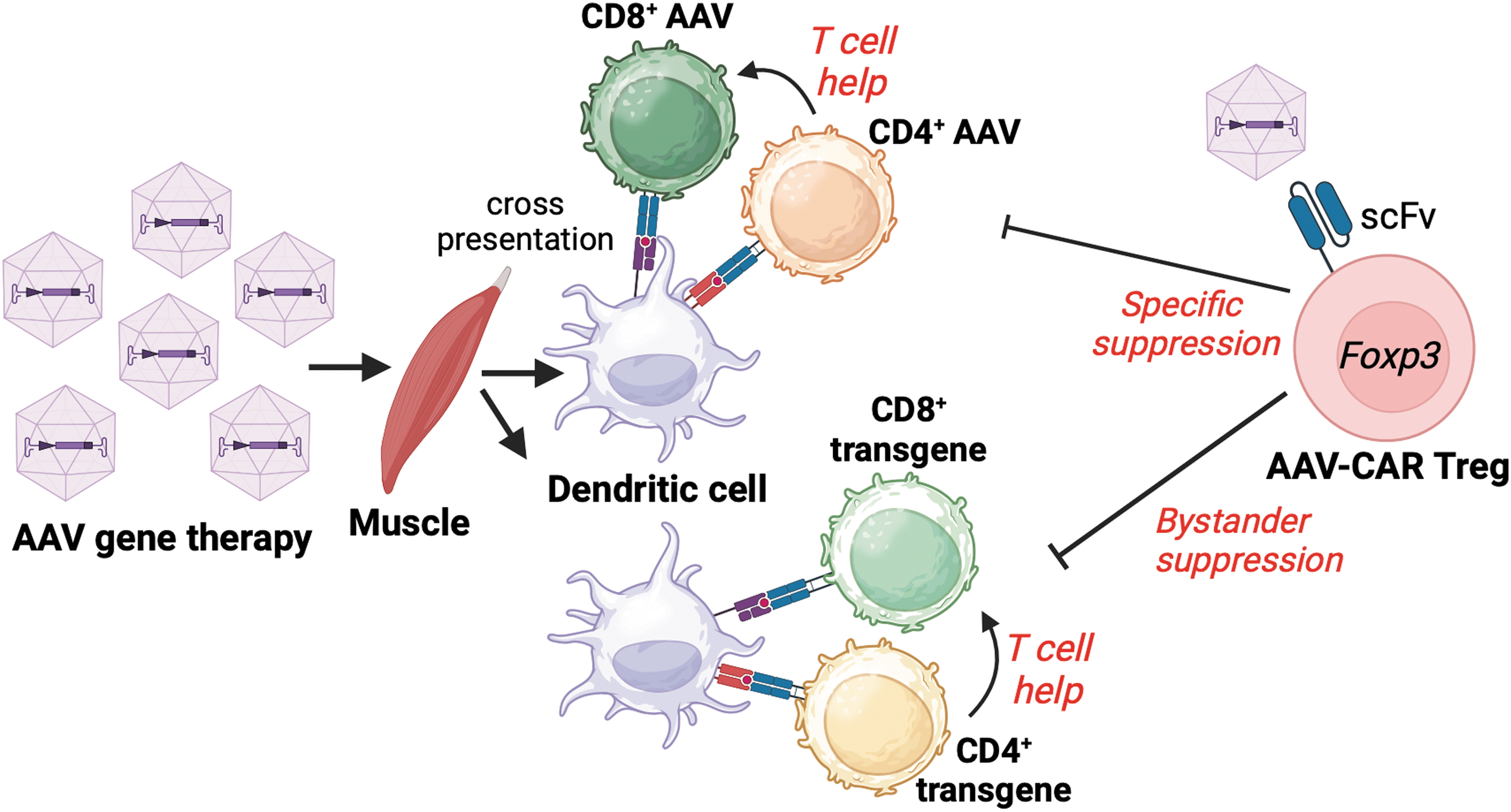
Graphic outlining the development of AAV capsid and transgene-specific CD4+ and CD8+ T cells in response to muscle-directed gene therapy. AAV-CAR Tregs expressing a scFv to intact AAV capsid can suppress capsid-specific CD8+ T cell responses and CD4+ T cell help, decrease tissue inflammation, and potentially mediate bystander suppression against cellular responses to the transgene. AAV, adeno-associated virus; scFv, single chain variable fragment.
A reduction in cellular infiltration into the injected muscle and sustained therapeutic transgene expression was observed in the AAV-CAR Treg-treated mice. This work highlights the important role of capsid-specific T cell responses in the clearance of transgene expressing cells, but also shows a role for bystander suppression of immune responses to the transgene. Although the authors propose that bystander suppression occurs owing to the generation of a local immunosuppressive environment mediated by adoptively transferred activated Tregs, more studies are required to understand the in-depth mechanisms by which bystander suppression is mediated and whether it can lead to the development of endogenous Treg directed toward multiple antigen specificities.
Finally, given the risk of Treg plasticity to convert into Th1-like or Th17-like T cells under an inflammatory environment, questions remain on whether phenotype plasticity associated with loss of FoxP3 expression in Tregs can lead to conversion into antigen-specific “effector” T cells that might exacerbate pathogenic responses; in such cases, whether “forced” expression FoxP3 by lentiviral transduction or HDR approaches would improve stability, and if so, whether these “Treg-like” cells will be as potently suppressive as naturally derived tTregs. Studies are ongoing to assess the efficacy of these FoxP3 reprogrammed CAR Tregs (QEL-001, NCT05234190) in liver transplant patients.
IL-2–based therapies
Given the absolute dependence of Tregs on exogenously derived IL-2 for their maintenance and suppressive function, Treg-specific expansion in vivo by IL-2 is a compelling therapeutic approach. Tregs abundantly express the IL-2R but are reliant on IL-2 produced by other cells, including activated T cells, for their signaling and function. 7,119 Therefore, selective IL-2 consumption by depriving IL-2 from activated CD4+ and CD8+ T cells plays a key role in the suppressive feature of Tregs. 120
The IL-2R subunits, IL-2Rα (CD25), IL-2Rβ (CD122), and the common γ chain (CD132) together interact with IL-2. Tregs constitutively express CD25 and are more sensitive to IL-2 stimulation compared with the other T and NK cell populations, which express CD25 only when activated. Based on this IL-2 binding selectivity, IL-2/anti-IL-2 antibody complexes have been developed with the purpose of reducing affinity of IL-2 to IL-2Rβ and to selectively stimulate CD25+ FoxP3 Tregs. 121 IL-2 complexed to the mouse IL-2 antibody (JES6-1A12) prevents clotting factor VIII antibodies in hemophilia A mice following plasmid FVIII gene therapy by transiently expanding thymic Tregs. 122 However, because IL-2 complexes are noncovalently bound, there is a risk of expanding CD4+ or CD8+ T or NK cells owing to dissociation of the complex and release of unbound IL-2. 123 This risk can be averted by engineering a single-chain fusion protein composed of IL-2 and its antibody, which showed superior Treg bias compared with the IL-2 complex. 123 Other approaches such as IL-2 muteins, which are designed to be Treg selective by either increasing IL-2 affinity to IL-2Rα (NCT04680637, NCT04987307, NCT04924114) or by decreasing affinity to IL-2Rβ, 124 coupled to an Fc silenced human IgG1 to enhance pharmacologic half-life have been developed. This approach was shown to be effective for lasting suppression of transgene humoral responses in a preclinical model of plasmid-mediated FVIII gene delivery for hemophilia A. 125
Finally, to selectively deliver IL-2 only to Tregs, further increasing the safety profile and specificity of IL-2 therapy, promising approaches to engineer IL-2Rs have been developed. Sockolosky et al. developed an orthogonal IL-2/IL-2R system, where a mutant IL-2 (ortho IL-2) is engineered to only bind to a mutated IL-2R (ortho IL-2Rβ). 126 Adoptive transfer of engineered Tregs expressing the ortho IL-2Rβ enables their selective expansion in vivo after administration of ortho IL-2, which led to suppression preclinical models of lethal graft versus host disease (GvHD). 127 Similarly, Cook et al. engineered a chemically inducible signaling complex (CISC), which takes advantage of the capacity of rapamycin (sirolimus) to bind simultaneously to two different proteins, FK506 binding protein (FKBP) and mTOR via FKBP–rapamycin binding (FRB) domain. 128
By replacing the extracellular domains of IL-2Rβ and γ with FKBP and FRB, they showed that an exogenously supplied subtherapeutic dose of rapamycin or its analog was able to induce dimerization and mimic IL-2R signaling in cells expressing these fusion proteins. When expressed in T cells that were constitutively engineered to express FoxP3 by homology-derived recombination (engTregs), these CISC engTreg cells showed improved in vivo persistence and limited GvHD development in mice. Future developments to engineer IL-2 specificity toward engineered CAR or TRuC Tregs should considerably improve the in vivo persistence and suppressive capacity of engineered Treg cellular therapy (Fig. 2). This may also be useful to improve the in vivo persistence of ex vivo expanded polyclonal Tregs, which are subject to IL-2 withdrawal and apoptosis following adoptive transfer. 129

Strategies to deliver IL-2 to engineered Tregs. Improved in vivo expansion and persistence of transgenic TCR, CAR, or TRuC expressing Tregs by administering either a single chain IL-2 immunocytokine or an IL-2 mutein, by engineering an IL-2 CISC complex, or an ortho IL-2-IL-2R complex. CAR, chimeric antigen receptor; CISC, chemically inducible signaling complex; IL-2R, IL-2 receptor; TCR, T cell receptor.
CONCLUSIONS AND FUTURE DIRECTIONS
Tregs offer a new therapeutic approach to establish tolerance in AAV gene therapy by suppressing cellular and adaptive responses to the vector and transgene product and resolving inflammation in targeted organs, all without compromising overall T cell host immunity. Approaches can involve pharmacological treatment regimens to in vivo induce and expand Tregs or cellular therapy with Tregs. Although cellular therapy with polyclonal or engineered Tregs is an attractive approach, questions remain such as the optimal schedule for dosing (concomitant with vector administration or coinciding with transgene expression), number of doses, as well as the number of cells to be dosed. Other questions include whether Treg therapy can be combined with IS therapies in a synergistic manner, and strategies to potentiate in vivo survival and persistence of the transferred Tregs.
At present, the majority of Treg products generated for clinical application are autologous. Using various CRISPR/Cas-mediated TCR and MHC KO approaches can enable the design and banking of allogeneic “third-party” Treg products for off-the-shelf administration. This can also be facilitated by generating induced pluripotent stem cell–derived Tregs. Altogether, these innovative technologies should provide the next generation of Treg therapeutics to augment various gene therapy applications.
Footnotes
AUTHORs' CONTRIBUTIONS
M.M.-M. and M.B. wrote the article. M.B. conceived the project and supervised the study.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by NIH grant R21 HL170146 to M.B.