
Abstract
Genome editing has the potential to treat genetic diseases in a variety of tissues, including the lung. We have previously developed and validated a dual adeno-associated virus (AAV) CRISPR platform that supports effective editing in the airways of mice. To validate this delivery vehicle in a large animal model, we have shown that intratracheal instillation of CRISPR/Cas9 in AAV5 can edit a housekeeping gene or a disease-related gene in the lungs of young rhesus monkeys. We observed up to 8% editing of angiotensin-converting enzyme 2 (ACE2) in lung lobes after single-dose administration. Single-nuclear RNA sequencing revealed that AAV5 transduces multiple cell types in the caudal lung lobes, including alveolar cells, macrophages, fibroblasts, endothelial cells, and B cells. These results demonstrate that AAV5 is efficient in the delivery of CRISPR/Cas9 in the lung lobes of young rhesus monkeys.
INTRODUCTION
Genome editing in somatic cells and tissues has the potential to provide long-term expression of therapeutic proteins to treat a variety of genetic diseases, such as cystic fibrosis and other inherited disorders of the lung. 1 –3 Nonhuman primates, particularly rhesus monkeys, play a crucial role as a translational animal model for investigating human diseases and evaluating the safety of new therapies. 4 However, delivering genome editing machinery to disease-relevant cell types in the lung of primates has remained a challenge. 3,5 Adeno-Associated Virus (AAV) is a common viral vehicle for gene therapy with tropism for various cell and tissue types. 6,7 AAV1 has been used to successfully transduce chimpanzee airways in vivo. 8,9 However, to our knowledge, genome editing using other AAV serotypes, such as AAV5, has not been previously reported in rhesus monkey airways.
The NIH Somatic Cell Genome Editing (SCGE) Consortium 10 was launched in 2018 to accelerate the development of safe and effective genome-editing methods and tools. The goal of our project, funded through the SCGE Program, was to develop technologies to deliver genome editing machinery to disease-relevant cells in the lung. 10 We had previously developed and validated a dual AAV5 platform that supports effective editing in mouse airways. 11 As a result of these findings, we scaled up AAV production to test genome editing delivery in rhesus monkeys. Herein, we demonstrate that intratracheal administration of a dual AAV5 vector encoding CRISPR/Cas9 can mediate genome editing in rhesus airways. Up to 8% editing was observed in lung lobes, including a housekeeping gene GAPDH and a disease-related gene angiotensin-converting enzyme 2 (ACE2). ACE2 is a key receptor for SARS-CoV-2 virus infection and is expressed in a subset of lung cells 12 ; GAPDH is ubiquitous. Using single-nuclear RNA-sequencing (snRNA-Seq), we systematically characterized cell types transduced by AAV5.
MATERIALS AND METHODS
Generation of plasmid
The creation of pAAV-sgRNA was achieved via Gibson assembly, involving the fusion of four distinct DNA fragments: (1) gBlock carrying sgACE2 or sgGAPDH under the control of the U6 promoter, (2) gBlock incorporating the chicken beta-actin promoter, 13 (3) gBlock containing sequences for rhesus chorionic gonadotropin (rhCG) and miR142 binding sites, and (4) an AAV backbone that had been digested with MluI and EagI (Supplementary information). S. pyogenes (Sp)Cas9 is expressed from our published single-stranded AAV vector with U1A promoter. 14
AAV vector production
AAV5 vectors were produced at the Viral Vector Core located at the Horae Gene Therapy Center within the University of Massachusetts Medical School. 11,15 Briefly, the rAAV vector plasmid, containing an expression cassette for the gene of interest flanked by AAV2 ITRs, was co-transfected into HEK 293 cells. This co-transfection was carried out with a packaging plasmid and an adenovirus helper plasmid. The recombinant viruses are subsequently purified using the standard CsCl gradient sedimentation method and desalted through dialysis. 16 To ensure their quality, the vectors undergo rigorous testing. This includes Droplet Digital PCR (ddPCR) titration to determine DNase-resistant vector genome 17 concentration, using probes and primers targeting the Poly A region of the vector genome. Additionally, silver-stained SDS-polyacrylamide gel analysis is employed to assess the purity of each production lot. 18
In vivo studies
All procedures conformed to the requirements of the Animal Welfare Act, and protocols were approved prior to implementation by the Institutional Animal Care and Use Committee (IACUC) at the University of California, Davis. Nine ∼3–4 month-old rhesus monkeys (Macaca mulatta) (∼1 kg; males and females) were screened then assigned to treatment groups. All animals were confirmed seronegative for AAV5 and SpCas9 antibodies prior to study assignment. Group 1 (n = 4; 2 males, 2 females) was included to evaluate editing of GAPDH 3’ UTR. Group 2 (n = 4) was designed to examine the editing of ACE2. One animal (male) served as a control.
Animals were sedated with Telazol intramuscular (IM; 5–8 mg/kg), and blood samples were collected (∼4 mL; hematology, clinical chemistry, serum, plasma). A CT scan was obtained immediately prior to vector administration. Instillation of the solution into the airways was performed using the MADgic laryngo-tracheal mucosal atomizer (Fisher Scientific, Catalog No. NC0924493, Teleflex LLC MADgic Laryngo-Tracheal Atomization Device MAD700) as previously described. 19 The atomizer was carefully inserted into the trachea using a laryngoscope, then a prefilled syringe containing 1 mL of the instillation solution was connected to the proximal end of the atomizer, and the content was expelled into the airways by applying pressure to the syringe pestle, creating a mist. This was immediately followed by 400 μL of ambient air to complete the instillation of material in the atomizer, which was then carefully removed. Each administration consisted of 1 mL sterile PBS containing 2x1013 vg of scAAV5-sgRNA or 2x1013 vg of AAV5-Sp Cas9. Animals were then positioned on the scan bed, and a post-administration CT scan was obtained (Fig. 2). All animals were monitored closely immediately after and during the 3 week post-administration period. All remained healthy and robust during the study period. Following euthanasia (overdose of pentobarbital), the chest cavity was opened, and the lung vasculature was perfused with PBS and 10 U/mL Heparin via the right ventricle. Specific sections of the lung lobes were removed for detailed analysis.
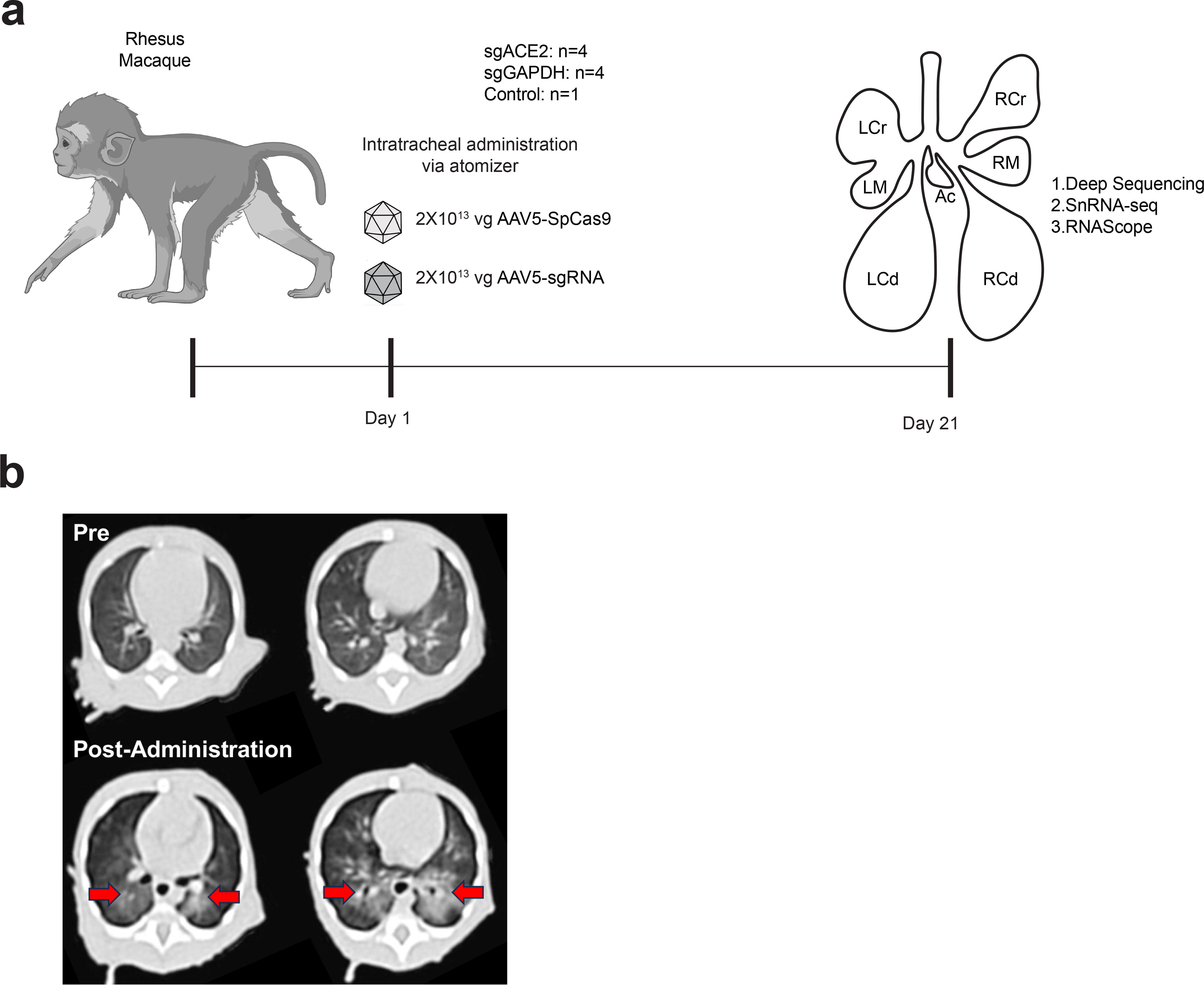
CT scans.
Lung collection and processing
The pluck (lung lobes and heart) was carefully removed from the chest cavity. The left caudal lobe was dissected, tied off with a suture at the main lobar bronchus, and cut below the suture, with fresh tissue as well as ∼½ inch of trachea collected. Three sections from the lower left and three sections from the airway of the left caudal lobe were snap-frozen for gDNA isolation. The rest of the lung lobes were immersed in 4% paraformaldehyde for 48 h, then placed in 30% sucrose in PBS for 48 h. Sections of the lobes were embedded in paraffin for subsequent immunostaining analysis.
Droplet digital PCR (ddPCR)
Vector DNA in tissue was quantified using a duplex Taqman-based ddPCR assay. One Taqman reagent targets SpCas9 (Assay ID: AIAA0SW, ThermoFisher Scientific); the other Taqman reagent targets RNaseP as a normalization control (Cat No. 4403328, ThermoFisher Scientific). ddPCR was performed with a QX200 system (Bio-Rad).
RNAscope
To detect SpCas9 mRNA by RNAscope, formalin-fixed, paraffin-embedded (FFPE) monkey lung samples were sectioned at 4 μm, followed by deparaffinization. The slides were then permeabilized with 70% ethanol for at least 1 h at room temperature. Subsequently, the slides were treated with a proteinase K solution (10 μg/mL) for 20 min at 37°C. Afterward, the slides were incubated with a Cas9 probe (10 nM) in hybridization buffer (mixture of 90 μL buffer from SMF-HB1-10, 10 μL formamide, and 10 nM Probe) overnight at 37°C. Visualization was carried out using the ACD RNAscope™ 2.5 HD Assay in accordance with the manufacturer’s instructions.
Sample preparation for nuclei extraction
All the following sample preparation steps were performed on ice when possible. Tissues were placed on dry ice in approximately 80 mg sections and stored at ≤ −80°C until nuclei extraction was performed. Prior to thaw, C-tubes were prepared with a 2 mL solution of CST or TST as described previously. 20 Tissues were removed from ≤ −80°C and immediately placed into the prepared C-tube, then dissociated with gentleMACS for 2 min. Dissociated tissue was filtered through a 40 μm filter and centrifuged at 500 g for 10 min (with brake on 5). The supernatant was removed and replaced with 1 mL of ST with RNAse inhibitors. This solution was filtered through a 35 μm filter and manually counted for nuclei loading for snRNA-seq.
Single-nucleus RNA-Seq (snRNA-seq) with Seq-Well
Seq-Well was performed as previously described 21,22 with minor changes to better preserve nuclear RNA. Briefly, Seq-Well arrays were pre-loaded with mRNA capture beads and stored prior to nuclei loading in array quenching buffer. Approximately one h before nuclei loading, storage buffer was aspirated from the arrays and replaced with 1x ST. Immediately prior to nuclei loading, ST buffer was aspirated, and approximately 15,000 nuclei in 200 μL of 1xST with RNAse inhibitors were loaded onto the arrays. The arrays were left to facilitate nuclei loading for five min, at which point they were washed twice with 5 mL of 1x ST buffer and sealed with a semipermeable membrane. Membrane bound arrays were left in specialized clamps at 37°C for 20 min to expedite sealing. Arrays were then left in 5 mL lysis buffer for 20 min, followed by 40 min in 5 mL of hybridization buffer to improve mRNA binding to the capture beads. Beads were removed from the arrays with wash buffer, collected, and reverse transcription was performed overnight. Beads were next treated with an exonuclease solution followed by second strand synthesis. Beads were then whole transcriptome amplified (WTA), and cDNA was selected for using a 0.6x and 1.0x SPRI clean up. Samples were prepared for sequencing with a Nextera reaction used for tagmentation of N700 and N500 indices. Post-Nextera libraries were SPRI purified at 0.6x volume, and DNA length and concentration were determined with high-sensitivity D5000 screen tapes. Libraries were prepared and sequenced on Illumina Nextseq 2000 instruments. The paired-end read structure for the 100 cycle kits used was 21 by 50 by 8 for reads one, two, and custom read one primer length respectively. Samples were loaded into the Nextseq cartridge at approximately 2.2 nM, and cartridge preparation followed the manufacturer’s suggestion.
Data matrix generation
Sequencing outputs were processed on the Broad Institute’s Terra platform. FASTQs were generated with bcl2fastq and aligned using STARsolo against a custom pre-mRNA inclusive macaque genome. This genome was generated using the Ensembl database. After an initial quality control analysis of the macaque data was performed, a whitelist of “high-quality” cell barcodes was generated. A second alignment of those barcodes was run against a genome of expected CRISPR sequences. This two-step, two-genome process was used to avoid multimapping clash of edited sequences. The small data matrix from the CRISPR genome was merged as metadata with the standard macaque genome prior to analysis.
Cell type and CRISPR edit calling
Data analysis was performed using R version 4.2.2 and Seurat version 4.3.0. Output digital gene expression (DGE) matrices were pruned and merged into one parent dataset. Only cells with between 500 and 3,500 genes, between 1,000 and 7,500 transcripts, and less than 10% mitochondrial expression were kept. The dataset was processed using the standard Seurat pipeline. Specifically, each transcript in each cell in the parent matrix was normalized against the rest of the dataset and scaled by 10,000, before being log-transformed. Next, to improve biological signal, the top 1,000 variable genes were identified and used for downstream analysis. Lastly, the data was scaled before performing dimensionality reduction. Principal component analysis (PCA) over the variable genes was performed, and the top 28 variable PCs, as determined by jackstraw, were used for SNN-based cell clustering. Clusters were annotated with canonical marker genes to identify cell types and visualized via UMAP plots. This method identified a cluster uniquely marked as low-quality cells. This cluster was further investigated and ultimately removed, and the above processes repeated with slight modification. Namely, the PCs used were lowered to 27 and the SNN-clustering results did not produce any uniquely low-quality clusters. Clusters with overlapping known marker genes were merged and labeled appropriately. Lastly, expression of our CRISPR edits was overlayed with our cell type markers to identify editing biases.
Statistical analysis
Statistical analyses were conducted with GraphPad Prism 8.4. Sample size was determined based on preliminary data rather than predefined statistical methods. Group allocation was carried out in a random manner. For all experiments, the data shown in the figure legends represent biological replicates (n) and are presented as the mean ± standard deviation (SD).
RESULTS
Validating single-guide RNAs (sgRNAs) in monkey cells
To demonstrate the feasibility of genome editing in lung airways of rhesus monkeys, we developed AAV vectors encoding sgRNAs targeting the ACE2 gene or the GAPDH 3′-UTR (Fig. 1a and Supplementary information). We screened for functional sgRNAs in the COS-7 African green monkey kidney cell line. Candidate sgRNAs were designed to target ACE2 or GAPDH sequences with 100% identity between rhesus and COS-7 cells (confirmed by sequencing) and were cloned into AAV5 vectors under the control of the U6 promoter (Fig. 1b). The AAV5-sgRNA vector also encoded the β chain of rhCG. CG is produced by cells in the placenta in early pregnancy and served as a reporter gene to aid in identifying cells that express the sgRNA—without inducing an immune response, as described previously. 23 We further inserted two mir-142 binding sites (miR-142BS) into the 3’UTR of rhCG to repress its expression in dendritic cells. 24

Validating sgRNAs target ACE2 and GAPDH in the African Green Monkey Kidney Fibroblast Cell line COS-7.
For GAPDH, we used a dual sgRNA approach to induce a 75-bp deletion, allowing us to directly assess the editing efficiency by PCR (Fig. 1a). Partial deletion of the GAPDH 3′-UTR is less likely to affect lung cell viability than sgRNAs targeting exons. We co-transfected COS-7 cells with the AAV5-sgRNA vector and an AAV5 vector encoding SpCas9 (pAAV5-SpCas9) under the control of the U1A promoter. 15 By PCR and densitometry analyses, we observed that 35%–50% of GAPDH 3′-UTRs contained the 75-bp deletion (Fig. 1c and d).
To identify sgRNAs that effectively edit ACE2, we designed nine sgRNAs (sg1-9) targeting different ACE2 exons. COS-7 cells were transfected with SpCas9 and individual ACE2 sgRNA plasmids, and editing was assessed using the T7 endonuclease 1 indel assay. 25 Sg3 (targeting exon 8) gave rise to the highest editing efficiency compared to other sgRNAs (Fig. 1a and e and Supplementary information). Deep sequencing of PCR amplicons confirmed that sg3 had the highest editing efficiency (∼47% indels; Fig. 1f); for example, sg3 produced 10% more indels than by the next best sgRNA (i.e., sg6, ∼37% indels). We therefore chose sg3 for studies in young rhesus monkeys.
In vivo studies
Nine ∼3–4 month-old rhesus monkeys (∼1 kg; males and females) were screened, then assigned to study groups. All animals were confirmed seronegative for AAV5 and SpCas9 antibodies prior to study assignment. We packaged SpCas9 and sgRNA vectors in AAV5 capsids, which are capable of infecting lung airways in mice. 15 Each animal was administered 2 × 1013 viral genome (vg) equivalents of each AAV particle using an intratracheal administration approach. 19 Nine monkeys were included in three groups: four received sgGAPDH; four received sgACE2; and one rhesus served as an untreated control (Fig. 2a). Group 1 (n = 4; 2 males, 2 females) was included to evaluate editing of GAPDH 3’ UTR. Group 2 (n = 4) assessed the editing of ACE2. All animals remained healthy and robust during the study period with no evidence of adverse findings. All body weights and hematology and clinical chemistry panels (prior to and post-administration) were within normal limits (data not shown). CT scans performed immediately prior to and post-administration revealed areas of vector deposition (consolidation) (Fig. 2b) after instillation of the vector into the airways via an atomizer (see the section Materials and Methods).
Biodistribution of SpCas9 in the lungs of young rhesus monkeys
To assess the biodistribution of SpCas9 and gene editing in the lungs, tissues were collected post-administration after 21 days, and lung samples were then analyzed. Paraffin-fixed lung sections from the right lung lobes (cranial, middle, caudal) were used to measure the biodistribution of SpCas9 mRNA by RNAscope. 26 SpCas9 positive cells were detected in all lung lobes analyzed (Fig. 3a, b).

Biodistribution of SpCas9 in lung lobes.
CRISPR editing of ACE2 and GAPDH in rhesus monkey lung lobes
To measure the levels of GAPDH and ACE2 editing in lung samples collected, we isolated genomic DNA from the left caudal lung lobe samples that were quick-frozen over liquid nitrogen (Fig. 4a) and then deep sequenced for GAPDH or ACE2 target region amplicons. As expected, we detected AAV genome by ddPCR in all tested samples (Supplementary Figure S1). In all four monkeys that received the AAV5 GAPDH sgRNA vector, we detected the expected 75-bp deletion in lung and airway samples, with the highest editing efficiency of 6.2% (Fig. 4b and c). The variation of editing in neighboring lung tissue (Fig. 4b) is due to the variable AAV vector distribution throughout the lung tissue, which was supported in the CT scans. With the method for administration, it would not be expected for editing to be consistent throughout all of the lung lobes or in all tissue sites. Similarly, we analyzed three monkeys administered with AAV5 ACE2 sgRNA vector and detected indels in exon 8 of ACE2. In addition to left caudal lobes, we analyzed ACE2 editing in left cranial and middle lobes as well as the accessory lobe. We observed up to 8% editing of ACE2 in the lung lobes (Fig. 4d and e). No editing of GAPDH or ACE2 was detected in samples from the control animal (Fig. 4b and d). Together, these data suggest that intratracheal instillation of CRISPR/Cas9 in AAV5 can edit genes in the lung lobes of rhesus monkeys.

CRISPR editing of GAPDH 3’ UTR and ACE2 in lung lobes.
Characterization of AAV5 transduced lung cells by snRNA-Seq
Cell types with important roles in respiration and lung function include type I and II alveolar cells (AT1 and AT2), macrophages, ciliated cells, basal cells, goblet cells, endothelial cells, and fibroblasts. 27 To determine which cell types were transduced by AAV5 in the caudal lung lobes, we isolated nuclei from frozen lung tissue of the left caudal lobe and performed snRNA-Seq (Fig. 5a; see the section Materials and Methods). After filtering for high quality cells, we performed variable gene selection, dimensionality reduction, clustering, and marker gene analyses to annotate the different captured cell types (Fig. 5b and Supplementary Figure S2). We then used the presence or absence of Cas9 or rhCG transcripts to identify rhesus lung cell types transduced by AAV5 in vivo.
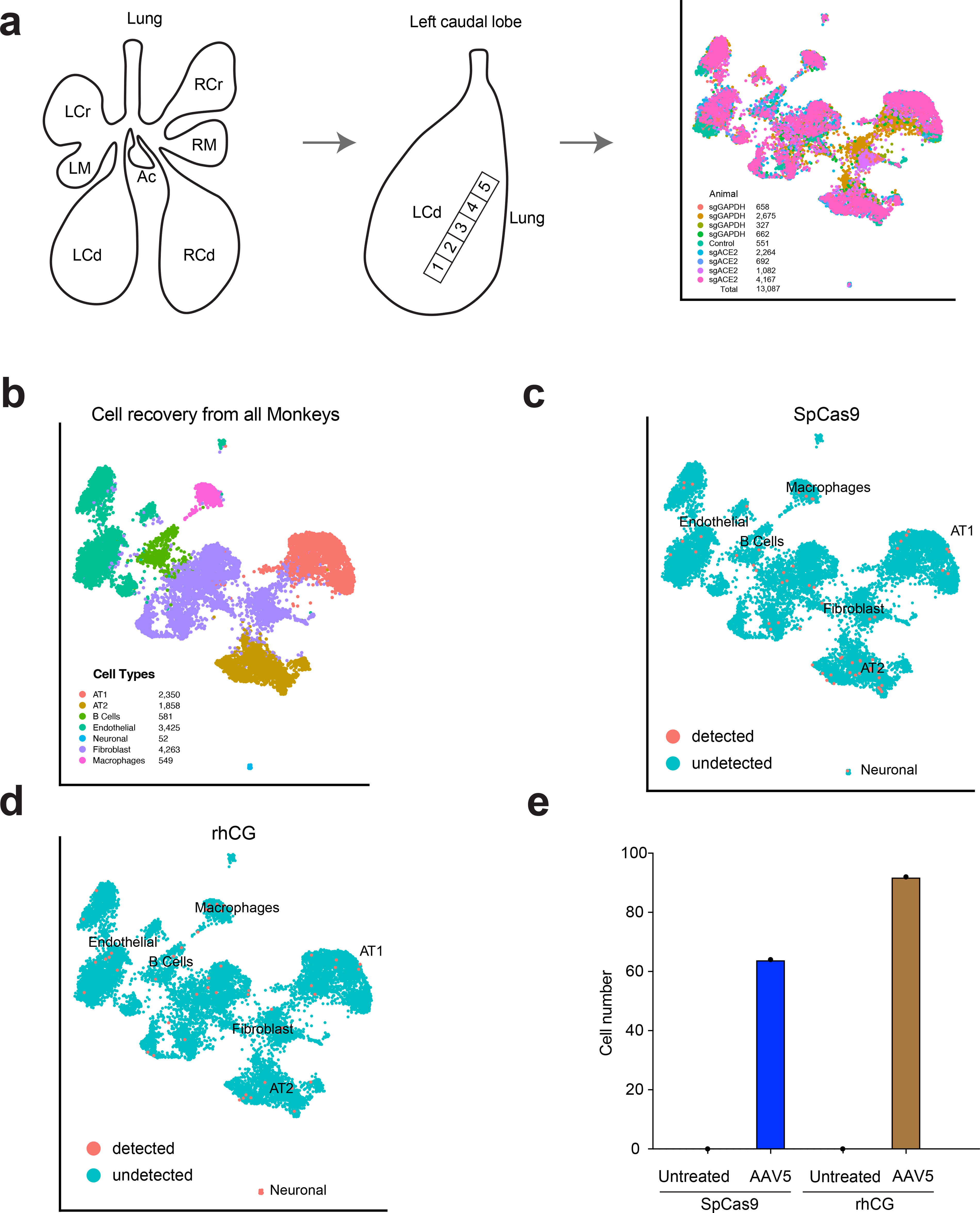
Characterization of lung cell types expressing SpCas9 and rhCG by single-nucleus scRNA-Seq.
Sampling a total of 13,078 nuclei, we detected Cas9 transcripts in 64 nuclei from AAV5-treated rhesus monkeys (Fig. 5c). In most of the Cas9-positive cells, we only detected one Cas9 transcript. The Cas9-positive nuclei were from AT1, AT2, macrophage, fibroblast, and endothelial cell types (Fig. 5c and Supplementary Figure S3). Interestingly, we also identified a few Cas9-positive B cells (4 nuclei). Similarly, we detected rhCG transcripts in multiple cell populations (Fig. 5d), whereas rhCG or Cas9 transcript were not detected in any nuclei from the control animal (Fig. 5e). A previous study has shown that AAV5 did not transduce resting primary B-cells in vitro, but the transduction was enhanced in Epstein–Barr virus-positive B-cells upon antigen activation. 28 We tested AAV5-GFP in a murine pro-B cell line, Ba/F3, and observed GFP signal at a multiplicity of infection of 105 (Supplementary Figure S4). Together, these data are consistent with the low frequency of editing but nevertheless indicate that AAV5 can transduce multiple cell types in the rhesus monkey lung.
DISCUSSION
Our results have demonstrated that AAV5-mediated delivery of CRISPR/Cas9 supports genome edits in the lungs of young rhesus monkeys. Two types of editing—deletions directed by dual sgRNAs and indels directed by individual sgRNA—occurred with similar frequencies. The frequency of editing was lower than outcomes observed in mice, 15 as has been demonstrated in other reports. Whereas the AAV study described herein analyzed editing by deep sequencing in bulk lung tissue, a recent study with nonviral vector delivery and a comparable administration approach analyzed airway epithelia samples using cytology brushes, 19 which differed from the whole tissue approach taken in these studies.
A limitation of the snRNA-Seq analysis is that we detected Cas9-expressing cells at a lower frequency than expected based on the frequency of editing. This may be due to the low amounts of RNA found in individual nuclei. To overcome this, future work could include scRNA-Seq analysis on fresh tissue which may provide more RNA reads. This may also aid in improving representation of different cell types typically undersampled by snRNA-seq, such as T cells, enhancing our ability to understand the full tropism of the vector. 20,29
A previous study used FACS and immunofluorescence to profile cell types transduced by an AAV2/8 GFP reporter in the mouse lung. 30 However, such methods are not feasible in the rhesus because of the strong immune response against GFP. Our study using snRNA-Seq demonstrates that AAV5-CRISPR/Cas9 transduces multiple lung cell types in rhesus monkeys. The insights gained from this work at the single-cell level contribute to our understanding of the diversity of AAV-transduced cell types in the rhesus monkey lung.
The choice of AAV5 has the advantage of being an FDA-approved platform for hemophilia A gene therapy, 31 and among the most efficient serotypes for lung transduction in primates. 9 AAV5 also has a lower overall rate of seropositivity in humans 32 and rhesus monkeys (Tarantal, personal communication). We observed that AAV5 transduces AT1 and AT2 cells (Fig. 5), suggesting the potential to edit disease genes in alveolar cells using this AAV5 platform. We also detected Cas9 RNA in B cells and macrophages. Persistent AAV expression of Cas9 can induce an anti-Cas9 immune response in mice. 33 For the rhesus studies described herein, we prescreened all animals to ensure that they were seronegative for antibodies to AAV5 and SpCas9 prior to vector administration. No anti-Cas9 immune responses were detected either prior to or in serum after intratracheal AAV administration. In future work it will also be important to characterize any potential immune responses within the lung lobes to AAV5-mediated delivery of CRISPR/Cas9 and the frequency of off-target editing.
In summary, our study demonstrates the feasibility of dual-packaged AAV5 delivery of CRISPR/Cas9 for genome edits in the lung lobes of young rhesus monkeys. This work paves the way towards devising strategies for improving AAV-based CRISPR delivery for inherited lung diseases across a range of age groups.
Footnotes
ACKNOWLEDGEMENTS
We thank S. Wolfe and E. Sontheimer for discussions and Marrah Lachowicz-Scroggins and PJ Brooks at NIH for advice and discussions. D. Conte edited this paper.
The authors thank Y. Liu in the UMass Morphology for histology.
DATA AVAILABILITY
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files or upon request. Information about these studies is also provided in the SCGE Toolkit as noted above.
AUTHORS’ CONTRIBUTIONS
S.Q.L. performed the in vitro experiments, analyzed data, and wrote the article with co-authors. A.W.N. and M.R. performed snRNA-seq. A.F.T., M.M., and C.L. performed the rhesus studies including selection and monitoring of animals vector administration and blood and tissue collection. C.Z., J.X., Q.S., and D.W. prepared the AAV., X.Z. and J.W. performed in vitro experiments. T.R.F., D.G.A., A.F.T., A.K.S., G.G., and W.X. designed the study and wrote the article with all co-authors.
AUTHOR DISCLOSURE
The authors declare no potential conflicts of interest. A.K.S. reports compensation for consulting and/or scientific advisory board membership from Honeycomb Biotechnologies, Cellarity, Ochre Bio, Relation Therapeutics, FL86, IntrECate Biotherapeutics, Bio-Rad Laboratories, Senda Biosciences, and Dahlia Biosciences unrelated to this work.
FUNDING INFORMATION
S.L., D.W., D.G.A., A.K.S., G.G., and W.X. were supported by NIH UH3-HL147367. W.X. was supported by grants from the National Institutes of Health (P01-HL158506, R01-CA275945, and R01-GM150279), and the Cystic Fibrosis Foundation. These studies were supported by the NIH SCGE Consortium. Information about these studies is provided in the SCGE Toolkit through the SCGE Dissemination and Coordinating Center (https://commonfund.nih.gov/editing), which is a platform housing data generated across all consortium initiatives.10 This was a collaborative study between an SCGE editor project (UH3-HL147367) and the Nonhuman Primate Testing Center for Evaluation of SCGE Tools (A.F.T.; U42-OD027094). Studies were also supported by the base operating grant for the California National Primate Research Center (P51-OD011107). In vivo imaging was performed with instrumentation funded by the NIH S10 High-End Instrumentation Program (A.F.T.; S10-RR025063 and S10-OD028713).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.