
Abstract
Maple syrup urine disease (MSUD) is a rare, inherited, metabolic disorder characterized by dysfunction of the multi-subunit, mitochondrial enzyme complex branched-chain alpha-keto acid dehydrogenase (BCKDH). BCKDH catalyzes the oxidative decarboxylation of branched-chain amino acids (BCAAs). BCAAs and their neurotoxic alpha-keto intermediates can accumulate in the blood and tissues in the absence of functional BCKDH. We evaluated a lipid nanoparticle (LNP)-based treatment approach to address all possible genetic mutations that can cause MSUD (BCKDHA, BCKDHB, and DBT). In the intermediate MSUD mouse model, which harbors a mutation in the dihydrolipoamide branched-chain transacylase E2 (DBT) subunit of BCKDH, repeated administration of LNP-encapsulated mRNA therapy significantly extended survival and reduced serum leucine levels. We also evaluated our LNP approach in several models of classical MSUD, namely DBT knockout (KO) mice and the new BCKDHA KO and BCKDHB KO mice. The latter two were generated by CRISPR/Cas9 gene editing and contain the highly prevalent classical MSUD-causing mutations seen in the Mennonite and Costa Rican populations. Intravenous LNP-encapsulated mRNA administration extended survival and increased body weight in the DBT KO and BCKDHA KO models of classical MSUD but was not effective in BCKDHB KO mice. Our data provide a promising proof-of-concept that a universal, mutation-independent approach to treating MSUD is possible and viable.
INTRODUCTION
Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder characterized by dysfunction of the multimeric mitochondrial enzyme complex branched-chain alpha-keto acid dehydrogenase (BCKDH). 1 BCKDH catalyzes the oxidative decarboxylation of branched-chain amino acids (BCAAs). In the absence of functional BCKDH, the BCAAs leucine, isoleucine, and valine as well as their neurotoxic alpha-keto intermediates can accumulate in the blood and tissues. MSUD derives its name from the distinctive sweet odor of excreted sotolone in affected patient’s urine during branched-chain ketoaciduria. 2
MSUD presents in approximately 1 in 150,000 live births in the general population. 3,4 The disease arises from mutations occurring within the three genes that encode the complex’s components (BCKDHA, BCKDHB, DBT). 5 –8 The majority of MSUD patients have mutations in DBT; the incidence of specific mutations is significantly increased in certain populations with founder effects. 9,10 For example, the frequency of the c.1312T>A, p.Tyr438Asn (Y393N) mutation in BCKDHA increases the incidence of MSUD to as high as 1 in 400 in the Pennsylvanian Mennonite population. 11,12
The severity of MSUD can range considerably. The immediate neonatal symptom onset seen in the classical form of the disease is also the most severe and affects the majority of MSUD patients. Classical MSUD (cMSUD) patients have little-to-no enzyme activity (0%–2% of normal), and the disease is characterized by potentially fatal neurological dysfunction and critical brain edema. Intermediate MSUD—in which 3%–30% BCKDH activity is retained—affects a smaller subset of patients who exhibit serious, albeit less severe, symptoms such as developmental delay, failure to thrive, neurological impairment, seizures, and ketoacidosis. There is also an intermittent form of MSUD in which patients have similar BCKDH activity levels to those in intermediate MSUD (5%–20% of normal) but have normal BCAA levels when asymptomatic. However, these individuals present with MSUD symptoms during times of stress or illness. Inclusion of MSUD in newborn screening programs, particularly in areas with known founder effects, has improved some outcomes and drastically decreased the time to treatment for patients with cMSUD with some individuals being diagnosed prior to symptomatic elevations in plasma leucine. 11 However, neurological damage following a metabolic crisis is still a major concern.
MSUD is currently incurable, and treatment is limited to strict dietary protein restriction. Even with close monitoring of BCAA levels, MSUD patients are susceptible to various neurological comorbidities and may ultimately require a liver transplant. BCAA levels in MSUD patients are reduced within hours of receiving a liver transplant but remain approximately two-fold higher than normal levels. 11,13 Although such patients are no longer required to restrict protein intake, 13 they may still be at risk of developing hyperleucinosis (elevated leucine levels) when facing intercurrent illnesses. 14 Considering that the liver provides 9%–13% of the total BCKDH activity in humans, 15 restoring this level of activity to the liver could represent an effective treatment strategy.
Treatment approaches developed by our laboratory and others have focused on one or two of the three genes involved in MSUD due to packaging limitations within virus vectors. 1,16 We evaluated a novel, comprehensive treatment approach for various mouse models of MSUD that utilizes lipid nanoparticle (LNP)-encapsulated messenger (m)RNA encoding human BCKDHA (hBCKDHA), hBCKDHB, and hDBT. Our data suggest that this LNP-based therapy could represent a universal treatment approach for all MSUD patients.
MATERIALS AND METHODS
mRNA/LNP production
mRNA encoding hBCKDHA, hBCKDHB, and hDBT was synthesized in vitro using an optimized T7 RNA polymerase-mediated transcription reaction with complete replacement of N1-methylpseudouridine, as previously described. 17 The reaction included a DNA template containing the immunogen open reading frame flanked by 5′ untranslated region (UTR) and 3′ UTR sequences and was terminated by an encoded polyA tail. After transcription, the Cap 1 structure was added to the 5′ end using vaccinia capping enzyme (New England Biolabs) and Vaccinia 2′ O-methyltransferase (New England Biolabs). The mRNA was purified by oligo-dT affinity purification, buffer exchanged by tangential flow filtration into sodium acetate, pH 5.0, sterile filtered, and kept frozen at –20°C until further use.
The mRNA was encapsulated in a lipid nanoparticle through a modified ethanol-drop nanoprecipitation process as described previously. 18 In brief, ionizable, structural, helper, and polyethylene glycol lipids were mixed with mRNA in acetate buffer, pH 5.0, at a ratio of 2.5:1 (lipids:mRNA). The mixture was neutralized with Tris-Cl pH 7.5, sucrose was added as a cryoprotectant, and the final solution was sterile filtered. Vials were filled with formulated LNP and stored frozen at –70°C until further use. The drug product underwent analytical characterization, which included the determination of particle size and polydispersity, encapsulation, mRNA purity, double-stranded RNA content, osmolality, pH, endotoxin, and bioburden, and the material was deemed acceptable for in vivo study.
Mice
All mouse colonies were maintained at the University of Pennsylvania under specific pathogen-free conditions. All animal procedures and protocols received approval from the Institutional Animal Care and Use Committee of the University of Pennsylvania.
We obtained breeding pairs of heterozygous (HET) Dbt tm1Geh Tg(Cebpb-tTA)5Bjd Tg(tetO-DBT)A1Geh/J mice from The Jackson Laboratory (Bar Harbor, ME); these mice are hereafter referred to as intermediate MSUD, i.e., ‘iMSUD.’ Postnatal day 21 (P21)–P28 iMSUD mice received an intravenous (IV) injection containing 0.2, 0.5, or 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT or 1 mg/kg of LNP-encapsulated mRNA encoding green fluorescent protein (GFP) via the retro-orbital or tail vein, depending on the size of the mouse at the time of injection. Mice were then administered LNP-encapsulated mRNA weekly thereafter. Mice were not randomized to groups. Mice were monitored for survival and changes in body weight throughout the in-life phase of the study. Blood collections were performed either as a terminal procedure if mice were to be euthanized for clinical signs or during the in-life phase once mice reached greater than 15 g in body weight (non-fasted). As the iMSUD mice are a hypomorphic mouse model of MSUD resulting from knockout (KO) of mouse DBT and knock in of the human version of DBT for low-level expression to prevent the neonatal lethality seen in the cMSUD or DBT KO mouse, evaluation of protein expression by both Western blot and IHC was hampered by increased background levels. Therefore, we focused our evaluations on in situ hybridization (ISH), which was specific for the administered version of DBT (Fig. 2), and leucine levels as a biomarker of BCKDH activity.
cMSUD (DBT KO) mice were generated by backcrossing iMSUD mice with wild-type (WT) C57BL/6J mice to generate offspring that lacked the tTA and tetO transgenes for expression of hDBT. 19 BCKDHA KO and BCKDHB KO mice were generated at The Jackson Laboratory (Bar Harbor, ME) by CRISPR/Cas9 gene editing. BCKDHA KO mice harbor a Y439N mutation, which corresponds to the highly prevalent Y393N (c.1312T>A, p.Tyr438Asn) mutation in BCKDHA seen in the Mennonite population of Pennsylvania. 7,12,20 BCKDHB KO mice harbor a R215X mutation, which corresponds to the R285X (c.853C>T, p.Arg285Ter) mutation in humans. 7,8
LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT was evaluated in litters of newborn BCKDHA KO, BCKDHB KO, and cMSUD (DBT KO) mice. KO mice and their HET and WT littermates received an IV injection containing 1 or 2 mg/kg LNP-encapsulated hBCKDHA, hBCKDHB, and hDBT mRNA within 24 h of birth and on day 3 via the temporal vein. Weekly IV administration started on day 7 via the retro-orbital or tail vein, depending on the size of the mouse at the time of injection. Mice were monitored for survival and changes in body weight throughout the in-life phase of the study.
Serum analyses
We collected blood in serum-separator tubes and allowed the blood to clot. Serum was isolated and analyzed for leucine levels by Charles River Laboratories (Wilmington, MA).
In situ hybridization
Liver samples were fixed in 10% neutral buffered formalin and used to determine hBCKHDA, hBCKDHB, and hDBT mRNA presence by ISH, as described previously. 1 Z-shaped probe pairs specific for the human versions of hBCKHDA, hBCKDHB, and hDBT were synthesized by the kit manufacturer. An additional probe was used for the detection of LNP-derived hDBT RNA to prevent cross-reaction with the low level of endogenous hDBT RNA expression in the iMSUD mouse strain.
Statistical analysis
All values are presented as mean ± standard error of the mean (SEM). We compared treatment groups by the following analyses: comparison of survival using a log-rank test and body weight using linear mixed effect modeling. All analyses were performed within the R program (version 4.0.0) with the appropriate function. p < 0.05 was considered significant.
RESULTS
Repeated administration of LNP-encapsulated mRNA therapy extends survival and reduces serum leucine levels in the iMSUD mouse
The majority of MSUD patients have mutations in DBT, so we utilized the iMSUD mouse model for initial evaluation of the LNP-encapsulated mRNA therapy. iMSUD mice are a hypomorphic mouse model of MSUD resulting from KO of mouse DBT and knock in of the human version of DBT for low-level expression to prevent the neonatal lethality seen in the cMSUD or DBT KO mouse. 19,21 Natural history studies performed by us and others 1,19,21 have found that iMSUD mice survive until early adulthood (albeit with a reduced lifespan compared to WT mice) and display elevated circulating BCAA levels (Supplementary Fig. S1). At the time of weaning (P21–P28), iMSUD mice have reduced body weights compared to their HET and WT littermates.
P21–P28 iMSUD mice received an IV injection containing 0.2, 0.5, or 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT or 1 mg/kg of LNP-encapsulated mRNA encoding GFP via the retro-orbital or tail vein, depending on the size of the mouse at the time of injection (Fig. 1). No iMSUD mice administered LNP-encapsulated mRNA encoding GFP survived after study day 27 (Fig. 1A). Mice in this control group did not gain weight over the study duration (Fig. 1B) and had serum leucine levels of >2,500 µmol/L (Fig. 1C).

Repeated administration of LNP-encapsulated mRNA therapy extended survival and reduced serum leucine levels in the intermediate mouse model of MSUD. iMSUD mice received weekly intravenous doses of 0.2, 0.5, or 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT starting at P21–P28. An additional group of mice injected with 1 mg/kg of LNP-encapsulated GFP mRNA were included as negative controls. Mice were followed during the in-life phase for survival
Weekly administration of LNP-encapsulated mRNA therapy in weaned iMSUD mice increased survival in a dose-dependent manner, with all treatment groups exhibiting significantly increased survival compared to the GFP control group (Fig. 1A). Although body weights were significantly increased in all treatment groups compared with the GFP control, no dose response was observed (Fig. 1B). Serum leucine levels were reduced following administration of the LNP-encapsulated hBCKDHA/hBCKDHB/hDBT mRNA in iMSUD mice relative to the GFP control group (average of 2,675 µmol/L, standard deviation of 1,842 µmol/L), suggesting the improved survival following LNP treatment was due to a reversal of the elevated peripheral BCAA levels observed in MSUD (Fig. 1C). iMSUD mice administered 1 mg/kg displayed 100% survival (Fig. 1A) and relative normalization of serum leucine levels to an average of 427 µmol/L (standard deviation of 184 µmol/L), which is similar to average WT levels of 154 µmol/L (Fig. 1C).
Evaluation of RNA pharmacokinetics following repeated administration of LNP-encapsulated mRNA therapy in iMSUD mice
We harvested livers from iMSUD mice that received 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT in weekly IV injections starting at P21–P28 and evaluated RNA levels via ISH (Fig. 2A). Untreated iMSUD mice served as controls to determine the background level of hDBT expression in this hypomorphic mouse model (∼20% of cells were ISH positive for DBT, Fig. 2B). Mice were necropsied 24 h or more than 7 days following the final injection of LNP-encapsulated mRNA. We individually detected hBCKDHA, hBCKDHB, and hDBT RNA (red staining in Fig. 2A). hBCKDHA RNA expression peaked at 82% of cells being positive by ISH at 24 h post-injection, before decreasing to 42% at more than 7 days following the final injection. We used two different probes to detect DBT, one that did and another that did not cross-react with the low level of endogenous hDBT expression in the iMSUD mouse strain. Using the probe that cross-reacted with both versions of DBT RNA (endogenous and expressed following LNP administration), we detected an average of 39% of cells that were ISH-positive at 24 h post injection. This expression is above the endogenous expression levels of DBT in the iMSUD model (∼20%) as indicated by 6.6% of cells being detected as positive with the hDBT-only probe. The levels of expression were undetectable 7 days or more following the final injection (Fig. 2).
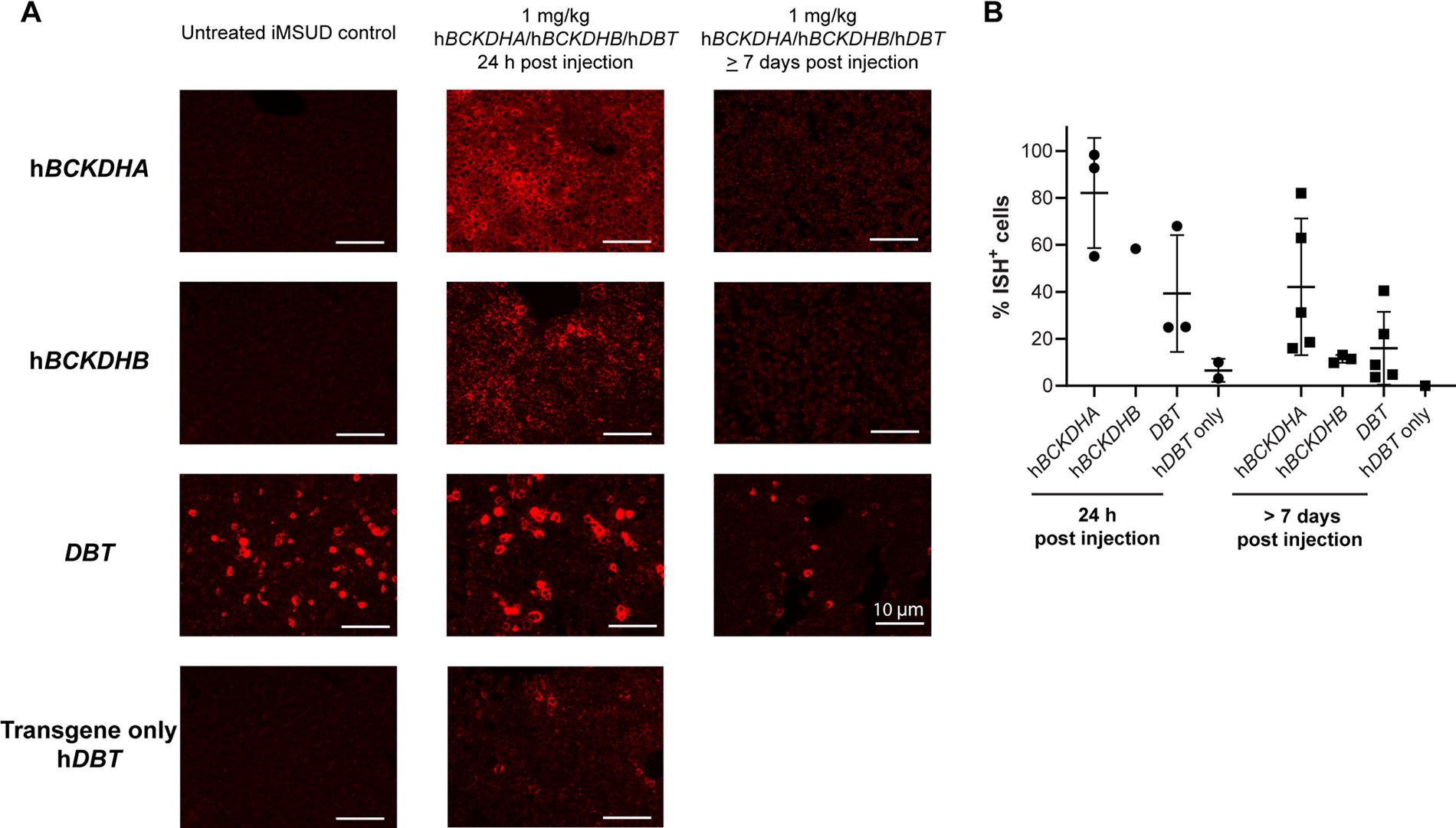
Evaluation of RNA pharmacokinetics following repeated administration of LNP-encapsulated mRNA therapy in the intermediate mouse model of MSUD. iMSUD mice received weekly intravenous doses of 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT starting at P21–P28. Untreated iMSUD mice were included as controls. Mice were necropsied 24 h or more than 7 days following the final injection of LNP-encapsulated mRNA and livers were harvested for in situ hybridization. hBCKDHA, hBCKDHB, and hDBT RNA were detected as red staining
IV administration of LNP-encapsulated mRNA extends survival, increases body weight, and reduces serum leucine levels of cMSUD mice
Based on our previous results in a mouse model of a less severe, intermediate form of MSUD, we evaluated the same treatment approach in a mouse model of the more common and severe cMSUD. The cMSUD or DBT KO mouse displays neonatal lethality within the first 1–2 days of life (Supplementary Fig. S1), as previously described. 19,21 Due to the severity of this model, we administered a higher dose of 2 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT to P0–P1 cMSUD mice, and HET and WT littermates via IV injection (temporal vein) with an additional injection on day 3 (Fig. 3). Starting on day 7, mice received weekly IV LNP administration via the retro-orbital or tail vein (depending on the size of the mouse at the time of injection).
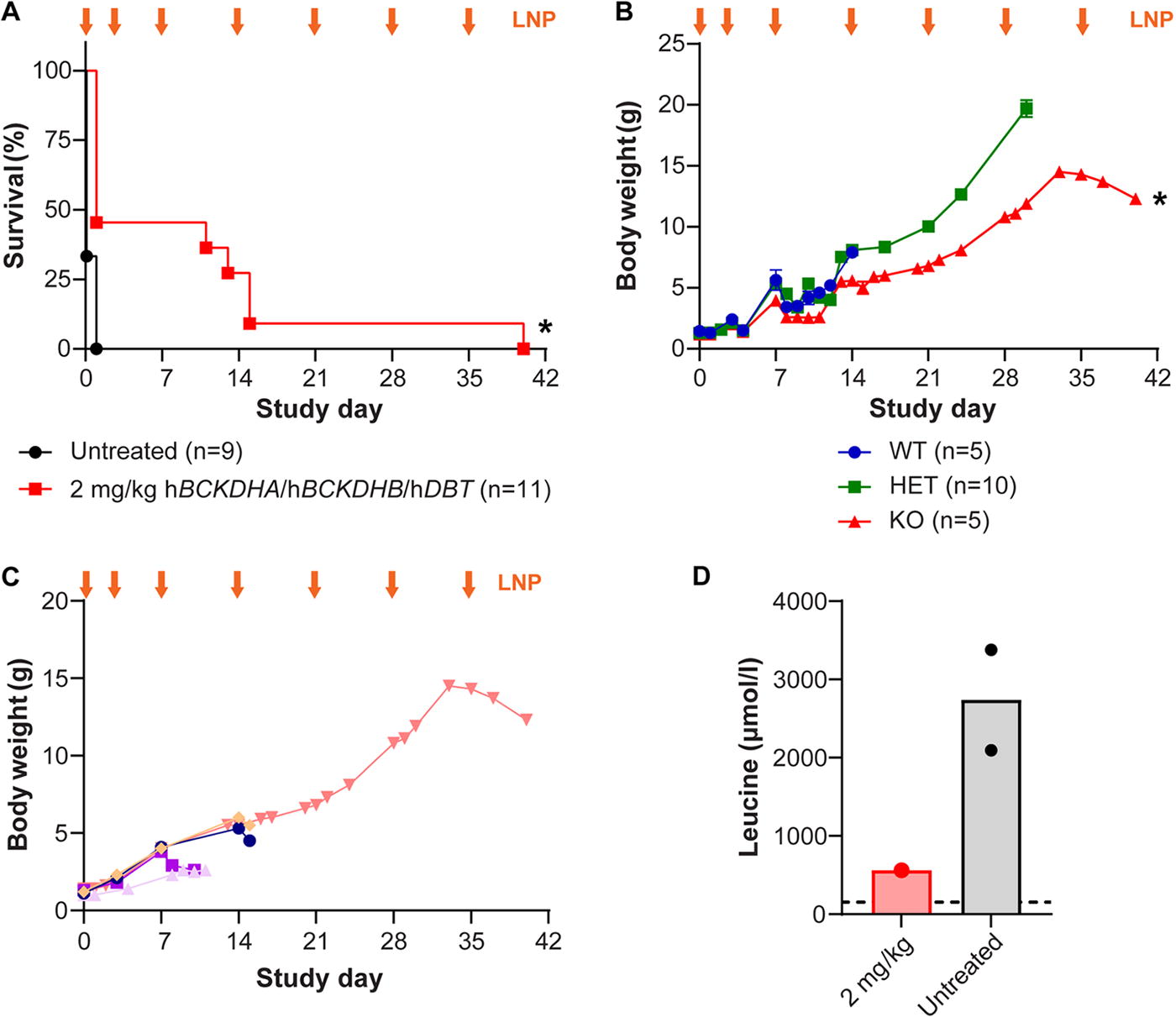
Intravenous LNP-encapsulated mRNA administration extended survival, increased body weight, and reduced serum leucine levels of cMSUD mice. Litters of newborn cMSUD (DBT KO) mice, and HET and WT littermates, were intravenously (IV) administered 2 mg/kg of LNP-encapsulated mRNA encoding the three BCKDH subunits (hBCKDHA, hBCKDHB, and hDBT) on days 0 and 3 of life via the facial vein. Starting on day 7, mice received weekly IV doses of LNPs via the retro-orbital or tail vein. Mice were followed for survival
Treatment with LNP-encapsulated mRNA significantly extended survival in this severe model of cMSUD, with some treated cMSUD mice surviving to days 11, 13, 15, and 40 (Fig. 3A). Although LNP-treated cMSUD mice exhibited enhanced survival rates, they failed to thrive, as highlighted by the significantly lower average body weight of treated cMSUD mice compared to their HET littermates (Fig. 3B). The body weight of treated cMSUD was a good predictor of survival, as mice that did not gain weight had to be euthanized earlier than others (Fig. 3C). Because of the severity of the KO mouse models and issues with failure to thrive, collection of blood at the time of euthanasia was highly difficult. Serum leucine levels in the treated cMSUD were lower compared to the levels in untreated mice, although they were not normalized to WT levels (Fig. 3D).
IV administration of LNP-encapsulated mRNA extends survival and increases body weight in two different classical models of MSUD
Our treatment approach comprises a single LNP therapy that contains mRNAs encoding all three genes that can be mutated in MSUD (i.e., BCKDHA, BCKDHB, and DBT). To evaluate the universality of our treatment, we evaluated our single LNP-encapsulated mRNA treatment in mouse models of each cMSUD form, not just that resulting from mutations in DBT. Therefore, BCKDHA KO and BCKDHB KO mice were generated at The Jackson Laboratory (Bar Harbor, ME) via CRISPR/Cas9 gene editing. BCKDHA KO mice harbor a Y439N mutation, which corresponds to the highly prevalent Y393N mutation in BCKDHA seen in the Mennonite population of Pennsylvania 7,12,20 and BCKDHB KO mice harbor a R215X mutation, which corresponds to the R285X mutation in humans. 7,8 Similar to the cMSUD (DBT KO) mouse, these mouse strains displayed neonatal lethality within the first 1–2 days of life (Supplementary Fig. S1).
P0–P1 BCKDHA KO, BCKDHB KO, and cMSUD mice (DBT KO), and HET and WT littermates, received 1 mg/kg of LNP-encapsulated mRNA encoding hBCKDHA, hBCKDHB, and hDBT via IV injection to the temporal vein, with an additional dose on day 3 (Fig. 4). Starting on day 7, surviving mice received weekly IV administrations of LNP via either the retro-orbital or tail veins (depending on the size of the mouse at the time of injection). For this study, we utilized the high dose evaluated in the iMSUD mouse study to see if rescue of more severe models was possible at a lower dose than administered in the cMSUD mouse study.
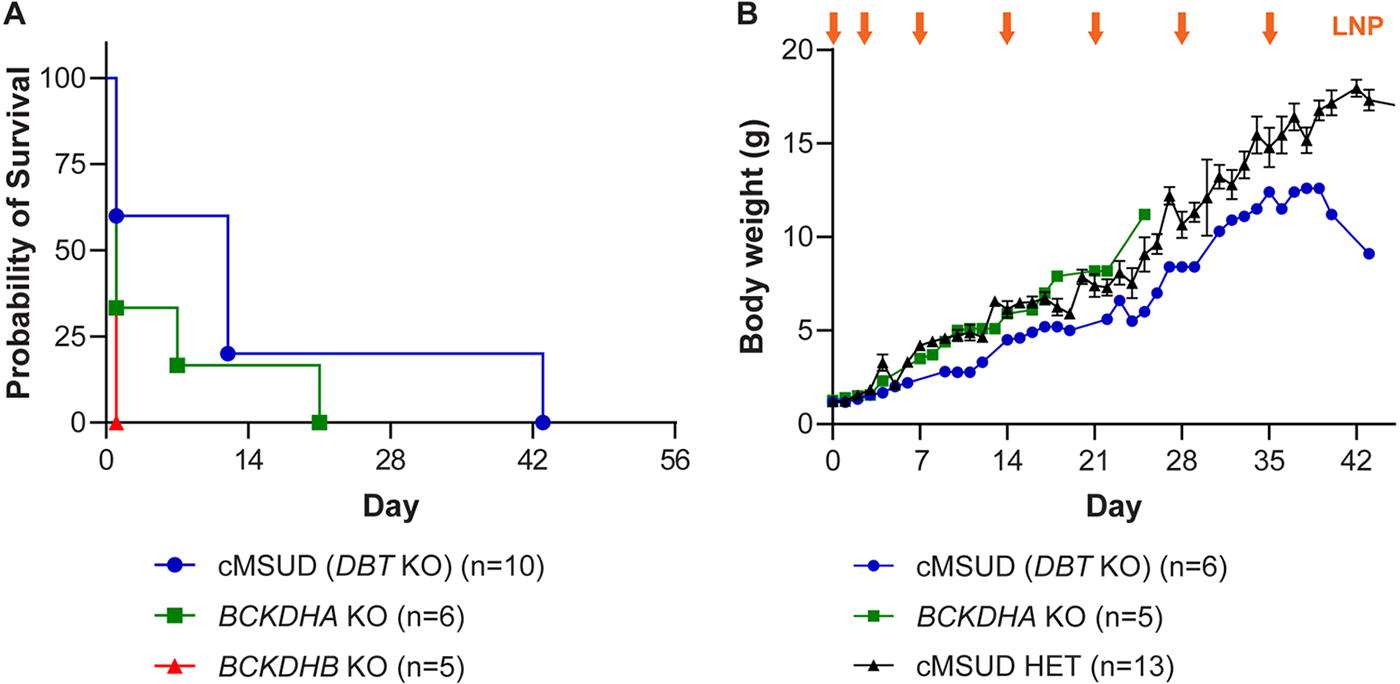
Intravenous LNP-encapsulated mRNA administration extended survival and increased body weight in two different classical models of MSUD. Litters of newborn BCKDHA KO, BCKDHB KO, and cMSUD (DBT KO) mice, and HET and WT littermates, were intravenously (IV) administered 1 mg/kg of LNP-encapsulated mRNA encoding the three BCKDH subunits (hBCKDHA, hBCKDHB, and hDBT) on days 0 and 3 of life via the temporal vein. Starting on day 7, mice received weekly IV doses of LNPs via the retro-orbital or tail vein. BCKDHA KO, BCKDHB KO, and cMSUD mice were followed for survival
Treatment with 1 mg/kg of LNP-encapsulated mRNA significantly extended survival in BCKDHA KO mice to 7 and 21 days but had no effect on the survival of BCKDHB KO mice, and significantly extended survival in cMSUD mice to 12 and 43 days (Fig. 4A, similar to that seen at the higher dose in Fig. 3A). We measured body weights of BCKDHA KO and cMSUD mice that survived past days 1–2 of life throughout the in-life phase of the study (Fig. 4B). Rescued BCKDHA KO and cMSUD mice rapidly increased in body weight with routine administrations of the LNP, and treated mice almost caught up with HET littermate controls until they were euthanized due to clinical signs (Fig. 4B).
DISCUSSION
We evaluated the potential of a mutation-independent approach for the treatment of MSUD across four different MSUD mouse models. Repeated IV delivery of LNP-encapsulated mRNAs encoding hBCKDHA, hBCKDHB, and hDBT increased survival and body weight, and decreased serum leucine levels in a hypomorphic MSUD mouse model that survives until weaning without clinical intervention. This mouse model recapitulates many aspects of the intermediate version of MSUD in patients, albeit without the documented neurological phenotype that has been described for cMSUD (complete KO) animals. Therefore, repeated administration of LNP-encapsulated mRNAs may represent a potential long-term universal treatment approach for MSUD.
Current clinical management of MSUD patients requires patients to follow a carefully monitored, restricted diet. This is highly burdensome to the patients, and the potential for neurological damage following a metabolic crisis remains a major ongoing concern. Recurrent IV administration of LNP-encapsulated mRNA therapy could significantly improve the standard of life for these patients. In these studies, mice received LNPs on a weekly basis due to the mRNA/protein pharmacokinetics. Although recurrent IV administration of an LNP-encapsulated mRNA therapy at intervals shorter than a month would be associated with some minor inconveniences, the stability of the individual mRNA could potentially be improved to increase the interval between dosing.
For many MSUD patients, there is the potential for undergoing liver transplantation, despite the risk for morbidity and, in rare cases, mortality, associated with the surgical procedure and long-term immunosuppression. Although BCAA levels are reduced within hours of a successful liver transplant, transplanted MSUD patients retain BCAA levels that are approximately twofold higher than normal. 11,13 Such patients are no longer required to restrict protein intake, 13 but they may still be at risk of developing hyperleucinosis (elevated leucine levels) when facing intercurrent illnesses. 14 The rapid rescue of cMSUD mice following administration of LNP-encapsulated mRNA suggests that this therapy could also be effective for treating acute metabolic crises in MSUD patients (either pre- or post-liver transplant) resulting from intercurrent illnesses. Therefore, this approach may represent not only a chronic treatment but also an as-needed rescue or “sick-day” therapy to prevent dangerous excursions in plasma leucine levels.
Treatment approaches developed by our laboratory and others have focused on one or two out of the three genes involved in MSUD due to packaging limitations within virus vectors. 1,16 We previously evaluated an adeno-associated virus vector-based approach for replacement of the DBT subunit in the iMSUD mouse model. High-dose vector substantially increased survival, maintained near-normal BCAA levels throughout the study, and protected the mice from a lethal high-protein diet challenge. 1 However, this approach requires a different gene therapy product to be developed and approved for mutations in each gene. Our LNP-based strategy utilizes mRNAs that encode hBCKDHA, hBCKDHB, and hDBT with the aim of developing a single therapy capable of treating any MSUD patient, regardless of genetic mutation. In addition, by providing the full-length mRNA for each gene, this approach does not rely on potential patients having a specific mutation (as in the case of certain gene editing approaches) as the majority of MSUD patients are compound heterozygotes.
By developing two new cMSUD mouse strains (BCKDHA KO and BCKDHB KO mice), we were able to evaluate the same LNP-encapsulated mRNA treatment across all models of classical MSUD-causing mutations in BCKDHA, BCKDHB, and DBT for the first time. The treatment approach was less effective in the three mouse models of classical or immediate neonatal onset utilized here, namely the BCKDHA KO, BCKDHB KO, and cMSUD (DBT KO) mice, but provides a promising proof-of-concept that a universal approach is possible and viable. This could result in a single therapy being manufactured, evaluated, and approved for all MSUD-causing mutations. Although we only observed a partial rescue in treated cMSUD mice, we can speculate that improvements in LNP formulation and/or mRNA stability could increase the survival rate and duration. In particular, the reduced detection of the hBCKDHB RNA and the lack of effect in the BCKDHB KO mice suggest this was the case. The ability of the LNP-encapsulated mRNA to rapidly exert a therapeutic effect in such a severe condition also suggests that this therapy could be deployed to treat acute crisis situations to reverse dangerously high serum leucine levels. We conclude that LNP mRNA therapy may represent an effective treatment option for both classical and intermediate forms of MSUD.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank the Program for Comparative Medicine and Histopathology Core at the Gene Therapy Program. The authors would like to thank Hanying Yan for performing the statistical analysis. The authors would also like to thank Christine Draper, Rucha Fadnavis, and Melanie K. Smith for invaluable technical assistance.
AUTHORS’ CONTRIBUTIONS
J.A.G.: Conceptualization, funding acquisition, investigation, methodology, supervision, writing-original, writing—review and editing; M.J.: Investigation, methodology; A.D.: Investigation; J.K.C.: Investigation; N.P.: Investigation; M.S.: Investigation; M.N.: Investigation; P.B.: Investigation; K.C.: Conceptualization, methodology, writing—review and editing; M.C.: Conceptualization, methodology; P.H.G.: Conceptualization, methodology, supervision, writing—review and editing; P.G.V.M.: Conceptualization, methodology, supervision, writing—review and editing; J.M.W.: Conceptualization, funding acquisition, supervision, writing—review and editing.
AUTHOR DISCLOSURE
The authors declare having potential competing financial interests. J.M.W. is a paid advisor to and holds equity in iECURE, Passage Bio, and the Center for Breakthrough Medicines (CBM). He also holds equity in the former G2 Bio asset companies and Ceva Santé Animale. He has sponsored research agreements with Alexion Pharmaceuticals, Amicus Therapeutics, CBM, Ceva Santé Animale, Elaaj Bio, FA212, Foundation for Angelman Syndrome Therapeutics, former G2 Bio asset companies, iECURE, and Passage Bio, which are licensees of Penn technology. J.M.W. and J.A.G. are inventors on patents that have been licensed to various biopharmaceutical companies and for which they may receive payments. K.C., M.C., P.G., and P.G.V.M. are current or former employees of
FUNDING INFORMATION
This work was supported by a sponsored research agreement with
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.