
Abstract
Mucopolysaccharidosis IVA (MPS IVA) is an autosomal recessive disease caused by a mutation in the N-acetylgalactosamine-6-sulfate-sulfatase (GALNS) gene resulting in progressive systemic skeletal dysplasia. There is currently no effective treatment available for this skeletal condition. Thus, the development of a new therapy stands as an unmet challenge in reversing or alleviating the progression of the disease. Our research, which could be a game-changer, hypothesizes that ex vivo lentiviral (LV) gene therapy (GT) could produce the supraphysiological level of active GALNS enzyme by hematopoietic stem cells (HSCs) transduced with LVs carrying the native GALNS gene under two different promoters (CBh and COL2A1), impacting bone and cartilage abnormalities in MPS IVA. We conditioned newborn knock-out (Galns−/−) MPS IVA mice with busulfan and intravenously transplanted LV-modified HSCs isolated from the bone marrow of Galns−/− donor mice. Transplanted mice were autopsied at 16 weeks, and tissues were collected to assess the therapeutic efficacy of modified HSCs in MPS IVA mice. Although HSC-LV-CBh-hGALNS provided a higher GALNS enzyme activity in plasma, HSC-LV-COL2A1-hGALNS stably corrected heart and bone abnormalities better under a low level of GALNS enzyme. Our findings suggest that ex vivo LV-GT may potentially treat MPS IVA.
INTRODUCTION
Mucopolysaccharidosis IVA ([MPS IVA]; Morquio A syndrome) is an autosomal recessive inherited metabolic disease caused by the deficiency of lysosomal N-acetylgalactosamine-6-sulfate-sulfatase (GALNS) enzyme due to mutations in the GALNS gene. Deficiency of this enzyme leads to the accumulation of glycosaminoglycans (GAGs), keratan sulfate (KS), and chondroitin-6-sulfate (C6S), in multiple tissues, including cartilage, bone, connective tissues, heart, and cornea. 1,2 The disease presents with systemic skeletal involvement, including the most severe growth impairment among all types of MPS, leading to difficulties in activities of daily living (ADL) and high morbidity. 2,3 Even though patients with MPS IVA look normal at birth, progressive systemic skeletal involvements appear by the age of 3 years. The common signs and symptoms of the disease in severe patients represent systemic skeletal dysplasia, comprising short stature with short neck and trunk, odontoid hypoplasia with unstable neck, pectus carinatum, tracheal narrowing, obstructive, restrictive lung, kyphoscoliosis, laxity of joints, coxa valga, genu valgum, and abnormal gait. The patients also feature valvular heart disease, hearing loss, corneal clouding, hepatomegaly, coarse face, and widely spaced teeth. 2 –4 MPS IVA patients, if untreated, become disabled and wheelchair-bound between the ages of 13–19 years. These patients mostly die of respiratory, cardiac, or cervical spinal cord complications in their 20s or 30s. 2,5
Current treatments for MPS IVA include enzyme replacement therapy (ERT) with the elosulfase alfa (Vimizin®; recombinant human GALNS enzyme) and hematopoietic stem cell transplantation (HSCT). 6 ERT improves endurance, exercise capacity, and oxygen utilization, resulting in improved ADL to some extent; however, its impact on bone remains limited due to the avascularity of cartilage and the growth plate of bone. Other limitations include the immune reactions of infused enzymes and the short half-life of infused enzymes in the circulation. 2,6,7 Allogeneic HSCT (Allo-HSCT) has been considered a promising treatment for MPS IVA. It improves pulmonary function, cardiovascular involvement, ADL, bone mineral density (BMD), and laxity of joints, in addition to reducing surgical interventions. 2,8,9 However, it is not standard of care for patients with MPS IVA due to the lack of evidence in bone and cartilage, risk of transplantation rejection, graft-versus-host-disease, limited age of transplantation, and the issues of finding human leukocyte antigen (HLA)-matched donors. 6,10 Overall, ERT and HSCT have limited or no impact in ameliorating skeletal complications, although they improve ADL. 2 Surgical interventions may still be required to improve clinical outcomes even after ERT or HSCT. Preclinical studies have extensively evaluated alternatives to ERT and HSCT: pharmacological chaperones, substrate reduction therapy, gene therapy (GT) with adeno-associated viral vectors (AAV), clustered regularly interspaced palindromic repeats (CRISPR/Cas9), or nanoparticles. 11 –15
Lentiviral (LV) mediated HSC-GT has become an appealing approach in LSDs, including MPS, Pompe, Fabry, Gaucher diseases, metachromatic leukodystrophy (MLD), as well as other disorders, particularly blood-related hemophilia, sickle cell, Wiskott–Aldrich syndrome, and β-thalassemia since it provides a one-time permanent treatment due to integration of the vector into hematopoietic stem cells (HSCs). 16 –23 LV-GT has been assessed via in vivo direct infusion and ex vivo stem cell-mediated infusion applications for many diseases. However, in vivo, direct infusion leads to off-target effects and potential immune reactions against infused LVs. 24,25 Given that HSCT, to some extent, impacts skeletal abnormalities in MPS IVA patients, 8,9 it is critical to exploit the potential effect of LV-modified HSCs on skeletal involvements. This aspect of our research is exceptionally intriguing and has the potential to impact the field significantly. Briefly, HSCs are collected from patients’ donor cells (autologous) and are modified with LVs in vitro. Patients are preconditioned with an applicable method (busulfan, fludarabine) to eliminate existing blood cells before transplanting modified HSCs to engraft in the bone marrow. Following transplantation, engineered HSCs are delivered into circulation and, thus, into many tissues at different rates. 26 Ex vivo HSC-GT by LVs has been demonstrated to achieve relatively stable and curative production of enzymes in patients with MPS IH, MPS IA, and MLD diseases, 27 –31 and mice with MPS II. 32
However, delivering enough therapeutic enzymes to hard-to-reach tissues such as bone and avascular cartilage remains challenging. 33 –35 Therefore, the current research aims to address the potential of LV-modified HSCs in treating skeletal involvements in MPS IVA patients and holds great promise for effectively treating this condition.
MATERIALS AND METHODS
Murine model studies
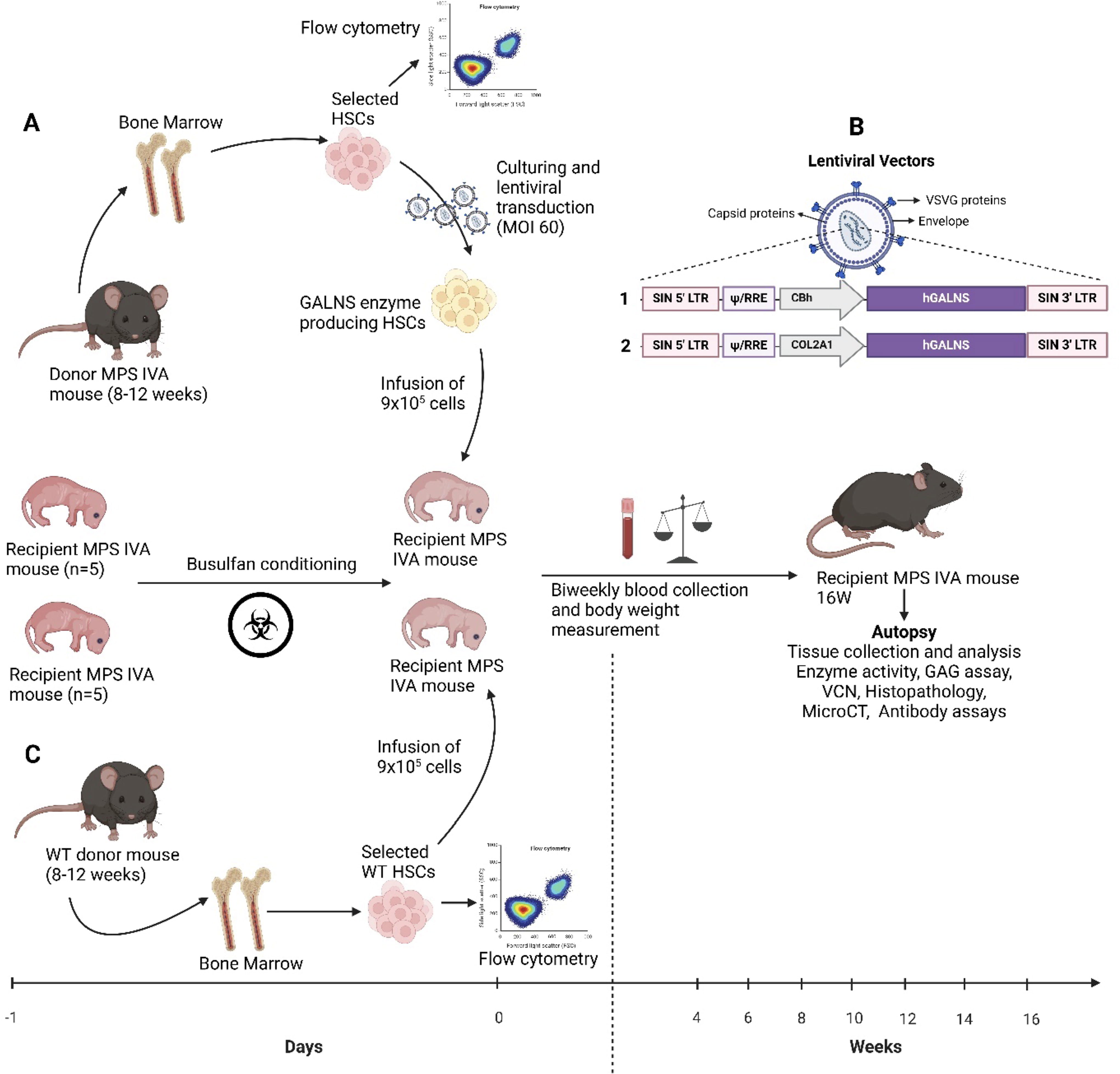
MPS IVA knock-out (KO) mice (Galns−/−, MKC2) were generated at Inotiv (West Lafayette, IN) and reported in our previous research. 36,37 This MPS IVA mouse model had a large deletion of ∼6,300 bp (exons 2–5: 6,359 bp) at genomic coordinates. We designed, generated, and tested cellular assay sgRNAs for CRISPR-Cas9 mediated KO in C57BL/6J mouse zygotes. Briefly, we designed sgRNAs targeting upstream of exon 2 and upstream of exon 6 of mouse Galns, which disrupted mGalns and made the MPS IVA murine model. The most potent sgRNA with minimal off-target potential was assembled into a ribonucleoprotein complex with Cas9 endonuclease and delivered into zygotes from C57BL/6J mice, followed by embryo transfer into pseudo-pregnant females. Viable progeny was analyzed for the desired mutation by genomic PCR and DNA sequencing. Twenty-three transgenic founder mice with KO have been identified to be positive. The two founders of choice were backcrossed to wild-type mice to generate F1 heterozygous progeny for the Galns−/− mice, as confirmed by PCR-mediated genotyping and DNA sequence analysis. Nine heterozygous mice (2 males) were identified (Galns+/−). Cohort breeding, cryopreservation, in vivo assays, and molecular analyses were made for these mice. After mating heterozygous male and female mice, we obtained homozygous mice (Galns−/−, MKC2). The age-matched male (wild-type and untreated) mice were used as controls. The colony was housed in a pathogen-free facility on a 12 h light/dark cycle. All mouse care and handling procedures were by the rules of the Institutional Animal Care and Use Committee of Nemours Children’s Health, Delaware Valley, under the protocol RSP21-12482-001—“Ex vivo GT with LV vectors.”
Construction of LVs and their efficiency in MPS IVA patients’ fibroblasts
We designed LVs under two different promoters (ubiquitous CBh and collagen targeting COL2A1) driving a human native GALNS expression (Fig. 1). LVs were provided by VectorBuilder (Chicago, IL). The final constructs for the transduction of HSCs were LV-CBh-hGALNS and LV-COL2A1-hGALNS.
Ex vivo gene therapy procedures. Isolation and modifications of HSCs removed from MPS IVA mice
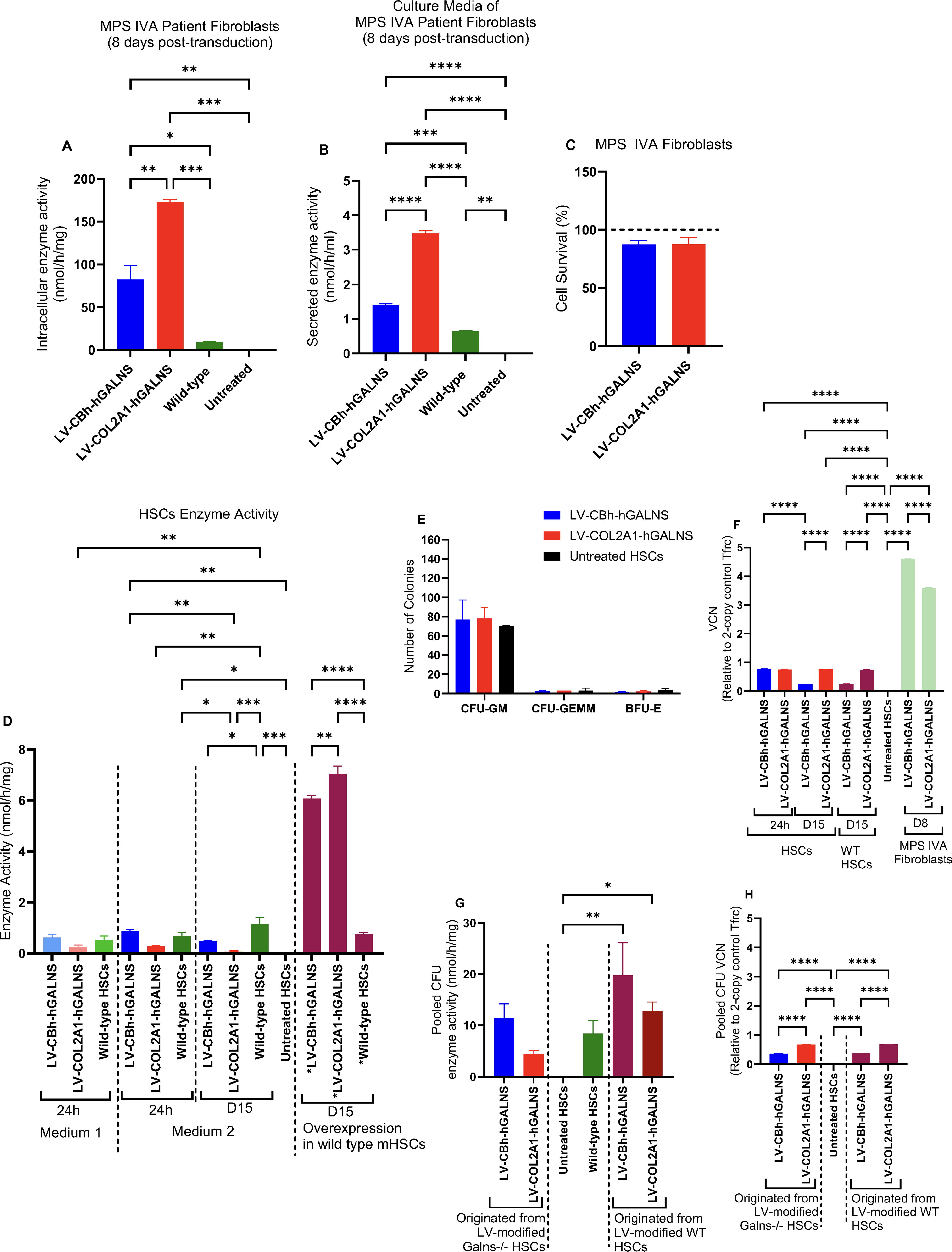
Before moving HSC experiments, we tested our LV constructs in MPS IVA patients’ fibroblasts. Patients’ fibroblasts (3 × 105 cells/well) were seeded in complete Dulbecco’s modified Eagle’s medium nutrient mixture F-12 (DMEM/F12, Gibco#11320033, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS; Gibco#10082147), 1% streptomycin/penicillin. Then, we transduced these cells at the multiplicity of infection (MOI) of 20 according to the manufacturer’s instructions. On day 8, transduced cells were collected from each well and analyzed to confirm their effectiveness in the intracellular and secreted GALNS enzyme activities and vector copy numbers (VCNs) (Fig. 2).
GALNS enzyme activity in MPS IVA patients’ fibroblasts and media
Isolation and transduction of mouse HSCs
Donor bone marrow was harvested from the femur and tibia of 8- to 12-week-old male Galns−/− mice for treatment groups (LV-HSC-GT) and from wild-type donors for allo-HSCT control group (Fig. 1). Galns−/− and wild-type donor HSCs were purified using EasySepTM mouse hematopoietic progenitor cell isolation kit (Stem Cell Technologies#18000, Vancouver, Canada). We utilized two different medium recipes to culture isolated HSCs: Medium 1. Dulbecco’s modified Eagle’s medium nutrient mixture F-12 (DMEM/F12, Gibco#11320033, Grand Island, NY) supplemented with 1% insulin/transferrin/selenium (Gibco#41400045), 1% penicillin/streptomycin (Pen/Strep; Sigma-Aldrich#P4333, Burlington, MA), 0.1% recombinant human albumin (SeraCare Life Sciences#18600016, Milford, MA), 10 mM 2-[4-(2-hydroxyethyl) piperazin-1-yl] ethane-1-sulfonic acid (HEPES; ThermoScientific#J16924.AE, Waltham), and the growth factors (PeproTech, Cranbury, NJ)-10 ng/mL murine stem cell factor (SCF; #25003, PeproTech), 100 ng/mL murine thrombopoietin (TPO; #31514), and 100 ng/mL Fms-related tyrosine kinase-3 (Flt-3; #25031L). Medium 2. Ham’s F12 nutrient mix medium (Gibco#11765-054, Waltham) supplemented with 10 mM HEPES, 1% penicillin-streptomycin-glutamine (PSG; Gibco#10378-016, Waltham), 1% insulin-transferrin-selenium-ethanolamine (Gibco#51500–056, Waltham), 1 mg/mL polyvinyl alcohol (PVA; Sigma-Aldrich#P8136), 100 ng/mL TPO, and 10 ng/mL SCF as described in the published protocol. 38 Then, lin− HSCs were seeded at a density of 0.9 × 106 cells per well into both complete mediums 1 and 2 for further LV transduction.
To modify Galns−/− HSCs by LVs, we followed the MOI of 60 based on the literature. 32,39 Galns−/− HSCs were transduced with LVs expressing the GALNS transgene at the MOI of 60 and incubated for 20 h at 37°C 5% CO2. The isolated HSCs were stained with HSC markers c-Kit, Sca-1, and SLAM. Following selected antibodies and isotypes were used (Biolegend, San Diego, CA): APC anti-mouse c-Kit (CD117; 0.2 mg/mL; #105811, Biolegend), FITC anti-mouse Sca-1 (Ly-6A/E; 0.5 mg/mL; #122506, Biolegend), Pacific Blue anti-mouse SLAM (CD150; 0.5 mg/mL; #115924), APC Rat IgG2b κ (#400612, Biolegend), FITC Rat IgG2a κ (#400506, Biolegend) and analyzed through flow cytometry to confirm the purity (Supplementary Fig. S1).
Busulfan conditioning and transplantation of HSCs into MPS IVA mice
Newborn recipient male Galns−/− mice (n = 5 per group) on day 1 were intraperitoneally (IP) injected with busulfan (60 mg/10 mL; Accord Healthcare Inc/Intas Pharmaceuticals Ltd., Ahmedabad, India) in a dose of 10 mg/kg body weight. The busulfan-conditioned recipients were housed in the biosafety room for 24 h before transplantation. The next day, LV-HSCs and allo-HSCs were individually collected via trypsin, and 0.5–0.9 × 106 were individually transplanted in the facial vein of busulfan-conditioned newborn male Galns−/− mice (n = 5 per group). The health of the newborn mice was monitored daily, the body weight was measured weekly starting from the age of 4 weeks, and the plasma samples and white blood cells (WBCs) were collected biweekly from weeks 4 to 16. Mice were euthanized 16 weeks post-transplantation, and all tissues, including brain, heart, lung, liver, kidney, spleen, tibia, arm, trachea, and bone marrow cells (BMCs), were collected for further analysis (Figs. 2 and 3).
Enzyme activities in tissues after LV-modified HSCs transplantation and wild-type allo-HSCT
4-Methylumbelliferone (4-MU) assay
The GALNS enzyme activity in cells, plasma, and tissue extracts was meticulously assessed using a 4-MU assay (Melford Laboratories Ltd, Suffolk, UK). MPS IVA patients’ fibroblasts, wild-type mice HSCs, and Galns−/− mice HSCs post-transduction were lysed in 60–100 µL of homogenization buffer (25 mM Tris–HCl, pH 7.2, 1 mM PMSF) via sonication for 30 s and 10% amplitude. Then, cell lysates were centrifuged for 10 min at 4°C. The supernatant was transferred into a new tube and assayed for the GALNS enzyme activity. As stated above, tissue was dissected and immediately homogenized with Bead Mill Homogenizer (OMNI International, Kennesaw, GA) in homogenization buffer. Homogenates were centrifuged for 30 min at 4°C, and the supernatant was transferred into a new tube. Both supernatants of cells/tissues and plasma samples underwent 4-MU enzyme assay and were incubated with 22 mM 4-methylumbelliferyl-β-galactopyranoside-6-sulfate (Research Products International, Mount Prospectm, IL) at 37°C for 16 h. The next day, 10 mg/mL of β-galactosidase from Aspergillus oryzae (Sigma-Aldrich, Saint Louis, MO) was added to the reaction and incubated for 1 h at 37°C. The reaction was stopped with 1 M of glycine buffer (pH 10.5 adjusted by NaOH). We used the FLUOstar Omega plate reader (BMG LABTECH Inc. NC) to measure the enzyme activity at an excitation wavelength of 336 nm and an emission wavelength of 450 nm. The activity was expressed as nanomoles of 4-MU released per hour per milligram of protein (nmol/h/mg). Protein concentrations were determined using a PierceTM BCA protein assay kit (Thermo Fisher Scientific #23225, Waltham, MA) (Figs. 2 and 3, Supplementary Figs. S2, Figs. S6, and Figs. S7).
WST-1 toxicity assay
The WST-1 toxicity assay was a pivotal part of our research, as it allowed us to test if LV transduction and further GALNS enzyme expression have a cytotoxic effect on MPS IVA patients’ fibroblasts at the MOI of 20. We seeded these cells at 1 × 104 cells/well in 100 µL culture medium and transduced for 48 h at 37°C and 5% CO2. Then, 10 µL of cell proliferation reagent WST-1 (Abcam#ab155902, Cambridge, UK) was added to each well, mixed by pipetting gently, and incubated for 4 h at 37°C and 5% CO2. We used blank control: 10 µL of WST-1 reagent was added to 100 µL of culture medium prepared parallel to experimental groups. The absorbance of each group was measured in a plate reader (FLUOstar Omega plate reader; BMG LABTECH Inc, NC) at an optical density of 450 nm, and the reference wavelength was set to 650 nm. The results were evaluated by averaging the duplicate reading for each sample and then subtracting the culture medium-related background from each sample to obtain the final absorbance. The equation to calculate the percentage of cytotoxicity using final absorbance is as follows: % Cytotoxicity = (100 × [Control – Samples])/Control (Fig. 2C).
KS assay by liquid chromatography–tandem mass spectrometry
The liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis was a significant part of our research, as it allowed us to evaluate the reduction of accumulated mono-sulfated-KS. The plasma and tissues of treated, untreated, and wild-type mice were analyzed by LC–MS/MS according to the previously described protocol (Fig. 4A–C). 40 –47

Mono-sulfated keratan sulfate (KS) concentrations in plasma
VCN analysis
To confirm the biodistribution of LVs in cultured Galns−/− HSCs 24 h and 15 days post-transduction and in the liver 16 weeks post-transplantation, DNA was extracted from the cultured mouse HSCs and liver samples with the Qiagen Gentra Puregene Tissue Kit, and DNA was digested with Proteinase K at 55°C. Following Rnase treatment, DNA was resuspended in Tris-EDTA buffer, and DNA concentration was measured. Quantification of VCN was performed by digital PCR (dPCR; ThermoFisher QuantStudio Absolute Q) as single plex (LV and Tfrc on 2 separate chips) using the primers (Integrated DNA Technologies) specific to the LV vector psi gene, resulting in an 82-bp PCR fragment (Supplementary Table S1). QuantStudio™ Absolute Q™ MAP16 Plate Kit and Master Mix (ThermoFisher#A53301) were used to prepare the reaction. The quantification was done with the following formulation: (LV cps/µL*DNA dilution)/(Tfrc cps/µL/2) (Figs. 2F and 4D).
Colony forming unit (CFU) assay
To set up the CFU assay for triplicate cultures, we thawed a vial of previously aliquoted 4 mL MethoCultTM GF M3434 complete media (StemCell Technologies#03434, Vancouver, Canada) overnight at 4°C. A total of 1 × 105 cells/mL from each group (untreated, LV-CBh and LV-COL2A1-treated groups) was diluted in the Iscove’s Modified Dulbecco’s medium (Gibco#12440053) with 2% FBS. Afterward, 400 µL of diluted cell suspensions were added to a 4 mL MethoCult tube previously thawed and mixed thoroughly via vortexing. Following removal of bubble formation, MethoCult mixture containing cells were distributed evenly in 3 wells of 6-well plates (9.6 cm2/well; FalconTM 353046; Fisherscientific#08-772-1B, Waltham) and incubated at 37°C, 5% CO2 with ≥95% humidity for 7–10 days according to the manufacturer’s instructions. Colonies were identified and counted by visual inspection using STEMgridTM-6 (StemCell Technologies#27000) via an inverted light microscope at 4× and 10× magnification (Fig. 2E and Supplementary Fig. S2B). After completing colony counting, HSCs in each well were collected in separate vials to confirm the GALNS enzyme activity and VCN of these pooled CFU colonies (Fig. 2F–H).
Pathology
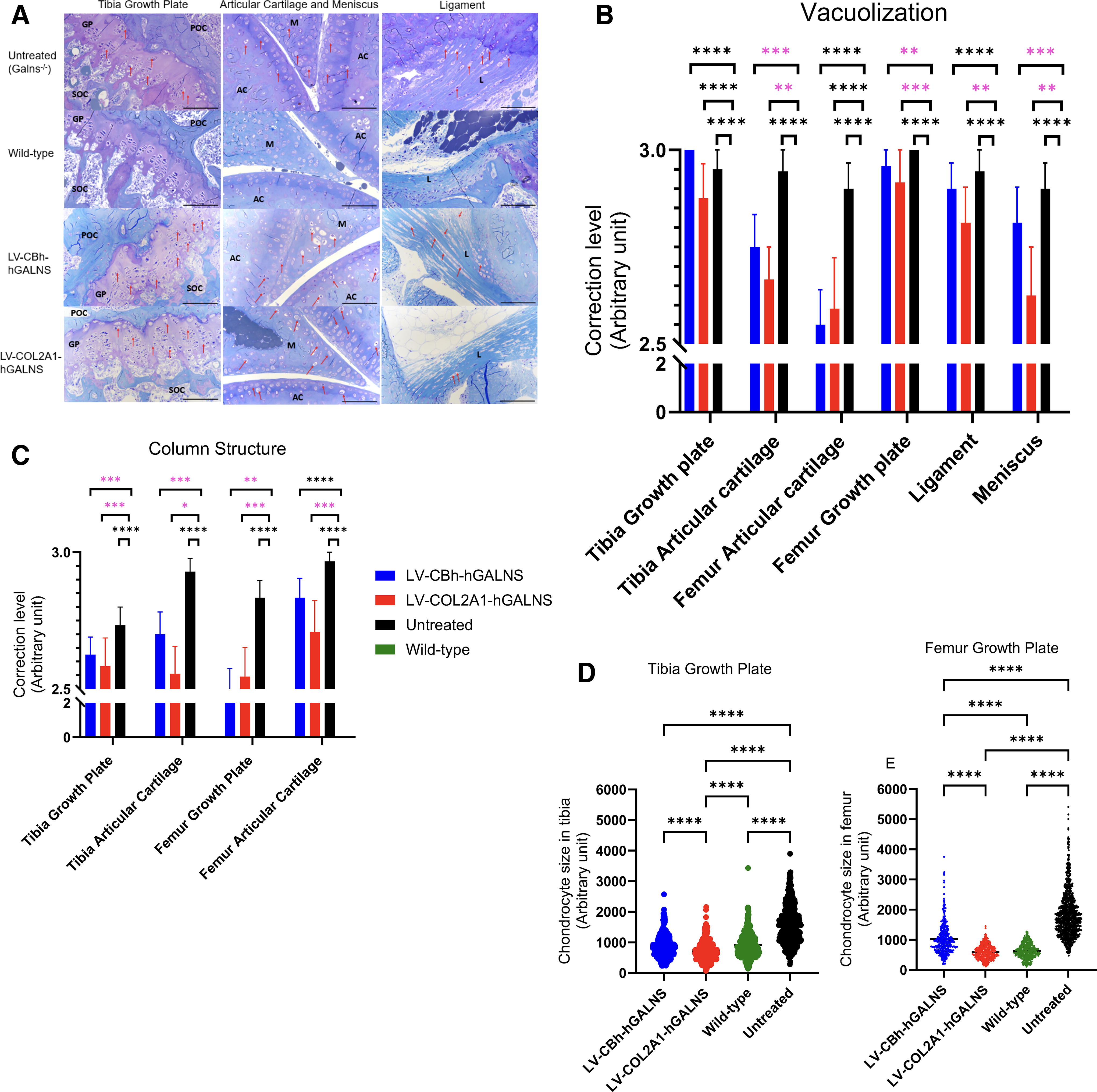
Mice were euthanized with CO2 gas 16 weeks post-transplantation, and then the heart, liver, and knee joints were collected in 10% formalin. To evaluate lysosomal storage by light microscopy, these tissues were then fixed in 2% paraformaldehyde, 4% glutaraldehyde, and toluidine blue-stained 0.5-µm-thick sections were prepared. Bone and heart pathologies were assessed in a double-blinded manner by giving scores to vacuolization and column structures in relevant tissues from 0 (the best) to 3 (the worst) (Supplementary Tables S2 and S3), and then statistical analysis was performed (Figs. 5 and 6). To evaluate chondrocyte storage, we used Image J (NIH) software and measured the cell size arbitrarily (namely by using a software tool to select/mark the perimeter of each chondrocyte only in the proliferative region of the growth plate to reflect the status of cell size) in nearly 100 cells per slides. 14,48
Bone pathology of lentiviral vector (LV-CBh-hGALNS or LV-COL2A1-hGALNS)-treated and control groups
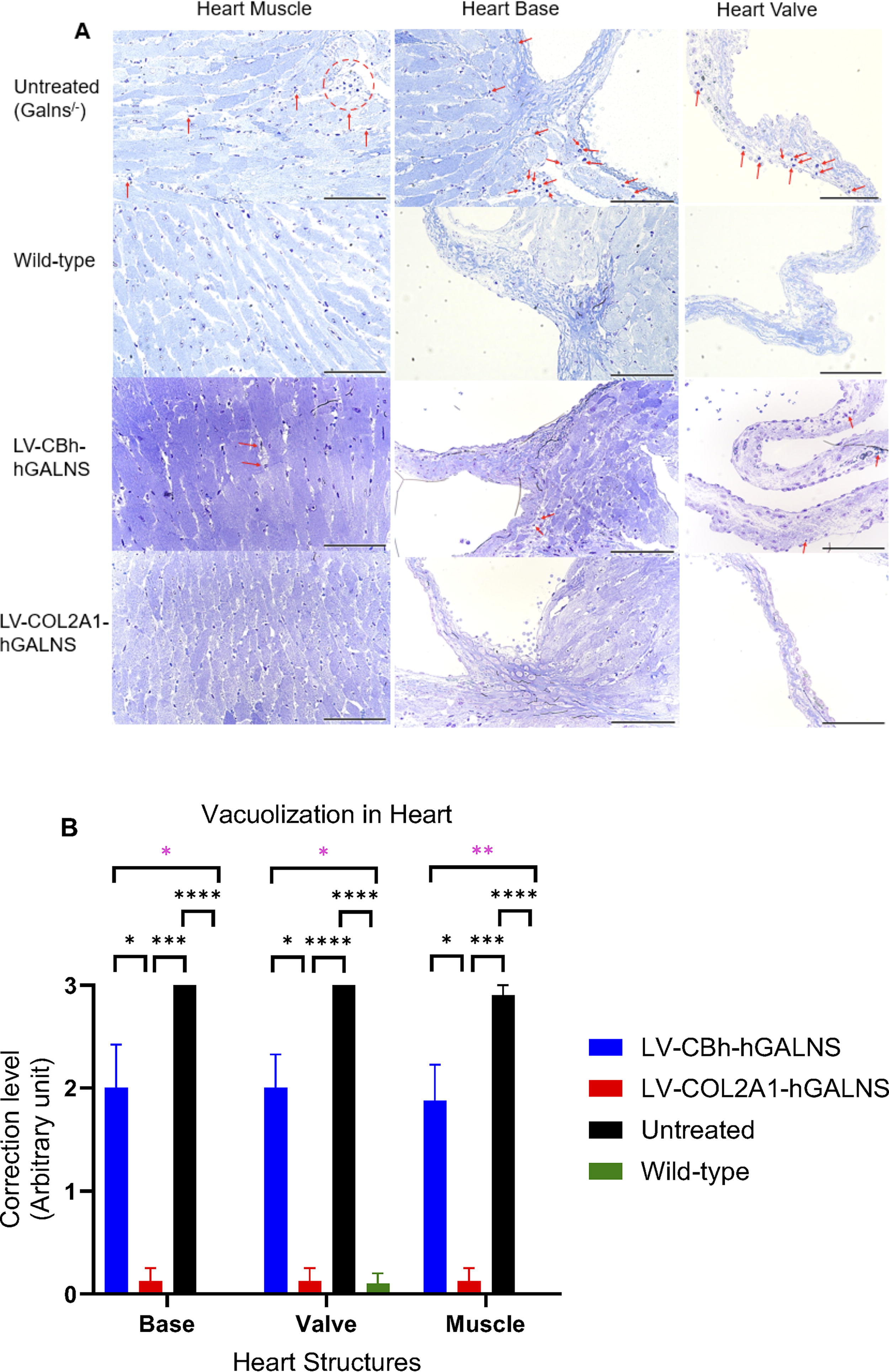
Heart pathology after lentiviral vector (LV-CBh-hGALNS or LV-COL2A1-hGALNS)-treated and control groups
Micro-CT
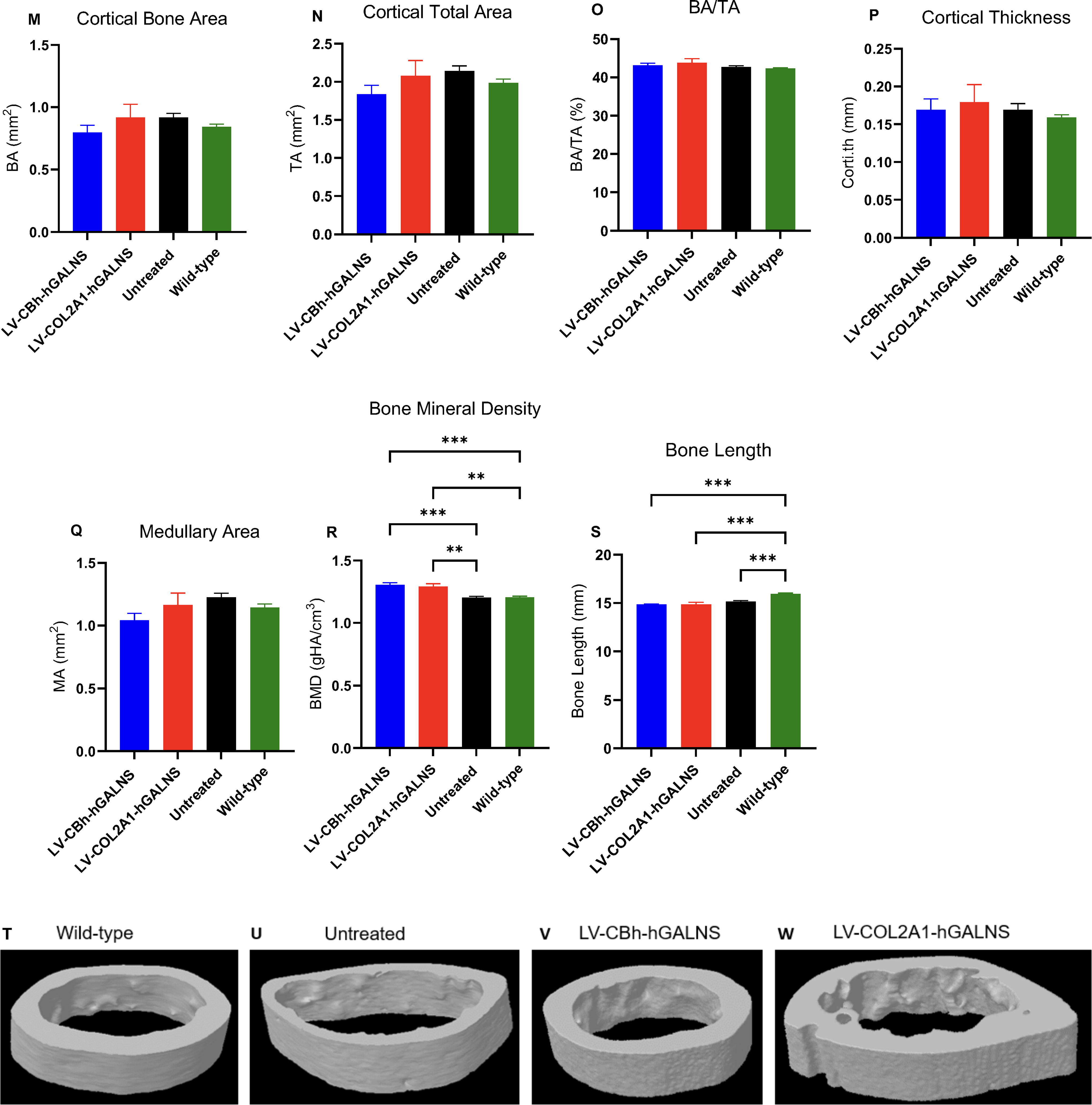
A micro-CT scan was performed on the right femur of wild-type, Galns−/−, and LV-HSC-treated groups using SkyScan 1276 micro-CT System (Bruker, Manning Park, MA). The femur samples were collected in 100% EtOH at 16 weeks post-transduction, and they were further wrapped in salinated (0.9% saline) gauze in preparation for the micro-CT imaging. The scanning was performed with high spatial resolution down to 8.0 µm pixel size, 512 projections, exposure time of 2,300 ms, photon energy of 85 keV, and current of 47 μA. A three-dimensional reconstruction of each bone was made. 43,49,50 We evaluated bone structure (trabecular and cortical bones) by measuring the following parameters: BMD, total volume, bone volume (BV), and thickness (Fig. 7). 49,50

Bone morphometric analysis. Trabecular bone morphology
Continued.
Immunohistochemistry
We confirmed the GALNS enzyme expression in the tibia and liver by performing immunohistochemistry (IHC) of GALNS (Fig. 8 and Supplementary Fig. S3). In addition, we analyzed KS and procollagen II levels via IHC. Collagen, KS, and GALNS were stained by anti-procollagen (Invitrogen#BTE0030202, Waltham, MA), anti-KS (Santa Cruz Biotechnology#sc-73518, Dallas, TX), and custom-made monoclonal anti-GALNS antibodies (Creative Biolabs, NY). 51 The liver and tibia were fixed in 10% formalin and sectioned with 5 µm-thickness for IHC. KS, GALNS, procollagen II distribution, and intensity patterns were investigated immunohistochemically to determine any correlation with therapeutic effects. To evaluate the expression of GALNS and collagen and the reduction of KS, we used Image J (NIH) software and analyzed each slide, selecting only the proliferative region of the growth plate using software tools. After selection, we utilized the software color intensity tools to quantify each section and compare it with untreated and wild-type controls.
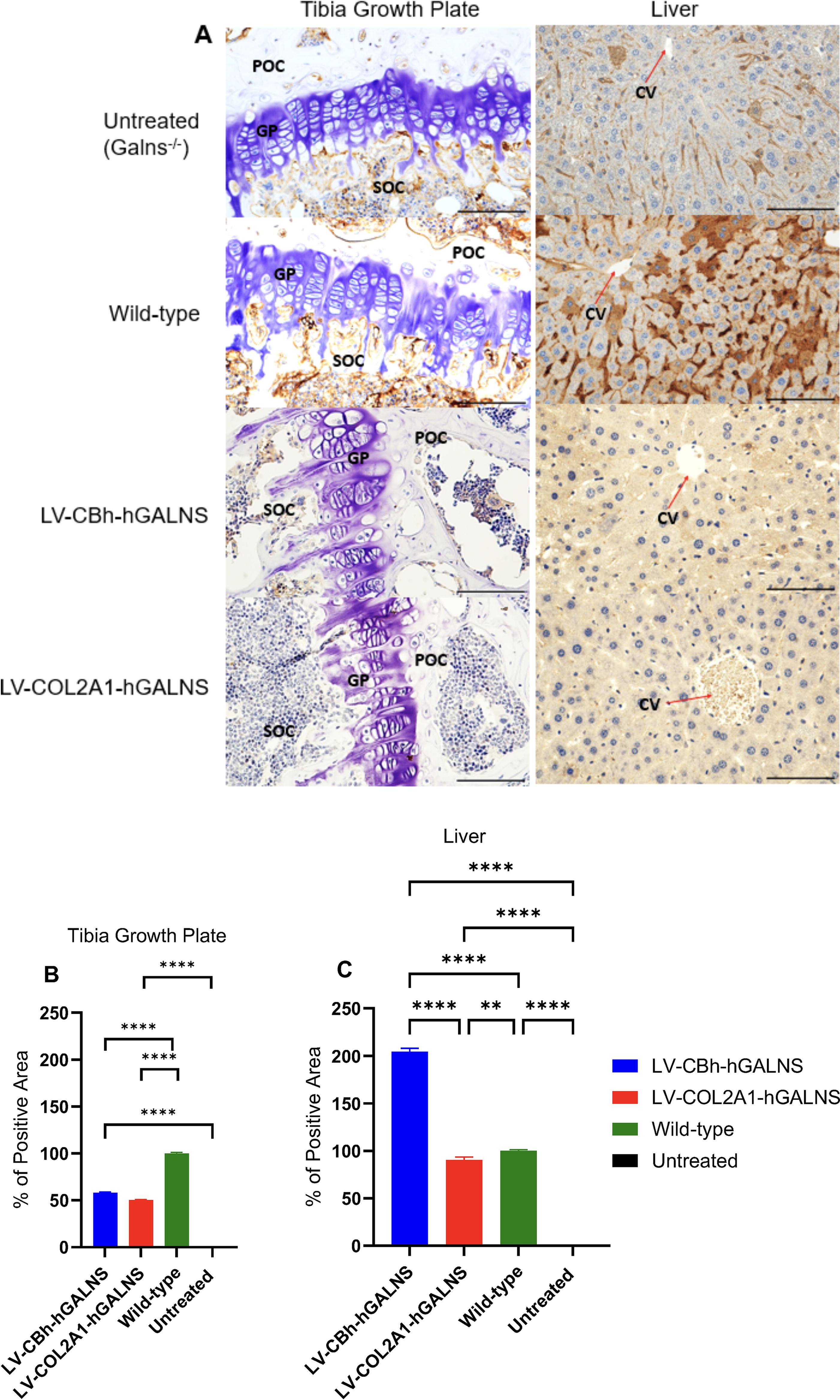
The GALNS enzyme expression in bone (tibia growth plate) and liver under anti-GALNS staining; 40× magnification with a 100-µm scale
Enzyme-linked Immunosorbent Assay (ELISA)
To determine the anti-GALNS antibody response, we modified the previously described indirect ELISA protocol. 52 Briefly, 96 well polystyrene microplates were coated with 2 ug/mL of Vimizin® enzyme in the coating buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.021% NaN3, pH 9.6) and incubated overnight at 4°C. Then, the coated plates were blocked with 3% BSA in PBS for 1 h at room temperature and washed first with TTBS (10 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.5) and second with TBS (10 mM Tris, 150 mM NaCl, pH 7.5). Biweekly collected plasma samples (1:100 dilution in TBST) and monoclonal anti-GALNS antibodies (Custom-made clone 2F5F2, Creative Biolabs, NY) were added into each well individually and incubated for 2.5 h at 37°C. After 5 times washing the plates with TTBS (×3) and TBS (×2) successively, the plates were incubated with peroxidase-conjugated goat anti-mouse secondary antibodies (Invitrogen#656120, Waltham) in TBST at room temperature for 1 h according to manufacturer’s instructions. Following four washes, first with TBST and the rest with TBS, 100 µL of peroxidase substrate 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS; Invitrogen#002024) was added into the reaction and incubate at room temperature for 30 min to develop the signal. The enzyme reaction was stopped with 100 µL of 2% sodium dodecyl sulfate, and the absorbance was measured at 450 nm using a FLUOstar Omega plate reader stated above. 25,53,54 Plasma concentrations of anti-GALNS antibodies were derived by extrapolating the absorbance values from a calibration curve using monoclonal anti-GALNS antibodies mentioned above (Supplementary Figs. S4–S6).
Statistical analysis
The quantitative data having normal distribution were stated as mean ± standard error, whereas quantitative data not having normal distribution were expressed as median (95% confidence interval). Shapiro–Wilk and Kolmogorov–Smirnov tests were performed to determine the normal distribution. In statistical tests, one-way analysis of variance (ANOVA) with Tukey post hoc or Kruskal–Walli’s test with Dunn’s multiple comparison tests was used to analyze more than two groups considering normal distribution. A p value of 0.05 or less was considered statistically significant. Comparisons are as follows: Treatment versus wild-type, treatment versus untreated, and treatment versus treatment. All statistical analyses were performed in GraphPad Prism 9.5.0 (GraphPad, San Diego, CA). The number of replicates (n) was five for the in vivo experiments (n = 5) and three for in vitro experiments (n = 3), independent biological replicates for mice and culture experiments.
RESULTS
Successful validation of LVs in MPS IVA patient fibroblasts has provided a strong foundation for our research and reassuringly confirmed the reliability of our findings
To develop LV-HSC-GT for patients with MPS IVA, we designed third generation self-inactivating LVs with native human GALNS transgene driven by the ubiquitous CBh (LV-CBh-hGALNS) or collagen-specific COL2A1 (LV-COL2A1-hGALNS) promoters. To confirm whether each LV has a therapeutic efficacy, we transduced fibroblasts (Galns−/−) derived from MPS IVA patients at the MOI of 20 according to the company instructions (Fig. 2A, B) and HSCs derived from MPS IVA mice. We evaluated enzyme activities before moving to ex vivo experiments compared to healthy controls (healthy human skin fibroblasts and wild-type mice HSCs) (Fig. 2C).
Intracellular enzyme activity in MPS IVA patients’ fibroblasts 8 days post-transduction showed a 9-fold increase under LV-CBh-hGALNS and a 19-fold increase under LV-COL2A1-hGALNS compared to wild-type (Fig. 2A). Also, we confirmed the secretion of produced GALNS enzyme in these culture media, in which LV-CBh-hGALNS showed 2.3-fold, and LV-COL2A1-hGALNS showed 5.7-fold elevation in the GALNS enzyme activity compared to wild-type (Fig. 2B). No cytotoxicity regarding LVs and overexpression of hGALNS enzyme was detected under each treatment and cell survivals were above 87% for both treated groups compared to untreated MPS IVA fibroblasts (100%) (Fig. 2C). Moreover, VCN was 4.6 ± 0.05 and 3.6 ± 0.01 relative to 2-copy control of Tfrc for LV-CBh-hGALNS and LV-COL2A1-hGALNS, respectively (Fig. 2F).
Evaluation of galns−/− (lin−) HSCs transduced with LVs
Lineage-negative (Lin−) HSCs were isolated from the bone marrow of GALNS−/− donor mice and wild-type donor mice via magnetic selection (Fig. 1). We measured the purity of lin− HSCs immediately after isolation via flow cytometry by staining c-Kit (CD117), Sca-1, and SLAM (CD150) surface markers, in which the final population of lin− cells was ∼21% of all isolated cells. Then, we gated lin−/c-Kit+/Sca-1+hematopoietic progenitor cells among lin−(20.57%), 95.75% of which were positive for CD150 marker (Lin−Sca1+c-kit+CD150+) (Supplementary Fig. S1). Here, we transduced the isolated cells in media 1 and 2 according to our transduction method and analyzed the GALNS enzyme activity and VCN 24 h and 15 days post-transduction. In medium 1, transduced or untransduced HSCs were maintained up to 36–72 h; however, they were either differentiated, aggregated, or primarily dead after 72 h post-transduction. Only enzyme activity and VCN were analyzed in 24 h post-transduction in medium 1 (Fig. 2D). In medium 2, we followed the same transduction method and MOIs, in which the stemness of transduced HSCs was well-maintained and cultured for long term to test the enzyme activity of GALNS, VCN, mono-sulfated KS levels, and other relevant experiments (Fig. 2D–H). The data showed no statistical differences in GALNS enzyme activity, VCN, or mono-sulfated KS levels in both media after 24 h; however, the maintenance of lin− HSCs was well-established without any differentiation in medium 2 during procedures. Therefore, we continued the rest of the experiments in medium 2 to culture and analyzed these cells for the long term. We then evaluated the purity and proliferation of cultured lin− HSCs on 20 days post-LV-transduction as described above. The data showed that the percentage of untreated Galns−/−, LV-CBh-hGALNS, and LV-COL2A1-hGALNS treated lin− HSCs reached 52.39%, 46.62%, and 59.74%, respectively, among which Sca1+c-kit+cells accounted for 97.57%, 96.39%, and 97.93%, respectively (Fig. S1D). In all groups, lin−Sca1+c-kit+CD150+cells were found at 97.33%, 96.44%, and 96.28% for untreated, LV-CBh-hGALNS, and LV-COL2A1-hGALNS, respectively (Supplementary Fig. S1D). These lin− cells were then evaluated for the production and secretion of the GALNS enzymes, which might result from a possible engraftment potential. However, we did not perform the engraftment experiment in the present study. In medium 2 culturing conditions, LV-transduced HSCs were cultured for up to 30–45 days with no differentiation (Supplementary Fig. S2C); however, such long-term cultures may have poor engraftment efficiency. Thus, we did not perform further analysis regarding this process. In murine experiments, all conditioned Galns−/− newborn recipients received LV-transduced HSCs 20–24 h post-transduction. Overall, even though the culturing conditions and maintenance of HSCs were improved, transduction with our LVs was not efficient in cultures as expected at the MOI of 60.
Ubiquitous CBh promoter elevated the GALNS enzyme activity rather than tissue-specific COL2A1
HSCs derived from MPS IVA mice were transduced with LVs to verify the GALNS enzyme activities compared to untreated and wild-type control HSCs. In transduced Galns−/− HSCs, we compared the enzyme activity of GALNS at 24 h and 15 days post-transduction. Following 24 h of transduction, the enzyme activity increased up to 0.9 ± 0.04 nmol/h/mg under LV-CBh-hGALNS as compared to untreated Galns−/− HSCs, and no statistical difference was found between LV-CBh-hGALNS-transduced and wild-type HSCs (0.7 ± 0.1 nmol/h/mg). At the same time, LV-COL2A1-hGALNS elevated enzyme activity by 0.3 ± 0.03 nmol/h/mg. The data confirmed that LV-CBh-hGALNS had ∼3-fold more enzyme activity than LV-COL2A1-hGALNS compared to untreated HSCs. However, with 15 days of culturing LV-transduced cells, the enzyme activity reduced to 0.5 ± 0.03 and 0.06 ± 0.03 nmol/h/mg under LV-CBh-hGALNS and LV-COL2A1-hGALNS treatments. No statistical significance was found regarding this reduction between 24 h and 15 days.
We also evaluated the overexpression of the GALNS enzyme by utilizing HSCs isolated from wild-type mice. The results showed that the activity of the GALNS enzyme increased by 86.8% and 88.6% under LV-CBh-hGALNS (6.07 ± 0.1 nmol/h/mg) and LV-COL2A1-hGALNS (7.02 ± 0.3 nmol/h/mg), respectively compared to untransduced wild-type HSCs (0.8 ± 0.06 nmol/h/mg) (Fig. 2D).
Confirming the GALNS enzyme activity in Galns−/− donor HSCs under LVs, we intravenously transplanted the LV-HSCs or allo-healthy HSCs into busulfan myeloablated Galns−/− newborn recipients.
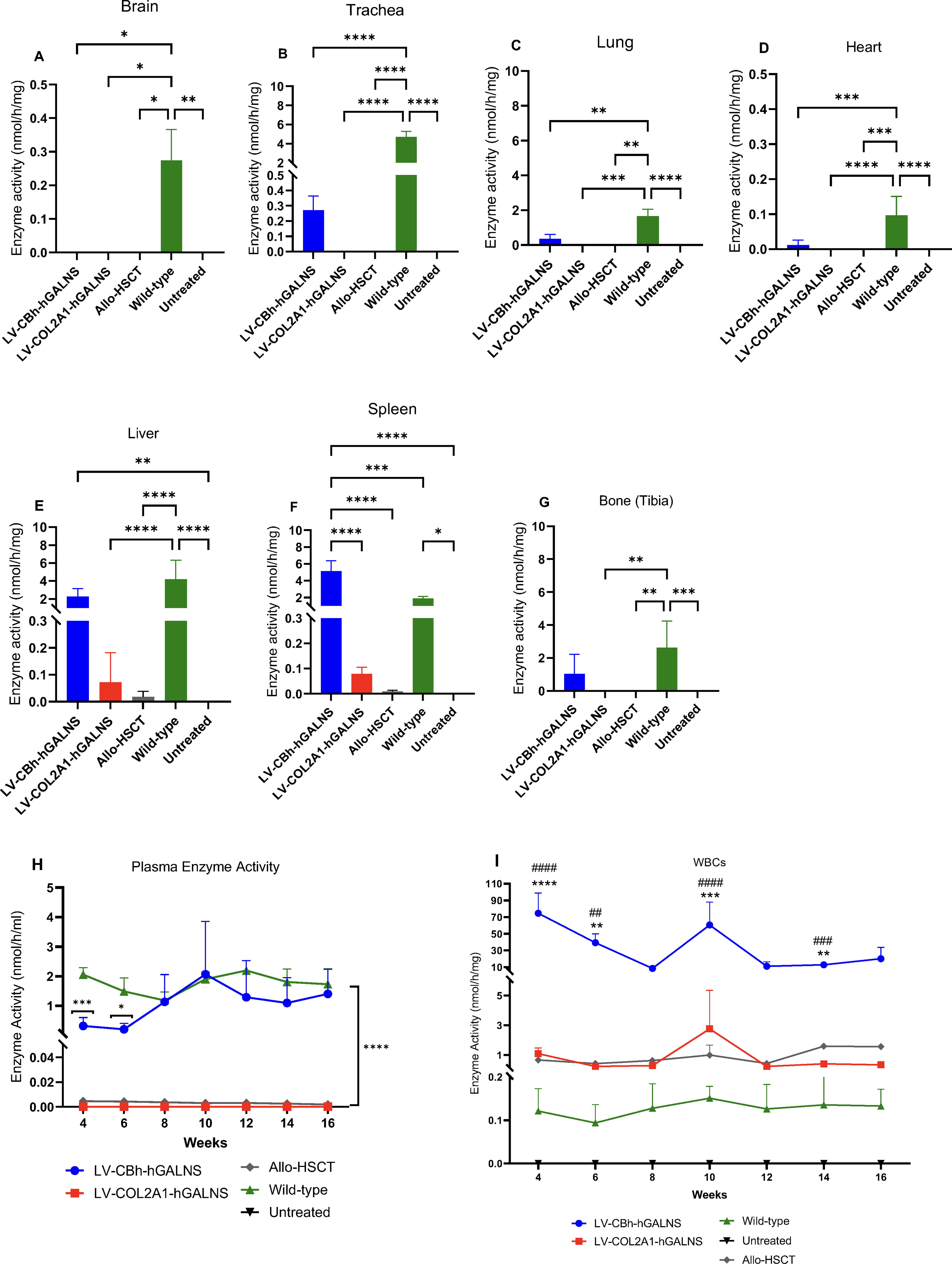
After transplantation and autopsies at 16 weeks, we analyzed plasma, WBCs, and tissue enzyme activities, including brain, trachea, lung, heart, liver, spleen, and bone (tibia) (Fig. 3A–I). In the brain, the enzyme activity under each treatment was undetectable compared to that of wild-type mice (2.7 ± 0.09 nmol/h/mg) (Fig. 3A). The enzyme activity was elevated in the trachea under LV-CBh-hGALNS (0.3 ± 0.09 nmol/h/mg) compared to that of untreated MPS IVA mice. However, it did not reach the enzyme activity of wild-type mice (4.7 ± 0.6 nmol/h/mg) (Fig. 3B). The enzyme activity via LV-COL2A1-hGALNS treatment was insufficient to detect in the trachea. Similar results were found in the lungs, bone (tibia), and heart under LV-COL2A1-hGALNS treatment (Fig. 3C, D, G). However, LV-CBh-hGALNS treatment increased the GALNS enzyme activity in the lungs, bone (tibia), and heart by 0.4 ± 0.2, 1.03 ± 0.5, and 0.01 ± 0.006 nmol/h/mg, respectively, compared to wild-type levels (1.6 ± 0.4, 2.6 ± 0.6 and 0.1 ± 0.02 nmol/h/mg, respectively) (Fig. 3C, D, G). In the liver, the GALNS enzyme activity increased under both vectors. Treatment with LV-CBh-hGALNS showed an increase by 2.3 ± 0.4 nmol/h/mg, while LV-COL2A1-hGALNS treated group showed a slight elevation by 0.07 ± 0.05 nmol/h/mg as compared to that of untreated group (Fig. 3E). The GALNS enzyme activity was normalized to the wild-type level (4.2 ± 0.9 nmol/h/mg) in the liver and bone (tibia). No statistical significance was found between LV-CBh-hGALNS and wild-type groups (Fig. 3E–G). In the spleen, both LVs drove the GALNS expression, increasing enzyme activity. Treatment with LV-CBh-hGALNS showed a significant increase in the GALNS enzyme activity by 5.1 ± 1.2, which was ∼2.7 times higher than the wild-type level (1.9 ± 0.2 nmol/h/mg). Additionally, the enzyme activity under treatment with LV-COL2A1-hGALNS (0.08 ± 0.02 nmol/h/mg) was significant compared to the wild-type level (Fig. 3F).
In plasma, treatment with LV-CBh-hGALNS normalized the enzyme activity to wild-type level starting from 6 to 16 weeks, and no statistical difference was found between LV-CBh-hGALNS and wild-type mice, except for 4 and 6 weeks. The enzyme activity under LV-COL2A1-hGALNS was not detectable in plasma. Furthermore, the allo-HSCT group was found to be ∼0.003 in plasma all over the experiment compared to LV-HSC-GT (Fig. 3H).
In WBCs, the GALNS enzyme was highly expressed under LV-CBh-hGALNS over 16 weeks (the highest, 74.6 ± 24.1 nmol/h/mg, and the lowest, 8.4 ± 1.5 nmol/h/mg), which was found statistically significant compared to wild-type levels (∼0.12 ± 0.05 nmol/h/mg). In addition, HSC-transplanted mice with LV-COL2A1-hGALNS elevated the GALNS enzyme activity in WBCs by ∼0.7 ± 0.5 nmol/h/mg. The GALNS enzyme activity was normalized to the wild-type level (Fig. 3I). This expression of the GALNS enzyme under treatment with LV-COL2A1-hGALNS was further detected in BMCs (Supplementary Fig. S2A).
In the LV-HSC-GT group, the enzyme activity of BMCs was analyzed under only LV-COL2A1-hGALNS and allo-HSCT groups because we missed the collection of BMCs in the group treated with LV-CBh-hGALNS. We detected relatively low enzyme activity levels in BMCs and slightly higher levels in WBCs of the same individual mice (Supplementary Fig. S2A).
In conclusion, our findings suggest that most enzymes might have been captured in the spleen due to their role in the local and systemic regulation of immunity. 55,56 This may negatively affect the delivery of the GALNS enzymes to the targeted tissues, specifically the bone.
Tissue-specific COL2A1 LVs reduced the accumulation of GAGs under lower expression of hGALNS
To confirm the effectiveness of LV-GT in KS reduction, we performed LC–MS/MS to analyze the bone (humerus) and plasma mono-sulfated KS levels (Fig. 4). In the plasma, we found statistical significance with a 50.1% reduction in mono-sulfated KS level in mice treated with LV-CBh-hGALNS (38.82 ± 1.9 ng/mL) and 44.4% reduction with LV-COL2A1-hGALNS (43.86 ± 4.8 ng/mL) treatment compared to untreated MPS IVA mice (78.9 ± 6.1 ng/mL). There were no statistical differences between the LV-treated and wild-type groups (23.79 ± 3.02 ng/mL). In addition, allo-HSCT group had no statistical difference compared to untreated, which results in no change in mono-sulfated KS levels in plasma (Fig. 4A). In WBCs, we found that LV-COL2A1-hGALNS (6.09 ± 0.8 ng/mL) reduced KS level by 56.2% compared to untreated control (13.93 ± 1.02 ng/mL) and no statistical significance was found between group treated with this vector and wild-type (7.40 ± 1.07 ng/mL). In contrast, there was no statistical significance in mono-sulfated KS concentrations between the LV-CBh-hGALNS group (14.90 ± 3.50 ng/mL) and the untreated group (Fig. 4B). In the bone, we demonstrated the differences in KS level by 40.7% between untreated Galns−/− (0.05 ± 0.003 ng/mg) and wild-type group (0.03 ± 0.003 ng/mg). The group treated with LV-CBh-hGALNS showed a 14.1% reduction in mono-sulfated KS levels (0.04 ± 0.004 ng/mg), but it was insignificant compared to the untreated group. On the contrary, KS level decreased by 40.2% under treatment with LV-COL2A1-hGALNS (0.03 ± 0.002 ng/mg) compared to untreated MPS IVA, which was statistically significant. These data showed that LV-COL2A1-hGALNS treatment normalized mono-sulfated KS level to that of wild-type (Fig. 4C). Additionally, the mono-sulfated KS level of WBCs and bone were found to be similar under each treatment compared to untreated and wild-type controls at 16 weeks. Concerning mono-sulfated KS levels under allo-HSCT, neither WBCs nor bone showed a significant reduction (Fig. 4B, C). As a result, LV-HSC-GT under each promoter significantly reduced mono-sulfated KS levels in plasma and bone. Importantly, LV-COL2A1-hGALNS significantly reduced mono-sulfated KS levels in bone compared to those in LV-CBh-hGALNS.
LVs under ubiquitous or tissue-specific promoters were similarly inserted into the genome of HSCs
To determine the VCN, we performed dPCR targeting the psi gene, and LV packaging signal sequence, in HSCs 24 h and 15 days post-transduction following each treatment. In LV-modified Galns−/− HSCs after 24 h, LV VCNs were found by 0.75 ± 0.02 and 0.74 ± 0.02 relative to 2-copy control of Tfrc under LV-CBh-hGALNS and LV-COL2A1-hGALNS, respectively while VCN on day 15 was 0.24 ± 0.003 and 0.74 ± 0.003 per relative to 2-copy control of Tfrc, respectively. This reduction in the LV-CBh-hGALNS group on day 15 was significant compared to 24 h post-transduction. No change was detected in LV-COL2A1-HGALNS-HSC group (Fig. 2F). Then, VCN was performed in LV-transduced wild-type HSCs on day 15, which was found that LV-CBh had 0.25 ± 0.005 while LV-COL2A1 was 0.73 ± 0.003 relative to 2-copy control of Tfrc (Fig. 2F). Furthermore, we analyzed VCN in pooled CFU colonies originated from both LV-modified Galns−/− and LV-modified wild-type HSCs (Fig. 2H). The results confirmed that the VCN was 0.36 ± 0.001 and 0.7 ± 0.005 relative to 2-copy control of Tfrc in LV-CBh and LV-COL2A1-modified Galns−/− HSCs, while it was 0.37 ± 0.005 and 0.7 ± 0.003 relative to 2-copy control of Tfrc in LV-CBh and LV-COL2A1-modified wild-type HSCs (Fig. 2H). Overall, VCN was similar in Galns−/− HSCs and wild-type HSCs and did not show significant alterations before and after the CFU assay.
We further analyzed VCN in liver samples at 16 weeks, which was 0.03 ± 0.007 and 0.03 ± 0.01 relative to 2-copy controls of Tfrc under LV-CBh-hGALNS and LV-COL2A1-hGALNS, respectively (Fig. 4D).
LV-GT did not affect the colony formation of transduced HSCs while increasing the GALNS enzyme activity in the pooled CFU colonies
To determine whether transduction of LVs negatively affects the colony formation of Galns−/− and wild-type HSCs, we seeded 7 days post-transduced HSCs at the recommended number and incubated them for 7–10 days. Following colony counting, we found CFU-GM, CFU-GEMM, and BFU-E under each condition. Further evaluations showed no significance in colony formation between untreated and LV-HSC-GT groups (Fig. 2E, Supplementary Fig. S2B). Furthermore, we pooled the CFU colonies and analyzed the GALNS enzyme activity. The results confirmed that the enzyme activity of pooled colonies originated from LV-modified Galns−/− HSCs was determined by 11.4 ± 2.8, 4.4 ± 0.7, and 8.4 ± 2.5 nmol/h/mg for LV-CBh-hGALNS, LV-COL2A1-hGALNS, and wild-type group, respectively. In contrast, it was found in LV-modified wild-type HSCs by 19.7 ± 6.3 and 12.8 ± 1.8 nmol/h/mg under LV-CBh-hGALNS and LV-COL2A1-hGALNS, respectively (Fig. 2G). Overall, LV-HSCs continuously produced the GALNS enzyme before and after differentiation.
COL2A1 LVs completely corrected heart pathology and partially improved bone pathology
To evaluate the effect of expressed GALNS enzyme, we examined the heart and knee joint by toluidine blue staining. In the bone, both treatments under CBh and COL2A1 promoters driving hGALNS expression showed partial correction in vacuolization and column structure of chondrocytes compared to untreated groups. However, they did not reach normalization (Supplementary Table S2 and Fig. 5A–C). Furthermore, we measured the size of chondrocytes in the growth plates of the tibia and femur (Fig. 5D, E) to confirm whether LV treatment reduces the storage materials. The data showed that the size of chondrocytes was normalized to the wild-type level with both LV-CBh-hGALNS and LV-COL2A1-hGALNS vectors.
In the heart, the treatment with LV-COL2A1-hGALNS showed complete correction of disease progression except for one mouse, with partial correction among all groups, compared to untreated MPS IVA mice. No statistical differences were found between the group treated with LV-COL2A1-hGALNS and the wild-type control group. Moreover, the group treated with LV-CBh-hGALNS showed a partial correction in the vacuolization of heart structures, which did not reach that of the wild-type level (Supplementary Table S3 and Fig. 6A, B).
Trabecular and cortical bone morphology
To analyze the trabecular and cortical bone structure following treatments with LVs, micro-CT was performed in the femur of treated and untreated Galns−/− and wild-type control groups at 16 weeks (Fig. 7). The representative group of data demonstrated that the LV-treated and control groups did not show apparent statistical differences in trabecular BV, trabecular number, trabecular separation, trabecular thickness, BMD, and degree of anisotropy (Fig. 7A–E). Similarly, no statistical difference was determined between LV-treated and untreated Galns−/− groups regarding the cortical bone parameters (Fig. 7M–R), except for an increase in the BMD of cortical bone, which was found statistically significant (Supplementary Fig S7S). Overall, the treatment with LV-COL2A1-hGALNS showed a slight improvement in trabecular and cortical bone formation compared to untreated and wild-type groups; however, no statistical significance was found (Fig. 7I–L and U–W).
The expression of the hGALNS enzyme reduced the accumulation of KS, increasing the expression of procollagen in bone and liver
To explore the expression of GALNS enzymes and alterations of KS and collagen levels under each LV, we stained the liver and bone (tibia) samples with anti-GALNS (Fig. 8A–C), anti-KS, and anti-collagen antibodies (Supplementary Fig. S3). In the tibia growth plate, we found that the GALNS positive area was ∼50% under both LV-CBh-hGALNS and LV-COL2A1-hGALNS compared to the wild-type level (100%) (Fig. 8B). However, this expression was not sufficient to fully correct bone pathology. According to protein concentration in bone, the LV-COL2A1-hGALNS-treated group had an elevation in the protein concentration compared to the untreated group, and this slight elevation was found to be significant compared to the wild-type (Supplementary Fig. S3). In the liver, the expression of the GALNS enzyme significantly increased under treatment with LV-CBh-hGALNS (∼204%), which was followed by LV-COL2A1-hGALNS by ∼90% compared to the wild-type level (Fig. 8C). This increase was found correlated with the enzyme activity in liver, but not with VCN since both vectors had similar copy numbers per genome.
A total anti-GALNS antibody elevation was observed under LV-HSC-GT
To investigate the immune reaction following the GALNS transgene expression, ELISA was performed using plasma samples of wild-type, treated, and untreated MPS IVA mice. Treated groups with LV-COL2A1-hGALNS showed significant elevation in the circulating anti-GALNS antibodies overtime starting from 6 weeks of age by 3.06 ± 0.7 compared to control groups (untreated MPS IVA mice: ∼0.01 ± 0.02 and wild-type mice: ∼0.11 ± 0.12). However, mice treated with LV-CBh-hGALNS showed significant up-and-down variations during treatment. Moreover, anti-GALNS antibodies were undetectable in the allo-HSCT group (Supplementary Figs. S4 and Figs. S5). We thought that the undetectable/weak production of the GALNS enzyme might not trigger a strong immune reaction in allo-HSCT compared to LV-HSC-GT groups.
Body weight did not represent the effectiveness of the treatment
After treatment, the body weight of mice from each group was measured weekly. The data showed that the LV-COL2A1-hGALNS or LV-CBh-hGALNS modified HSCs treated group significantly increased the body weight from the day of injection to the third week. This initial increase in body weight could indicate an initial positive response to the treatment. However, the subsequent decrease in body weight compared to untreated and wild-type controls from the 4th to the 16th week suggests that the treatment may not sustain body weight. In addition, no significant difference was found between the LV-COL2A1-hGALNS modified HSCs treated or allo-HSCT group and an untreated group from the 4th to the 16th week. LV-CBh-hGALNS modified HSCs treated group remained under the body weight level of the untreated group (Supplementary Fig. S8). We assumed that busulfan administration might have a detrimental effect on the low body weight over 16 weeks.
DISCUSSION
We have conducted the first preclinical study of ex vivo LV-GT on the MPS IVA mouse model. In this study, we designed third generation SIN LVs under ubiquitous (CBh) or tissue-specific (COL2A1) promoters. To verify the effectiveness of each LV, MPS IVA patients’ fibroblasts were transduced, in which both LVs significantly increased the intra- and extracellular GALNS enzyme activity at supraphysiological levels. Among these vectors, LV-COL2A1-HGALNS was the best LV to drive the GALNS expression in human fibroblasts. However, these increases were not associated with higher VCN. Indeed, LV-CBh-hGALNS had significantly higher VCN than LV-COL2A1-HGALNS, although the GALNS enzyme activity was the opposite. Therefore, we concluded that high copies might not promote higher gene expression. Another critical question was whether high GALNS expression resulted in cytotoxicity in these fibroblasts. High levels of the GALNS enzyme did not trigger any cytotoxicity.
Previous studies assessed the effectiveness of ubiquitous and myeloid-specific promoters under retroviral vectors in different cell lines (HL-60, Jurkat, HeLa, and CD34+ cells), indicating high expression of transgenes driven by ubiquitous promoters (CMV, MoMuLV LTR, SV40, or PGK) rather than myeloid promoters (CD11b, CD18, CD19, or CD34). 32,57 It is crucial to consider the natural course of diseases when designing LVs. For example, ubiquitous promoter CMV and MoMuLV LTR, despite their elevated transgene levels, are lineage nonspecific. With such promoters, HSCs may not be efficiently transduced. However, the lineage-specific CD11b promoter has shown promising outcomes. This information engages us in the design considerations for LVs. 30,39
To set an appropriate culture condition and maintain the stemness of isolated HSCs in cultures during analysis, we tried several medium conditions, one of which helped maintain HSCs and better proliferate with no differentiation. In this way, we eliminated potential pitfalls during the period required to transduce HSC before transplantation. Upgraded medium 2 increased long-term culturing efficiency in HSCs and gave us a high HSC yield to perform all in vitro analyses, 38 but not a high expression of GALNS enzyme after LV transduction in Galns−/− HSCs. Encountering this obstacle guided us in assessing LVs in HSCs derived from wild-type mice. We suspected that LVs might not fully treat Galns−/− HSCs because a large deletion in the GALNS gene was introduced into this mouse model. Under the same transduction conditions, wild-type HSCs were transduced with each LV, and we confirmed a significant overexpression of the GALNS enzyme under both LVs compared to untransduced wild-type HSCs. Surprisingly, the LV-COL2A1-HGALNS was significantly higher than the LV-CBh-hGALNS in wild-type HSCs, while the opposite was for Galns−/− HSCs. Overall, the activity of the GALNS enzyme was detected in all experiments, even using Galns−/− HSCs having such a large deletion.
We found similar VCNs in Galns−/− and wild-type HSCs 24 h and 15 days after transduction with both LVs. However, on day 15, we observed a significant reduction in VCN under LV-CBh-hGALNS, while no change was seen in LV-COL2A1-HGALNS. Detecting VCN a few days after transduction is critical to eliminate false results, as VCN detected 24 h post-transduction may not accurately reflect the viral copies. The results in wild-type HSCs also confirmed that the LV-CBh-hGALNS had a low VCN compatible with the result in Galns−/− HSCs. These LV transductions did not affect the CFU colonies of Galns−/− and wild-type HSCs. The results of enzyme activities, VCNs, and CFU colonies were promising to pursue our aims in vivo. Although the GALNS enzyme activity increased under both treatments in the differentiated cells, VCNs stayed at similar levels as those in HSCs. This led us to consider that increased enzyme activity may not be associated with VCN. A recent study analyzed CD34+ HSCs transduced with LV-CD11b, showing the effect of high VCN on the IDS enzyme activity. 58 However, the question of how high VCN can trigger high expression remains, specifically in the context of LVs, as they threaten insertional mutagenesis. These findings underscore the need for further research and the potential for new insights and discoveries.
After confirming the effectiveness of LVs, we moved on to analyzing the results of the studies conducted on mice. The Galns−/− mice used in the study 59 exhibited a severe pathological condition at a later stage, similar to those of patients with severe MPS IVA. 60 Due to the age-related therapeutic response and the increased severity in MPS IVA mice, we introduced LV-engineered Galns−/− HSCs into busulfan-conditioned Galns−/− mice at the neonatal stage. Early treatment in this mouse model provided advantages in reaching targeted organs, particularly the bone, which is a key site of pathology in MPS IVA.
LV-CBh GT increased enzyme activity, but it was insufficient to fully correct the deficient enzyme, as it remained lower than the levels found in the wild-type mice. In the allo-HSCT group, we could only detect GALNS enzyme activity in the liver and spleen at traceable levels and not in other tissues. These findings were confirmed by testing mono-sulfated KS in plasma, leukocytes, and bone. There was a notable reduction in mono-sulfated KS levels in plasma under both LVs. However, only the LV-COL2A1 group exhibited a significant decrease in leukocytes and bone compared to untreated controls, and no significant differences were found in mono-sulfated KS levels between the LV-COL2A1 and wild-type groups.
It was crucial to investigate why mono-sulfated KS levels were reduced more in the LV-COL2A1 group than in the LV-CBh-HSC group, even though the GALNS enzyme activity was lower in LV-COL2A1 GT. On the contrary, allo-HSCT mice did not show significant changes in mono-sulfated KS levels in plasma, leukocytes, and bone. It is important to note that our analysis was limited due to certain unexpected limitations. For instance, we could only analyze the VCN in the liver after transplantation and found that the VCN was low in the liver. We could not analyze VCN in the bone marrow due to the limited amount available. These limitations should be considered when interpreting the results of the allo-HSCT group.
In summary, LV-HSC-GT demonstrated better outcomes than allo-HSCT but did not reach the normal physiological level of the GALNS enzyme. Current therapies such as ERT and HSCT have shown some improvement in disease progression but are limited in their ability to improve bone growth. 3,61 –63 Considering all these factors, our experimental design may still require improved LVs or busulfan conditioning to achieve supraphysiological levels. This study has unveiled a promising potential for the treatment of MPS IVA. We observed a partial correction of bone pathology and a complete correction in heart pathology (heart valves and muscle) in the MPS IVA mouse model. The determined number (0.5–0.9 × 106) of HSCs modified by the LV-COL2A1-hGALNS showed significant improvements, while slight enhancements were noted under treatment with LV-CBh-hGALNS. The life-threatening condition of impaired cardiac function in patients with MPS IVA, causing high morbidity and mortality, is a significant challenge. 14,64 However, our heart pathology data provides a ray of hope, encouraging us to enhance viral constructs to achieve better results in correcting bone pathology. The sustained and persistent GALNS enzyme expression, via modified HSCs with LVs, into the blood circulation could improve both soft- and hard-to-reach tissues by directly targeting and cross-correcting.
To assess the preconditioning with busulfan on immune tolerance induction, we analyzed total antibody levels to the GALNS enzyme. The data showed highly elevated anti-GALNS antibody titers at 16 weeks post-transplantation (Supplementary Fig. S4–S6). Recent reports suggest busulfan as a potential myeloablative agent with varying doses, injection routes, and administration periods depending on diseases. 32,65 –67 We believe that busulfan conditioning was insufficient to eliminate the existing cell niche. Therefore, an elevation in total anti-GALNS antibody levels may affect the activity of the GALNS enzyme (Supplementary Fig. S5–S7). Anti-transgene antibodies are an unmet challenge, particularly in GT approaches. Sawamoto et al. analyzed anti-GALNS antibodies in MPS IVA Galns−/− and immune-tolerant (MTOL) mice following direct infused AAV8 GT. Galns−/− mice had significantly higher circulating anti-hGALNS antibodies, while no antibodies were detected in MTOL mice. 14 Similarly, these anti-transgene neutralizing antibodies were reported in Pompe mice, detrimental to engraftment and vector copies. To overcome this, LVs-modified allogeneic stem cells expressing acid α-glucosidase (GAA) were transplanted at the subtherapeutic level in Pompe mice conditioned with total body irradiation 4 weeks before high doses of ERT administration. As a result, LV-GT allowed ERT to effectively reduce glycogen storage and restore skeletal function by inducing immune tolerance in Pompe mice. 68 We assumed that the elevation of the circulating total anti-GALNS antibodies might have eliminated the secreted GALNS enzyme. Further experiments are required to confirm the necessary preconditioning dose with busulfan or derivatives. In our study, we could not analyze the chimerism of the engraftment after transplantation, which is a limitation of the study; however, the lower enzyme activity in LV-transduced HSCs compared to literature indicates that the efficiency of transduction is a critical factor in the elevation of enzyme activity. 30,31 Therefore, our future goal is to design better LV vectors to increase transduction efficiency. However, some recent studies revealed that HSCs constitutively express interferon (IFN)-stimulating genes (ISGs) to eliminate viral infections, and these ISGs mediate anti-viral resistance in vivo and ex vivo settings of therapies. 69 IFN binding transmembrane protein (IFITM3) was described in HSCs, stating that IFITM3 inhibited the entry of LVs into HSCs. The degradation of this membrane protein significantly enhanced gene transfer and gene editing. 70,71 Furthermore, clinically relevant VCNs were shown as 1 per genome in HSCs in which innate immune-associated IFITM3 was degraded by cyclosporin H, and the culture plates were supplemented with deoxynucleotides. With all these ways described here, transduction efficiency and engraftment potential increased while preserving the quiescence and stem cell properties of HSCs. 69 –71
The current data has not only encouraged us but also reinforced our unwavering commitment to investigating and improving our approach. With this determination, we have taken further steps to modify our LVs by replacing an appropriate mammalian promoter. This will be followed by the transduction of HSCs and the transplantation of modified HSCs into preclinical models, including MPS IVA mice and rats. This research suggests that further development of LV-HSC GTs as a treatment for MPS IVA patients in a clinical setting is warranted.
CONCLUSIONS
Ex vivo LV-HSC-GT is a potential approach to treating genetic diseases. Our study is the first report on administering LVs-modified HSCs expressing the GALNS transgene in MPS IVA mice. We demonstrate the therapeutic effects of ex vivo LV-GT with the IV infusion of transduced HSC, providing crucial information for optimizing future preclinical studies in MPS IVA.
Footnotes
DATA AVAILABILITY STATEMENT
Data generated for this project is included in the Supplementary Material. Additional data are available from the corresponding author upon reasonable request.
AUTHORS’ CONTRIBUTIONS
B.C.: Conceptualization. E.R. and S.T.: Methodology. B.C., E.R., S.K., and N.S.: Validation. B.C.: Formal analysis. B.C.: Investigation. B.C.: Resources. S.T.: Data curation. B.C. and S.K.: Writing—original draft preparation. B.C.: Writing—review and editing. S.T.: Visualization. B.C.: Supervision. S.T.: Project administration. S.T.: Funding acquisition. All authors have read and agreed to the published version of the article.
AUTHOR DISCLOSURE
All authors declared no conflicts of interest.
FUNDING INFORMATION
B.C. was supported by the National MPS Society (ID 4892) and the Turkish Ministry of Education (YLSY2017-Doctoral Scholarship). This work was also supported by grants from the Austrian MPS society, A Cure for Robert Inc., The Carol Ann Foundation, Angelo R. Cali & Mary V. Cali Family Foundation, Inc., The Vain and Harry Fish Foundation, Inc., The Bennett Foundation, Jacob Randall Foundation, and Nemours Funds. S.T. was supported by an Institutional Development Award from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (NICHD) (
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.