
Abstract
Severe combined immunodeficiency (SCID) encompasses rare primary immunodeficiency disorders characterized by deficient T-cell development, which leads to a severely compromised immune system and susceptibility to life-threatening infections. Among SCID subtypes, IL7RA-SCID is caused by mutations in the interleukin 7 receptor alpha chain (IL7RA) and represents a significant subset of patients with limited treatment options. This study investigated the efficacy of a self-inactivating (SIN) alpharetroviral vector (ARV) engineered to deliver a codon-optimized IL7RA cDNA to restore T-cell development in Il7r-knockout mice. We compared the elongation factor 1 alpha short (EFS) promoter and the lymphoid-restricted Lck promoter for their ability to drive IL7RA expression and found that the EFS promoter enabled robust and sustained IL7RA expression that led to the functional rescue of T-lymphopoiesis in vitro and in vivo. Conversely, though effective in vitro, the Lck promoter failed to produce viable T-cell populations in vivo. Our results highlight the potential of using SIN-ARVs as a gene therapy (GT) strategy for treating IL7RA-SCID. Importantly, sustained production of T-lymphocytes was found in both primary and secondary transplant recipient animals with no adverse effects, supporting the safety and feasibility of this approach. Overall, this study provides valuable insights into the development of GT for IL7RA-SCID and underscores the clinical potential of an EFS-driven SIN-ARV to restore IL7RA-deficient immune function.
INTRODUCTION
Severe combined immunodeficiency (SCID) represents a group of rare, life-threatening primary immunodeficiency disorders that predominantly affect young children, with an occurrence of approximately one in every 54,000 newborns in Germany (similar to global rates). 1 SCID is characterized by a profound defect in T-cell development or function and is often associated with deficiencies in B and/or natural killer (NK) cells. Due to compromised adaptive immunity, patients with SCID are highly susceptible to recurrent infections from a variety of pathogens, including bacteria, viruses, fungi, and protozoa. Clinical manifestations typically appear by 6 months of age or earlier, and without intervention such as bone marrow transplantation, these children rarely survive beyond their first year. 2
Most SCID forms result from inherited monogenetic defects involving multiple genes (e.g., IL2RG, ADA, IL7RA, RAG1, RAG2, and JAK3), 3 –6 which can be addressed with gene therapy. 7 Recent and promising clinical trials conducted in several successfully international clinical trials for SCID-X1 (IL2RG-SCID) showed that gene therapy is a valid alternative for SCID treatment. 8 –11 Furthermore, in 2016, a gammaretroviral-based vector to treat adenosine deaminase deficiency-SCID (ADA-SCID) received marketing authorization in Europe, 12 –16 and a trial for recombinase activating gene 1 (RAG1-SCID) has just started, but the development of a clinical gene therapy strategy addressing IL7RA-SCID remains to be shown.
In humans, loss-of-function mutations in the IL7RA gene lead to IL7RA-SCID, which accounts for ∼10–20% of all SCID cases worldwide and is the most common form of T−B+NK+ SCID (OMIM: 608971), 3,17 highlighting the urgent medical need to create a gene therapy option for these patients. The IL7RA gene encodes the α chain of the interleukin-7 receptor (IL7R), which, together with the IL2 common gamma chain (IL2RG), is crucial for T-cell development and function. IL7R signaling promotes T-cell differentiation, survival, maturation, and T-cell receptor (TCR) rearrangement. 18 Disruption of IL7 signaling in Il7r-knockout (Il7r−/−) mice arrests T-cell development at the thymic progenitor stage and B-cell development at the pro-B cell stage, leading to severe immune deficiencies. 19 While hypo-expression can cause immunodeficiency, IL7RA overexpression may arrest cells at progenitor stages, potentially contributing to leukemia/lymphoma formation. 20 –22
Previous preclinical studies using gammaretroviral vectors to introduce functional murine Il7r cDNA in Il7r−/− mice showed that unregulated and ectopic expression could lead to adverse effects, such as neutrophil expansion and splenomegaly, possibly due to high IL7RA expression levels driven by the LTR of the oncogenic gammaretroviral vector used in the study. 23 Overexpression of IL7R has been linked to thymoma and other malignancies, highlighting the necessity for regulated IL7RA expression in therapeutic applications.
This study focuses on the development of novel self-inactivating (SIN) alpharetroviral vectors (ARVs) with a safer integration profile 24,25 and a codon-optimized human IL7RA cDNA to treat IL7RA-SCID. We demonstrate the functional rescue of IL7RA using a clinically relevant vector in Il7r−/− mice, driven by the elongation factor 1 alpha short (EFS) promoter, and observed no adverse events in primary and secondary recipient mice, further expanding the gene therapy options for IL7RA-SCID and similar diseases.
MATERIALS AND METHODS
ARV production
ARVs were produced by cloning codon-optimized human IL7RA cDNA into a SIN ARV(pAlpha.SIN.noTATA.promoter.transgene.wPRE). 26 The IL7RA cDNA was codon-optimized to improve expression by adaptation to favored human codon usage and removing cryptic splice and polyadenylation sites. This vector contained either a human elongation factor-1 alpha short promoter (EFS) 27 or a mouse lymphocyte-specific protein tyrosine kinase (Lck) proximal promoter 28 to drive IL7RA expression. Viral supernatants were generated by transient transfection of the ARV along with the codon optimized alpharetroviral gag/pol helper plasmid (pcDNA3.alpha.gag/pol.co) 29 and the vesicular stomatitis virus envelope glycoprotein envelope plasmid (pMD.G) into 5 × 106 293T cells via a standard calcium phosphate transfection method. 25,26 Viral supernatants were concentrated and stored at −80°C after filtration through Millex-GP 0.22 µm (Millipore). Viral supernatants were titrated on K562 cells, and virus titers were determined by vector copy number (VCN).
Enrichment, cultivation, and transduction of murine lineage-negative cells
Bone marrow (BM) cells from B6.129S7-Il7rtm1Imx /J (Il7r−/−) mice were extracted from femurs and tibias. Lineage-negative (Lin−) cells were isolated through magnetic sorting using lineage-specific antibodies (MojoSort™ Mouse Hematopoietic Progenitor Cell Isolation Kit; Biolegend). The Lin− cells were cultured in hematopoietic stem and progenitors cell (HSPC) expansion medium (StemSpan medium; Stem Cell Technologies) supplemented with STIF cocktail (50 ng/mL murine stem cell factor, 20 ng/mL murine thrombopoietin, 20 ng/mL murine insulin-like growth factor 2, 10 ng/mL human fibroblast growth factor 1; all from Peprotech), 2% penicillin/streptomycin, and 1% glutamine (PAA). For transduction, Lin− cells were stimulated overnight with STIF medium and then transferred to a 96-well round bottom plate incubated in 100 μL of HSPC medium and viral supernatant. To enhance transduction, 1 mg/mL Synperonic® F 108 (Sigma-Aldrich) were added. After 24 h, 90 μL of transduction medium was replaced with fresh HSPC medium, and the cells were harvested the next day for subsequent in vitro differentiation assays or transplantation.
In vitro T-cell differentiation assay
A total of 5 × 104 OP9-DL1 cells were seeded in 12-well plates one day before co-culture. 27 The following day, 2 × 105 transduced or control Lin− cells were co-cultured in minimal essential medium-α (α-MEM) with 20% fetal bovine serum, supplemented with 5 ng/mL human FMS-like tyrosine kinase 3 ligand and 1 ng/mL human interleukin-7 (hIL7). Cultures were harvested every 3–4 days, and hematopoietic cells were passaged onto new OP9-DL1 cell cultures. A small fraction of hematopoietic cells were used for flow cytometric analysis to monitor T-cell development. The concentration of hIL7 was reduced to 0.5 ng/mL 10 days after initiation of the culture.
Animal handling and transplantation
Animals were kept and cared for in accordance with the regulations of the animal facility at Hannover Medical School and the local authority. Recipient mice, aged 4–5 weeks (NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJZtm), were irradiated (1.5 Gy) 1 day before transplantation. Lateral tail vein injection was used to administer 1.0–1.5 × 106 transduced Lin− cells harvested 48 h after transduction. For secondary transplants, frozen total BM cells from primary recipients were thawed, and 1 × 106 BM cells were transplanted into secondary recipients. Recipient mice were provided with drinking water containing 100 mg/mL Ciprofloxacin (Bayer Vital GmbH) for 2 weeks to ensure antimicrobial protection. Peripheral blood (PB) was obtained every 4 weeks after transplantation (weeks 4, 8, 12, 16, and 20) before sacrificing the animals on week 20. Gross anatomy examination was performed to check for any organ abnormalities, including spleen (Spl), thymus, and liver size.
Flow cytometry analysis
Cells harvested from OP9-DL1 culture were blocked with CD16/32 FcγR antibody and stained with CD4 (PerCP-Cy5.5; eBioscience #45–0042-82), CD25 (APC; Biolegend #102012), Gr-1 (AF700; Biolegend # 108422), CD11b (AF700; Biolegend # 101222), CD8 (APC-Cy7; Biolegend #100714), CD44 (PE; Biolegend #103008), hCD127 (PE-Cy7; Biolegend #351320), and mCD127 (BV785; Biolegend #135037). To analyze leukocyte composition from PB, Spl, and BM, erythrocytes were lysed and leukocytes were blocked with CD16/32 FcγR antibody before staining with CD3e (FITC; eBioscience# 11–0031-85), B220 (PerCP-Cy5.5; eBioscience #45-0452-82), CD45.2 (APC; Biolegend #109814), Gr-1 (AF700; Biolegend # 108422), CD11b (AF700; Biolegend # 101222), CD8a (APC-Cy7; Biolegend #100714), CD45.1 (PE; eBioscience #12-0453-83), hCD127 (PE-Cy7; Biolegend #351320), NK 1.1 (PE-Dazzle594; Biolegend #108748), CD4 (BV605; Biolegend #100548), and mCD127 (BV785; Biolegend #135037). Thymocytes were stained with CD8a (PerCP-Cy5.5; BD Bioscience #551162), CD25 (APC; eBioscience #17-0251-82), CD45.2 (AF700; Biolegend #109822), CD45.1 (APC-Cy7; eBioscience #47-0453-82), CD44 (PE; Biolegend #103008), hCD127 (PE-Cy7; Biolegend #351320), CD4 (PE-Dazzle594; Biolegend #100566), and mCD127 (BV785; Biolegend #135037). Dead cells were discriminated with 1 μg/mL of 4′,6-diamidino-2-phenylindole, and cells were analyzed on a CytoFLEX S device (Beckman Coulter GmbH).
VCN determination
VCN was determined using quantitative PCR following established protocols. 30,31 In brief, quantitative real-time PCR was performed on genomic DNA extracted using the blood gDNA extraction kit from Qiagen. ABI Taqman Fast Advanced Master Mix (Thermo Fisher) was used for PCR amplification on a StepOnePlus Real-Time PCR instrument. The vector copies per cell were determined by quantifying the wPRE present in the integrated vector relative to the host gene PTBP2 (polypyrimidine tract-binding protein 2). The primers and probes used for this experiment were as follows: wPRE_fw: GAGGAGTTGTGGCCCGTTGT, wPRE_rev: TGACAGGTGGTGGCAATGCC; wPRE_probe: FAM-CTGTGTTTGCTGACGCAAC-BHQ1. PTBP2_fw TCTCCATTCCCTATGTTCATGC, PTBP2_rev: GTTCCCGCAGAATGGTGAGGTG; PTBP2_probe: JOE-ATGTTCCTCGGACCAACTTG-BHQ1.
RESULTS
Restoration of T-lymphopoiesis in Il7r−/− HSPC via ARV-mediated expression of human IL7RA
We employed either a ubiquitous EFS promoter or a lymphoid-restricted Lck promoter to drive the expression of human IL7RA. As IL7RA-deficiency only affects the T-lymphopoiesis in humans, the Lck promoter was utilized to limit the expression of IL7RA expression in non-T lineages to prevent undesired expansion of other cell types. These vectors were transduced into Il7r−/− HSPC, referred to as the EFS and Lck groups, respectively, prior to co-culturing with OP9-DL1 stromal cells for T-lymphopoiesis differentiation (Fig. 1A). Hematopoietic cells were harvested from the co-culture and transferred to a new stromal culture every 3–4 days, with a small fraction of cells harvested at each passage to analyze T-lymphopoiesis via flow cytometry. Mouse thymopoiesis involves distinct developmental stages characterized by the expression of specific surface antigens. Thymocytes lacking CD4 and CD8 antigens are referred to as double-negative (DN) cells, which can be further subdivided into distinct populations based on the expression of CD44 and CD25: DN1 (CD44+ CD25

IL7RA expression from alpharetroviral vectors restored thymopoiesis of Il7r−/− HSPCs in vitro.
OP9-DL1 differentiation assay recapitulates IL7RA expression dynamics across T-lymphopoiesis stages
The expression level of IL7RA (CD127) is not constant across various stages of T-lymphopoiesis in the thymus. Here, we investigated the expression of murine IL7RA in WT mice at different stages of T-lymphopoiesis using the OP9-DL1 differentiation assay (Fig. 2A), quantified by mean fluorescent intensity (MFI) measured by flow cytometry. We observed that cells at the DN2 stage exhibited the highest CD127 expression levels, which gradually decreased throughout differentiation, reaching a nadir at the DP stage before increasing again at the single-positive stage (at least for CD8 single-positive cells). These findings are consistent with gene expression data from the Immunological Genome Project (ImmGen) microarray datasets GSE15907 (Fig. 2B).

IL7RA expression dynamics across T-lymphopoiesis stages is recapitulated by the OP9-DL1 differentiation assay.
Conversely, human CD127 expressed from our ARVs (Fig. 2C, D) was consistently expressed throughout T-lymphopoiesis at similar levels. Despite differences in expression patterns, both vector designs successfully restored T-lymphopoiesis. CD127 expression levels across T-lymphopoiesis stages in OP9-DL1 cultures were visualized by overlaying fluorescent intensity values on flow cytometry dot plots (Fig. 2E).
Murine CD127 was exclusively detected in the WT group and was confined to the non-myeloid fraction (Gr-1−Mac-1−) in the culture. Notably, DP fractions lacked CD127 expression, with the highest expression observed in DN2 fractions on dot plots. As expected, human CD127 was absent in both WT and Il7r−/− groups, and the expression of human CD127 was observed across all fractions regardless of differentiation status in the EFS group. A similar expression pattern was seen for the Lck group, except for the absence of CD127 signal in Gr-1+ and Mac-1+ DP fractions and DN1 fractions, highlighting the restricted expression of the Lck promoter in T-lineage cells.
Similarly, in the total population of each differentiation assay, human CD127 expression was undetectable in the non-transduced groups (Supplementary Fig. S2A). The percentages of human CD127-positive cells varied across different time points and duplicate wells (Supplementary Fig. S2B), likely due to the biological expansion of various populations (DN1-DN4, DP, SP, and myeloid) at different stages of the culture. Furthermore, even though the EFS group had a slightly higher initial percentage of transduced cells as compared with the Lck group (Supplementary Fig. S2C), the Lck group produced a higher percentage of hCD127-positive progeny than the EFS group, as measured by flow cytometry (Supplementary Fig. S2B) and mean VCN (Supplementary Fig. S2C).
Ex vivo application of ARV gene therapy resulted in in vivo reconstitution of Il7ra−/− HSPC T-cell development
To evaluate the vectors in vivo, Il7r−/− HSPC (CD45.2) were transduced with the therapeutic ARVs and transplanted into NSG recipient (CD45.1) (Supplementary Fig. S3). CD3e+ (CD4 and CD8) cells were detected in the PB of EFS group mice between weeks 8 and 12 after transplantation. At week 20 after transplantation, T-lymphocyte levels in the EFS group (10–15%) were about half of those in WT (19–35%) animals (Fig. 3A), and the CD4 and CD8 T-lymphocyte ratios were about 1:1 in both the EFS group and the WT animals (Fig. 3B, C). Interestingly, the Lck-driven vector that slightly outperformed the EFS vector in restoring T-lymphopoises in vitro failed in vivo, yielding no CD3 cells in recipient mice.

Reconstitution of Il7r−/− HSPC T-cell development in vivo with ARVs. Donor-derived cells were stained for
Il7r-deficient mice had impaired B-lymphopoiesis as compared with WT mice; however, no increase in B220 cells was observed within our gene therapy groups (Fig. 3D). Gating on the limited B220+ cells revealed that, except in the WT group, the majority of these cells are NK1.1+, indicating their NK cell origins (Fig. 3E, Supplementary Fig. S4). In fact, without the B- and T-cells, NK cells occupied a significant portion of the PB as shown in the Lck-group and Il7r−/− PB (Fig. 3F). Recovery of the T-cell population in Il7r−/− mice with the EFS vector restored the proportion of NK1.1+ cells back to the level observed in WT PB.
Although increased percentages of myeloid cells (Gr-1+ Mac-1+) were observed in mice transplanted with the ARVs (both EFS and Lck) compared with WT animals (Fig. 3G), this was also observed in the Il7r−/− animals. However, the total white blood cell (WBC) count (Fig. 3H) in both vector transduced groups remained within the normal range (2 × 103−104 cells/mm3) 32 20 weeks after transplantation with gene-modified HSPC.
Restoration of thymus in NSG by repopulating with GT cells
Recipient NSG mice typically have a vestigial thymus due to the lack of interaction between thymocytes and thymic epithelial cells. However, upon transplantation with the GT ARV expressing IL7RA from an EFS promoter, a visible thymus was observed in the animals sacrificed at week 20 after transplantation (Fig. 4A). Subsequent analysis of these thymuses revealed the development of all stages during thymopoiesis (Fig. 4B, Supplementary Fig. S5), with the proportion of each population comparable to that of WT mice (Fig. 4C). Notably, no visible thymuses were found in NSG mice transplanted with HSPCs transduced with the Lck promoter-driven IL7RA vector, nor in Il7r−/− mice. Interestingly, the exogenously expressed human IL7RA (CD127) from the EFS promoter exhibited a similar expression pattern in various stages of thymopoiesis to that of WT thymocytes (Fig. 4D, E).
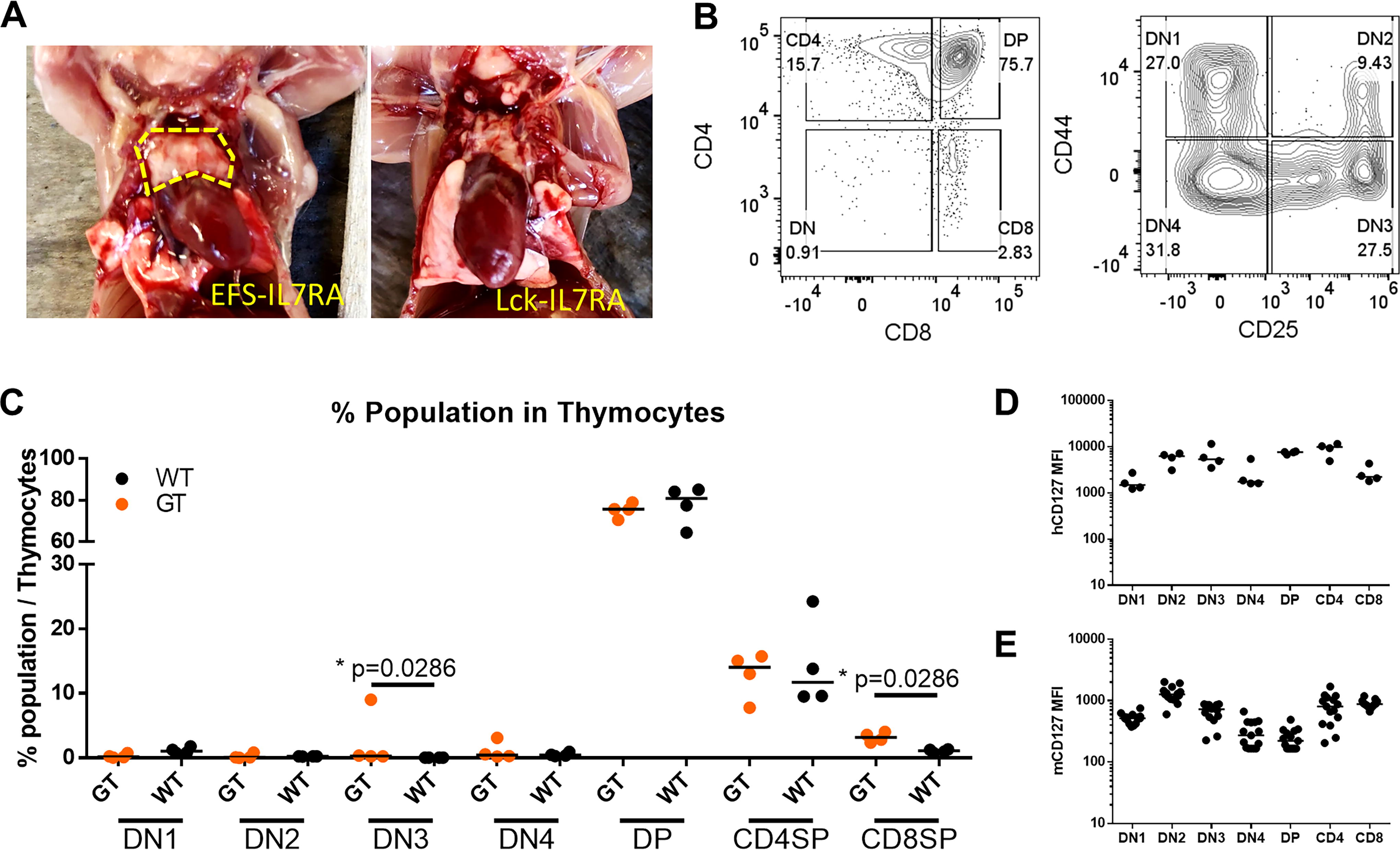
Restoration of NSG thymus after transplantation with gene therapy cells. Visible thymus recovery was seen in NSG mice from the
Persistence and T-lineage commitment of GT-cells in secondary NSG recipients
BM from primary recipients of the EFS group was transplanted into secondary NSG recipients. GT-cells successfully engrafted as evidenced by detection in multiple lineages in the secondary recipients, including T-lymphocytes, with the exception of the B-lineage. Most recipients rapidly achieved high chimerism levels, with donor cells predominantly responsible for hematopoiesis from week 4 post-transplantation onward (Fig. 5A). CD3e+ T-lymphocytes were detectable in PB as early as week 8 (median 1.25%) with continued expansion until the experimental endpoint at week 20 (median 9%) and contained both CD4+ and CD8+ cells (Fig. 5B–D). In one animal, the donor cells were below detection levels in PB and BM at week 20, but donor T-cells were detected in the Spl. The mean VCN in PB was 1 and 0.9 in the Spl and BM, indicating the persistence of GT cells in long-term hematopoiesis. Importantly, no adverse effects were observed in the secondary recipient animals.
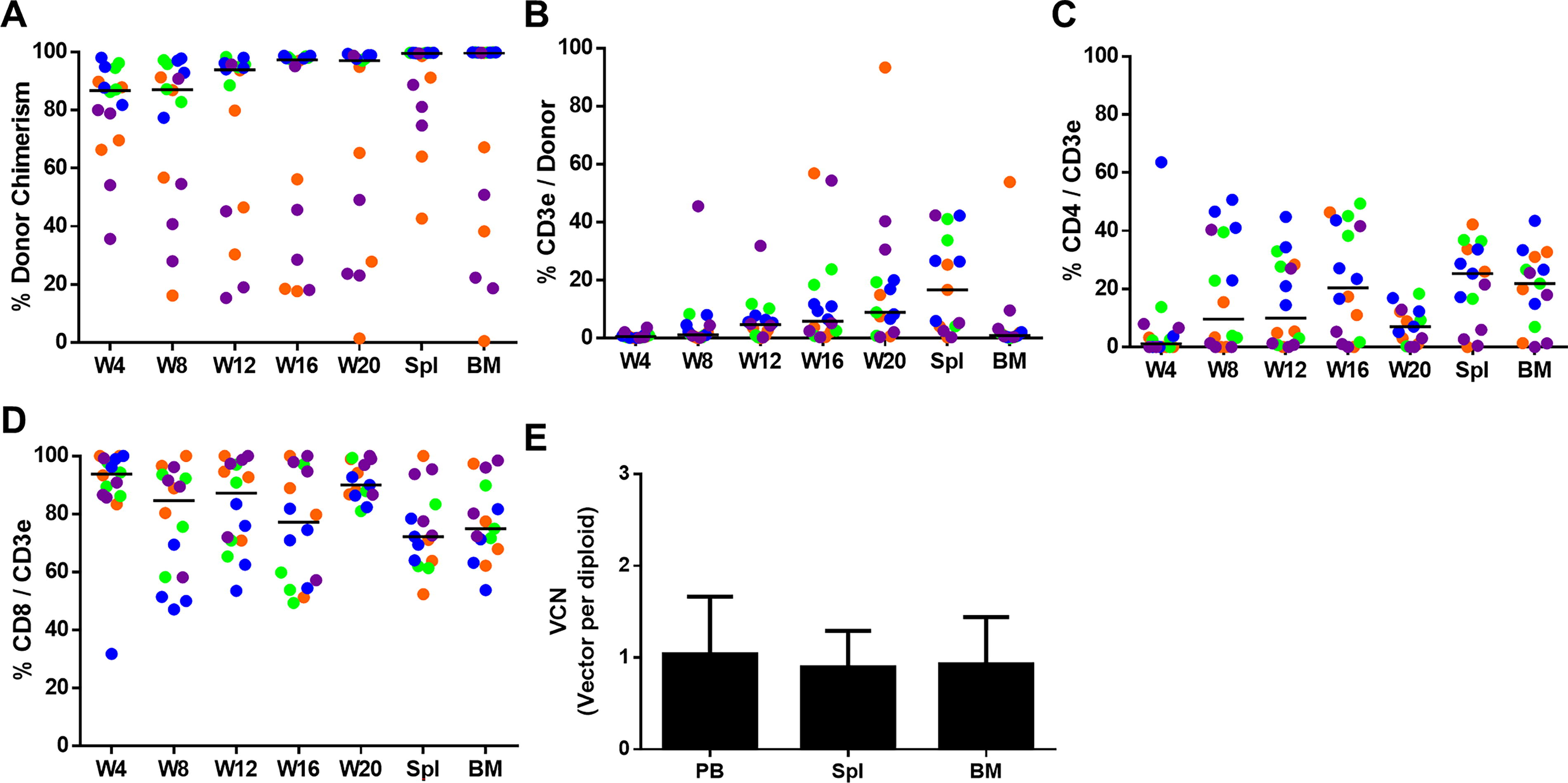
EFS-IL7RA-modified-HSPC contribute to long-term hematopoiesis.
DISCUSSION
We have demonstrated the functional rescue of murine Il7r-deficient HSPCs using SIN-ARVs 26 expressing human IL7RA. Using the EFS promoter, our SIN-ARV successfully restored T-cell development both in vitro and in vivo, demonstrating the feasibility of this novel gene therapy strategy for IL7RA-SCID.
Our in vitro OP9-DL1 differentiation assay recapitulated the dynamic expression of murine IL7RA across various stages of T-lymphopoiesis, revealing a peak in CD127 expression at the DN2 stage and a subsequent decline at the DP stage, which aligns with previously reported data from the Immunological Genome Project 33 (Fig. 2). These data indicate that in vitro assays, such as OP9-DL1-mediated T-cell differentiation assays 34,35 or artificial thymic organoid, 36 are capable of recapitulating the dynamic expression of IL7RA. Notably, vectors driven by the EFS and Lck promoter restored T-lymphopoiesis despite the almost constant IL7RA expression levels from these vectors throughout the T-cell differentiation stages (Fig. 2).
The in vivo experiments further substantiated the efficacy of the EFS-driven IL7RA expression to restore lymphopoiesis. EFS vector-transduced HSPCs led to the development of CD3e+ T-cells in PB, albeit at lower levels compared with WT mice (Fig. 3A). The ratio of CD4+ to CD8+ T cells remained comparable (Fig. 3B, C), indicating a successful physiological balance in the restoration of T-lymphocytes. The failure of the Lck-driven vector in vivo underscores the importance of conducting in vivo GT studies. As the Lck gene is mainly expressed in T-lymphocytes, we speculate that IL7RA expression under its promoter might not provide sufficient IL7 signaling during early stages of differentiation to support the development of HSPCs into lymphoid progenitors (e.g., common lymphoid progenitor) that populate the thymus.
Interestingly, B-cells were not detected in our GT mice (Fig. 3D), and the limited B220+ cells found in PB were mostly positive for NK1.1 (Fig. 3E), indicating that these are a subfraction of B220+ NK cells. Similarly, low B-cell recovery was also observed in an earlier study in which Lin− HSPC were transduced with a gammaretroviral vector to deliver murine Il7r cDNA, suggesting that delivery of the human IL7RA cDNA was not the reason for the low B-cell levels. 23 In contrast to their study, we did not observe evidence of splenomegaly in our transplanted animals (Supplementary Fig. S6), indicating that abnormal myeloid expansion does not occur in our transplanted animals. More importantly, despite observing an increased proportion of myeloid cells in GT mice (Fig. 3G), the total WBC counts remained within normal ranges (Fig. 3H). This increase in the myeloid proportion in GT groups (Fig. 3G) resulted from the lack of B-cells, which comprised a significant proportion in the WT PB (Fig. 3D) and is also reflected in the Il7r−/− animals. This suggests that the observed proportional changes are due to the lack of certain WBC populations rather than aberrant myeloid expansion, highlighting the potential low genotoxic nature of the SIN-ARV.
Thymic reconstitution in NSG mice transplanted with EFS-IL7RA HSPCs was notable, with visible thymus formation and the presence of all thymopoiesis stages, which closely reflects WT thymus development (Fig. 4). This finding highlights the potential of our gene therapy approach to restore thymopoiesis and generation of peripheral T-cell populations.
Lastly, the persistence of gene therapy cells in secondary recipients following serial transplantation suggests the potential long-term stability and self-renewal capability of the transduced HSPCs. The ability of these cells to contribute to multiple hematopoietic lineages and sustain T-cell production without noticeable adverse effects, such as abnormal blood counts, extreme lineage skewing, or irregular organ size and color, supports the promise of our gene therapy approach (Fig. 5).
However, our study is not without its limitations. The study utilized an SIN-ARV with a clinical configuration expressing human IL7RA cDNA. Although murine and human IL7 and IL7R are cross-reactive, the strength of downstream signaling activation may differ, as reported by Johnson et al. 37 Nevertheless, the mismatching of human IL7RA, murine IL2RG, and murine IL7 can confer adequate IL7 signaling in regenerating T-lymphocytes in Il7r−/− HSPCs, as demonstrated in this study and in an abstract by Triebwasser et al. published an elegant presented at the 2021 ASH meeting. 38
Further complicating the situation, IL7RA also functions as the co-receptor for thymic stromal lymphopoietin (TSLP), which signals through a heterodimeric receptor complex with the TSLP receptor encoded by the CRLF2 gene. 39,40 TSLP signaling is primarily active in B and myeloid cells, and its deregulation is associated with leukemia. 41 However, TSLP does not cross-react between mouse and human cells due to only 43% homology between the TSLPs of the two species. 42 Consequently, overstimulation of TSLP signaling is unlikely to occur in our current in vivo model, and thus, we would not observe adverse events such as leukemia driven by this pathway.
Restricting IL7RA expression to T-lymphocytes in gene therapy may further improve the safety profile. However, the T cell-specific Lck promoter did not lead to the recovery of T-cell development in this study. During the preparation of this article, Rai et al. published an elegant and promising CRISPR/Cas9-based homology-directed repair (HDR) strategy for IL7RA-SCID, which could represent a potentially translatable curative approach. 43 However, it should also be noted that recent studies have seen potential challenges with CRISPR/Cas9-based HDR, including low HDR efficiency and off-target effects. Additionally, there have been observations of reduced repopulating potential and graft clonality in CRISPR/Cas9-based HDR edited HSPCs, which should be addressed for translation. 44,45
Although these challenges can be mitigated with advancements in the field, alternative strategies could only benefit patients. Alpharetroviral SIN vectors were shown to have a comparatively neutral integration pattern compared with the gammaretroviral and lentiviral vectors that have been utilized in clinical settings. 24,25 With recent reports of rare cases of MDS and clonal expansion 46 –49 from clinical trials that used the relatively safe lentiviral vectors, the more neutral ARVs may provide a safer option, as they mainly target intergenic regions of the genome as opposed to gammaretroviral vectors that favor promoter/enhancer and transcription start sites and HIV-derived lentiviral vectors that favor introns and exons of genes. 24,25
In conclusion, our study represents a step forward in the development of gene therapy for IL7RA-SCID. The successful restoration of T-cell development using an EFS-driven SIN-ARV offers a promising therapeutic strategy with potential clinical applicability and further broadens the gene therapy options that can be developed to help these patients.
Footnotes
ACKNOWLEDGMENTS
The authors gratefully acknowledge Maike Stahlhut, Nicole Dörpmund, and Melanie John for their invaluable assistance in this study. Additionally, the authors would like to thank Tobias Mätzig, Michael Rothe, and Melanie Galla for their insightful suggestions and critiques.
AUTHORS’ CONTRIBUTIONS
T.-C.H.: Conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—reviewing and editing, visualization, and project administration. M.A.M.: Writing—original draft, writing—reviewing and editing, and visualization. A.J.T.: Supervision, funding acquisition, conceptualization, and writing—reviewing and editing. A.S.: Supervision, funding acquisition, conceptualization, resources, and writing—reviewing and editing.
AUTHOR DISCLOSURE
A.S. is a consultant for Apriligen and Boehringer Ingelheim and the co-inventor on a patent for ARV technologies (patent no. WO/2010/130844). A.J.T. is on the Scientific Advisory Board of Orchard Therapeutics and Rocket Pharmaceuticals. The other authors declare no competing interests.
FUNDING INFORMATION
This study received funding from EU H2020 grants
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.