
Abstract
All current market-approved gene therapy medical products for in vivo gene therapy of monogenic diseases rely on adeno-associated virus (AAV) vectors. Advances in gene editing technologies and vector engineering have expanded the spectrum of target cells and, thus, diseases that can be addressed. Consequently, AAV vectors are now being explored to modify cells of the hematopoietic system, including hematopoietic stem and progenitor cells (HSPCs), to develop novel strategies to treat monogenic diseases, but also to generate cell- and vaccine-based immunotherapies. However, the cell types that represent important new targets for the AAV vector system are centrally involved in immune responses against the vector and its transgene product as discussed briefly in the first part of this review. In the second part, studies exploring AAV vectors for genetic engineering of HSPCs, T and B lymphocytes, and beyond are presented.
INTRODUCTION
Gene therapy has entered clinical practice for distinct indications in the areas of rare monogenic diseases and chimeric antigen receptor (CAR) T cell–based immunotherapies for malignant diseases (i.e., B cell lymphomas and multiple myeloma). In both of these areas, therapeutic options have greatly expanded due to the progress in delivery tools and genome editing technologies. 1 Moreover, the field is clearly moving beyond its traditional “fields of action” and is starting to address more common monogenic, polygenic, and infectious diseases. Another prominent extension of the field is vector-based vaccine development, which has been driven largely by global health care challenges such as the COVID pandemic.
Analyses of gene therapy medical products (GTMPs) for in vivo gene therapy clearly indicate vectors based on the adeno-associated virus (AAV) as the dominant class of GTMPs on the market.
1,2
Alipogene tiparvovec—approved to treat patients with lipoprotein lipase deficiency—was the first GTMP that received market approval in the Western world.
1
Following this landmark event in 1 October 2012, seven further in vivo GTMPs entered the market in Europe and/or the United States.
2
These are either applied to the patients’ tissue locally, such as treatments of inherited retinal dystrophy caused by RPE65 gene mutations (Voretigen neparvovec; subretinal application) or aromatic

The AAV vector system. AAV vectors are composed of a nonenveloped protein capsid and a single-stranded DNA. The viral genome of approximately 4.7 kb as depicted here for AAV serotype 2 (AAV2) is flanked at both ends by inverted terminal repeats (ITRs) serving as origin of replication and packaging signals. 3 The rep gene encodes the viral Rep proteins (Rep78, Rep68, Rep52, and Rep40), responsible for transcriptional control of AAV specific promoters, for AAV replication, genome packaging, and integration of the wild-type AAV genome into the host genome. 3 Transcription of the rep gene is controlled by p5 and p19 promoters, respectively. The cap gene encodes the capsid proteins (VP1, VP2, and VP3) and—in alternative open reading frames—the nonstructural proteins assembly activating protein (AAP) 3 and membrane associated accessory protein (MAAP). While the former supports capsid assembly, MAAP is involved in control of viral replication and egress. 4 Transcription of the cap gene is controlled by the p40 promoter. In AAV vectors, viral genes are replaced by the transgene expression cassette or the HDR template sequence. Two vector genome configurations are explored 5 : (1) the single-stranded AAV vector genome configuration that resembles the natural genome configuration. Sense and antisense versions of this vector genome are packaged with equal efficiency into AAV capsids. Within the cell, single-standed AAV vector genomes have to be converted into a DNA double-strand either by second-strand synthesis or by hybridization of two strands of opposite polarity. (2) The self-complementary AAV vector genome configuration is packaged as single-stranded DNA, but forms a DNA double-strand upon uncoating. This is possible because sense and antisense sequences are contained on the same vector genome separated by an additional ITR. AAV, adeno-associated virus.
Since AAV serotypes differ in receptors used for cell attachment and entry and since the human population differs in the prevalence of serotype-specific anti-AAV antibodies, tissues of different species are screened for the presence of AAV sequences to expand the pool of capsids that can eventually be included into the AAV vector toolbox. 6 The capsid itself is a multiprotein complex formed by three AAV capsid proteins, termed viral protein (VP)1, VP2, and VP3, in an ∼1:1:10 ratio. 7 The capsid determines viral and, thus, also vector tropism and possesses enzymatic activities, as well as signal sequences involved in the intracellular processing of incoming vector particles. 8 In addition, there is increasing evidence that the capsid is involved in transcriptional regulation. 9
The capsid shows an impressive stability, which simplifies manufacturing, storage, and shipment of AAV vectors. 10 Capsid stability might have evolved as an advantageous feature for a virus that depends on the presence of unrelated viruses for efficient progeny production. 3 Specifically, when wild-type AAV infects a cell in the absence of a helper virus coinfection, the capsid protects the viral genome from being recognized by the innate immune system, thereby promoting AAV’s entry into a latent life cycle. 3 The latent phase is turned into a productive infection upon helper virus infection, which releases the repression of viral transcription and initiates viral genome replication. 3 Well-known helper viruses for AAV are members of the adenovirus or herpes virus family, but also other viruses such as papilloma viruses or human bocavirus 1 have been described. 3 In case of helper virus coinfection, which is assumed to be responsible for the seroconversion reported to frequently occur in early childhood, 11 AAV progeny production is immediately induced, interestingly, accompanied by a reduced helper virus progeny production. 3 Despite its advantage for vector manufacturing, capsid stability also has a downside as only a proportion of the incoming vector particles are capable of successfully releasing their genomes, a process termed vector uncoating. 12 –14
With this in mind, huge efforts in basic research and vector development focus on the AAV capsid to decipher the capsid–host interaction in greater detail and to tailor the capsid via protein engineering to better meet the requirement of the envisioned application. Equally important are efforts to engineer first generation vector genomes which are designed to improve transcriptional activity, enable cell type selective expression, reduce innate immune recognition, and address the issue of a limited packaging capacity as AAV capsids can only harbor vector genome sizes of ∼5 kb in a nontruncated manner. 2
AAV VECTORS AS TARGETS OF IMMUNE CELLS DERIVED FROM THE HEMATOPOIETIC SYSTEM
Most in vivo applications of AAV vectors are focused on the central nervous system, eye, muscle, and liver. Vector doses between 10e11 and 10e14 per kg body weight are applied to reach therapeutically relevant levels of transgene expression in the target cell type. These high doses are recognized by the host immune system and elicit de novo anti-vector responses, which are largely mediated by members of the toll-like receptor (TLR) family.
TLRs are pattern recognition receptors that have evolved to detect microbes, including viruses, based on conserved patterns termed pathogen associated molecular patterns (PAMP), which distinguish intruders from the host. 15 All TLRs, except TLR3, use the same cytoplasmic adaptor, myeloid differentiation primary response protein 88 (MyD88), to transmit information about the presence of PAMP to downstream mediators, such as nuclear factor kappa B (NFκB), adaptor protein complex 1, or interferon regulatory factors, thus mounting an innate immune response through upregulation of innate immune-related gene expression. 15
Zhu and colleagues were the first to report on the importance of TLR9 in sensing AAV vectors (Fig. 2). 16 This TLR is located in the endosomal compartment of specific cell types, such as plasmacytoid dendritic cells (pDC), and is activated by single-stranded (ss) DNA, preferentially ssDNA containing unmethylated CpG motifs. 19 Accordingly, Zhu and colleagues reported that pDC, but not conventional DC (cDC) or liver macrophages (i.e., Kupffer cells [KC]) reacted toward AAV vectors by generating a type 1 interferon (T1 IFN) response. 16 The latter is specific to pDC and involves interferon regulatory factor 7, interleukin-1 receptor–associated kinase-1, tumor necrosis factor receptor–associated factor 3, IκB kinase α, and osteopontin—all of which are recruited to MyD88. 17 In vivo studies with TLR9−/− and MyD88−/− mice revealed the central importance of this pathway for capsid-specific, as well as transgene product-specific T cell and humoral immune responses. 16 Human pDC isolated from peripheral blood mononuclear cells (PBMC) and stimulated with AAV serotype 2 (AAV2) also reacted by producing T1 IFN mRNA, an effect that could be prevented with TLR9 antagonizing oligonucleotides, which suggests the presence of similar AAV detection mechanisms in mice and humans. 16
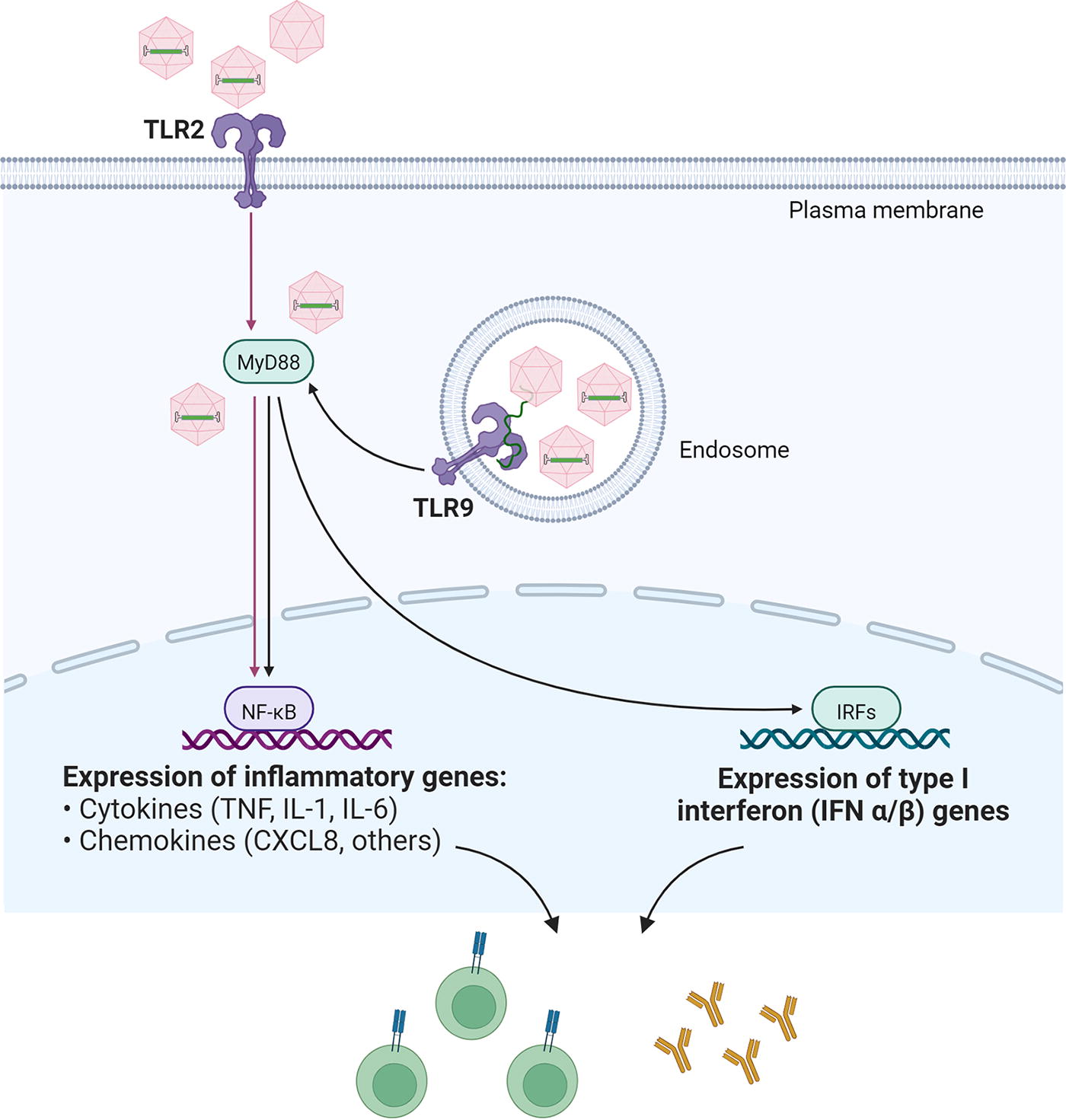
Innate immune sensing of AAV vectors by TLR2 and TLR9. Cells displaying TLR2 on the surface sense vector-containing, as well as empty, AAV capsids, while vector genomes are recognized by TLR9, which is located in the endosomal membrane of distinct cell types, including pDC. TLR2 and TLR9 use MyD88, an adaptor protein located in the cytosol. Depending on the factors recruited to MyD88, an inflammatory and/or type I interferon response is induced upon activation of respective immune response related genes. 16 –18 TLR, toll-like receptor.
In a subsequent study, Martino and colleagues confirmed the important role of TLR9 in triggering immune responses against AAV vectors and reported that vectors delivering a self-complementary (sc) genome configuration (Fig. 1), developed to improve transduction efficacy particularly for in vivo applications, 5 induced elevated adaptive immune responses against the capsid compared with the natural single-stranded (ss) genome configuration. 20 In this study, innate immune responses in mice were found to be at least partially dependent on KC, 20 confirming earlier observations by Zaiss and colleagues. 21 Moreover, neutrophils, macrophages, and natural killer (NK) cells were significantly elevated in the liver following systemic scAAV vector administration. 20 With regard to the latter, increased NK cell infiltrates compared to PBS-treated controls were also reported following local subretinal application of ssAAV8 in a murine model, indicating that NK cells were attracted to the injection site. 22 This may be related to a type 1 cell-mediated effector immunity, which is well known to occur in response to viruses. 23 Interestingly, scAAV vectors are also more potent inducers of transgene product-specific adaptive immune responses, 24 albeit the application route might have an impact. 25
Already early on, it became clear that innate immune responses are also invoked in a MyD88-dependent, but TLR9-independent manner. This includes humoral immune responses against the capsid 26,27 and transgene product, 26 with B cell intrinsic MyD88 signaling playing a major role. 26,27 Recently, a TLR9-independent induction of transgene product-directed CD8+ T cell responses that were dependent on interleukin-1 receptor (IL-1R)-MyD88 signaling was also identified in the liver. 28 However, in general, intact TLR9-MyD88 signaling appears to be crucial for induction of CD8+ T cell responses against the capsid and/or the transgene product. 26,29,30 In this innate immune recognition process, crosstalk between pDC and cDC is required with T1 IFNs serving as key immune mediators. 30,31
Since—as mentioned—CpG motifs in DNA function as important PAMP for TLR9, AAV vector genomes with reduced CpG content were explored, 32 which had significant impact on the anti-capsid specific CD8+ T cell responses. 33 Of importance in the context of clinical applications, effects differed depending on the T cell differentiation state. 34 Specifically, expansion of naive T cells directed against a capsid epitope was attenuated in the presence of AAV vector genomes with reduced CpG content, while capsid-specific memory CD8+ T cells expanded significantly better in the same setting. 34
Besides the vector genome, which needs to be detected while the AAV vector particle is trafficking through the endosomal compartment in TLR9-expressing cell types to trigger the above-mentioned innate immune responses, the capsid of AAV vectors has also been identified as a target of a pattern recognition receptor. Specifically, in primary human liver cell cultures, KC and liver sinusoidal endothelial cells were shown to sense capsids via cell surface-localized TLR2. 18 This sensing induced a NFκB-mediated inflammatory response with upregulation of interleukin-8 (IL-8), IL-1β, tumor necrosis factor-α (TNF-α), and IL-6, but without inducing a T1 IFN response. 18 Furthermore, Kuranda and colleagues observed innate and adaptive immune reactions in human PBMC after encountering empty AAV2 capsids. Interestingly, monocyte-derived DCs (moDCs) were identified as the main drivers of these immune responses, while pDC and cDC only showed a weak response. 35 Another important finding in that study was a striking correlation between the serology of the human PBMC donors and the type of immune cells in the blood that reacted to AAV capsids. 35 Specifically, individuals who had previously been exposed to the wild-type virus showed capsid-specific memory B cells, which were differentiated into antibody-secreting cells, a process driven by IL-1β and IL-6. Moreover, capsid-specific, TNF-α-producing memory CD8+ T cells were also detected in seropositive donors. 35 In contrast, in PBMC of seronegative individuals, neither B nor T cells responded to AAV capsids, while a NK cell subtype became activated and transiently produced cytokines, including IFN-γ and TNF-α. This result suggests that NK cells are involved in immune recognition of AAV capsids, specifically in seronegative individuals. 35
Lately, another part of the innate immune system received much attention, the complement system. Hepatotoxicity and thrombotic microangiopathy (TMA) events were observed in several clinical studies in which patients had received a high intravenous dose of AAV vectors. Complement activation is reported to be at least a contributing factor to the observed severe toxicities, including some fatal cases. 36 –38 The involvement of complement activation to the death of a young patient with Duchenne muscular dystrophy who received a high dose of AAV9 vectors to activate endogenous dystrophin expression by gene editing has remained unclear. 39 Since neither evidence of anti-AAV9 antibodies nor of effector T cell reactivity in the organs could be detected, innate immune responses are discussed as the triggering event. 39 The observed thrombocytopenia might indicate involvement of the complement system; however, no complement deposition was detected in the lung or heart, which argues against complement activation as cause of the fatality. Unusually, the patient developed an acute respiratory distress syndrome following a cytokine-mediated capillary leak syndrome. 39
Already in 2007, Zaiss and colleagues showed that complement factors can directly bind the AAV capsid. 40 In line, the alternative complement pathway was reported as cause for the AAV vector-related TMA observed in cynomolgus and rhesus macaques receiving a high dose (≥10e14 vector particles per kg body weight) of AAV9 vectors carrying enhanced green fluorescent protein (EGFP) or therapeutic transgenes. 41 Simultaneously, in the same animals endothelial cell damage was reported in the liver, which contributed to the fatal outcome. 41
Smith and colleagues reported that in case of pre-existing anti-AAV humoral immunity with high titers of neutralizing antibodies (NAb), elevated levels of the complement component C3a were detected in human blood spiked with AAV vectors. 42 West and colleagues added to this observation and demonstrated human complement activation by AAV vector-antibody complexes using sera from AAV seropositive donors, thereby suggesting a significant link between AAV vector neutralization and complement activation in individuals with high pre-existing anti-AAV immunoglobulin G (IgG) antibodies. 43 A study performed in dual-dosed mice by Emami and colleagues confirmed that complement is activated in the presence of high doses of AAV vectors and high pre-existing anti-AAV IgG titers. 44 Most recently, Salabarria and colleagues reported that the development of TMA in high-dosed patients was dependent on the presence of anti-capsid antibodies, confirming the link among neutralization, complement, and TMA observed in clinical trials. 45
The already-mentioned study by Smith and colleagues also reported uptake of AAV vectors into various cell types present in the blood, with neutrophils and moDCs showing the highest level, followed by monocytes, cDC, and pDC. 42 The uptake of AAV vectors into cDC triggered their activation, as indicated by CD86 upregulation. 42 In line, Emami and colleagues observed a correlation between complement activation and myeloid-derived pro-inflammatory cytokine production in mice, which were mainly generated by activated monocytes. 44 As one mechanism for immune stimulation, they proposed Fc-γ receptors and/or complement receptors to be activated in monocytes by complexes of AAV and antibodies. 44
AAV CAPSID ENGINEERING TO REDUCE DE NOVO HOST IMMUNE RESPONSES
Vector engineering is one strategy to address the issues of de novo immune responses caused by sensing AAV capsids or vector genomes. Modifying the viral capsid is also considered a valid strategy to develop capsids that escape recognition by anti-AAV antibodies induced by a prior contact with naturally occurring serotypes. Specifically, libraries of AAV capsid variants have been generated by introducing point mutations into the capsid protein-encoding cap gene through error prone PCR (Fig. 3). 2,7 These libraries are screened in a high throughput manner for variants that have maintained their tropism while having gained, through the introduced random mutations, the ability to transduce cells even in the presence of NAb. 48,49 Capsid shuffling is another strategy and is based on the generation of capsid chimeras from a set of different serotype cap genes that were fragmented by a nuclease and randomly rearranged. 2,7 Through these rearrangements, epitopes might have been destroyed to result in an immune escape phenotype. The third strategy, termed AAV peptide display library, relies on the insertion of peptides of a random sequence either as an additional sequence or to replace parts of the wild-type cap gene sequence. 2,7,46 Since these changes are made at surface exposed positions—as their primary objective is to retarget AAV capsid toward a novel receptor—an immune escape phenotype can be obtained in case the insertion itself masks or destroys an immune epitope. Simultaneously, the peptide insertion, which is a new sequence, is responsible for receptor binding and thus transduction is not dependent on the wild-type receptor binding motif, which might be blocked by binding of NAb. In addition to the direct effect of capsid engineering on the binding of pre-existing antibodies, capsid engineering performed to redirect vector tropism or to overcome post-entry barriers might also have an indirect impact on the vector–host immune response, as an increase in cell type selectivity and/or reduction in vector dose decreases the antigenic load and the risk of off-target transduction. 2,7,46

AAV capsid engineering. A number of capsid engineering strategies have been developed with examples as depicted here. Directed evolution approaches are based on libraries of capsid variants, which are screened in a high throughput manner ex vivo or in vivo for AAV capsids with desired features such as improved transduction efficacy, immune escape phenotype, and/or improved target cell selectivity. The capsid variants are generated by introducing point mutations into the cap gene (“random mutations”), by shuffling fragments of cap gene sequences of a set of serotypes or variants (“capsid shuffling”) or by introducing peptides of random sequence (“peptide display”). Rational design approaches introduce changes to the capsid protein amino acids by site-directed mutagenesis to improve transduction efficacy, for example, through reducing proteasomal degradation, or by adaptor molecules such as DARPins or nanobodies to redirect vector tropism toward a predefined cell surface molecule on the target cell. Likewise, peptides are introduced that function as receptor binding ligands. Moreover, adaptors or peptides can be chemically conjugated to the AAV capsid using either a wild-type capsid or following genetic engineering to confer a site-specific coupling. Moreover, capsid proteins of different serotypes can be combined to generate chimeric capsids that possess combined features of the parental serotypes. For further details, we refer to Wang et al., 2 Macdonald et al., 7 Büning and Srivastava, 46 and Pupo et al. 47 DARPin, Design Ankyrin Repeat Protein.
An elegant strategy to reduce anti-capsid T cell responses was developed by Arun Srivastava and his team. 50 They reported the exchange of distinct amino acid residues within the capsid proteins, that is, tyrosine to phenylalanine, threonine or serine to valine, lysine to glutamic acid, and reduced proteasomal degradation of capsids. 46 Consequently, transduction efficacy is improved and at the same time presentation of capsid protein-derived peptides on major histocompatibility class (MHC class) I proteins is lowered, resulting in a significant reduction of capsid-specific CD8+ T cells. 51 Recently, Bentler and colleagues reported on the insertion of a MyD88 dimerization blocking peptide into AAV2 capsids. 52 Blocking MyD88 dimerization and thus downstream TLR signaling is expected to impact on innate immune activation of incoming AAV vector particles. Indeed, Bentler and colleagues could demonstrate reduced expression of key mediators of innate immune responses, such as T1 IFN. Moreover, a delayed antibody response, as well as reduced CD8+ T cell responses against the capsid and the transgene product, were observed. Chan and colleagues specifically focused on TLR9 and introduced a TLR9 inhibitory oligonucleotide sequence into AAV vector genomes. 53 This strategy had an impact on adaptive immune responses, for example, by reducing infiltration of CD8+ T cells into vector-transduced tissues and at the same time increased the transduction efficacy of AAV vectors. 53 Moreover, microRNA targeting sites can be included as part of the vector genome to avoid off-target expression that might cause immune activation. 2 These strategies are preferable to a general immune suppression to reduce side effects. Moreover, they can easily be combined with one another to further reduce or possibly avoid de novo immune responses and to offer seropositive patients a path for receiving AAV vector-derived GTMPs.
Cell types involved in the anti-AAV immune response become targets for the AAV vector system
DC, NK cells, macrophages/monocytes, and B and T lymphocytes are key players in immune responses against AAV vectors and/or AAV vector-derived transgene products as briefly presented above. The same cell types, including their precursors, the CD34+ hematopoietic stem and progenitor cells (HSPCs), are important targets in gene therapy. Traditionally, integrating vector systems have been used to modify these cell types to avoid loss of therapeutic efficacy upon cell division. Thus, AAV vectors have not been considered as important gene delivery vehicles for HSPCs, T cells, B cells, or DC. This view has changed since gene editing entered the stage. In conventional gene editing approaches, designer nucleases such as zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), meganucleases, or the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) complex are used to introduce a DNA double strand break (DSB) in a sequence-specific manner. The DSB is repaired by the cellular DNA repair machinery. In the absence of a DNA sequence that serves as a DNA template for homology-directed repair (HDR), the lesion is repaired by nonhomologous end joining, which frequently results in insertions or deletions and therefore in a gene knockout. 1 In contrast, provision of a template for HDR enables gene correction or integration. 1 AAV vectors have emerged as promising tools to provide HDR templates because the “foreign” DNA is shielded from DNA sensors in the cytosol while being transported to the nucleus and because high HDR-meditated integration efficiencies at the DSB are achieved. 54 The latter is maybe due to the recognition of the AAV-inverted terminal repeat (ITR) structures that flank the AAV vector genomes (Fig. 1) by the cellular DNA repair machinery. 54 Despite this expansion of AAV’s conventional workspace, DC, NK, B, and T cells, as well as HSPCs, have remained difficult target cells for naturally occurring AAV serotypes as high vector doses are required, a challenge that is addressed by vector engineering.
AAV VECTORS AND HEMATOPOIETIC STEM AND PROGENITOR CELLS
The self-renewal capacity and potency to form all blood cell types have made HSPCs the primary target of ex vivo gene therapy approaches for treating patients with primary immunodeficiencies, metabolic disorders, or hemoglobinopathies. 1,2 Traditionally, ex vivo gene addition strategies employing gamma-retroviral or lentiviral vectors have been followed in which autologous HSPCs isolated from the patient are equipped with a functional copy of the missing or defective gene and reinfused. 1,55 Following engraftment, the functionally corrected HSPCs reconstitute the immune system of the patients with primary immunodeficiency without the risk of graft-versus-host disease (GvHD), which is a risk that accompanies allogenic HSPC transplantation that was the sole treatment option prior to the age of gene therapy. 56,57 Likewise, in case of sickle cell disease or beta-thalassemia, gene-modified HSPCs will give rise to functionally corrected erythrocytes. 1 Moreover, in patients with adrenoleukodystrophy, gene-modified HSPCs differentiate among other into myeloid-derived cells, including microglial cells after crossing the blood–brain barrier, 58,59 and produce the adrenoleukodystrophy protein, thereby reintroducing the ability to break down very long chain fatty acids. 60
The first proof of a functional cure by ex vivo gene therapy was reported in the landmark study by Cavazzana-Calvo and colleagues using first generation gamma-retroviral vectors to deliver the common gamma chain gene to CD34+ cells of patients with X-linked severe combined immunodeficiency (SCID-X1). 56 Equal success was reported by a parallel study performed by Adrian Thrasher and colleagues at University College London. However, insertional mutagenesis caused by the integration preference of the first generation gamma-retroviral vectors was also reported in both studies. 61,62 The huge dilemma of those early days of gene therapy had been the need to insert the functional copy of the gene into the patients’ HSPC genome to sustain therapeutic efficacy upon cell proliferation without having the tools to allow targeted vector genome integration or even a gene repair. To mitigate these risks in the pre-gene editing area, research focused on the development of safer vector designs and to embark on vectors with a safer integration profile, such as lentiviral vectors. 55 These advancements resulted in market approved GTMPs for adenosine deaminase deficiency, beta-thalassemia, metachromatic leukodystrophy, and early cerebral adrenoleukodystrophy. 1
Implementing gene editing is expanding the treatment options as a gene defect can be corrected, cells can be equipped with additional features through targeted gene insertion, expression levels of host genes can be modulated, or genes can be knocked out in a sequence-specific manner. 1 Indeed, the first market approved gene editing-based GTMP, Casgevy (Exa-cel), uses HSPCs as target cells and relies on a CRISPR/Cas9-mediated knockout of the erythroid specific enhancer region of the BCL11A gene. 1 Thereby, fetal globin expression is maintained in gene-edited erythrocytes with the ability to form a functional hemoglobin in patients with sickle cell disease and transfusion dependent beta-thalassemia.
Approaches to “repair” a gene defect or to add a novel gene in a stable manner require—as mentioned—the presence of a HDR DNA template. Among all tested naturally occurring serotypes, AAV6 emerged as a prime candidate for this function. 63 –65 Most frequently, vector-mediated HDR template delivery is combined with electroporation of cells to transfer the designer nuclease as mRNA or in case of CRISPR/Cas9 system as a ribonucleoprotein (RNP) complex (Fig. 4). This strategy has yielded targeted integration efficiencies of 60% in sickle cell disease and beta-thalassemia patient-derived autologous HSPCs and an engraftment efficiency of gene edited cells of over 25% in NSG mice. 66 –68 These results led to the first FDA-approved clinical trial for tackling sickle cell disease by an AAV vector-based gene editing strategy (NCT04819841).
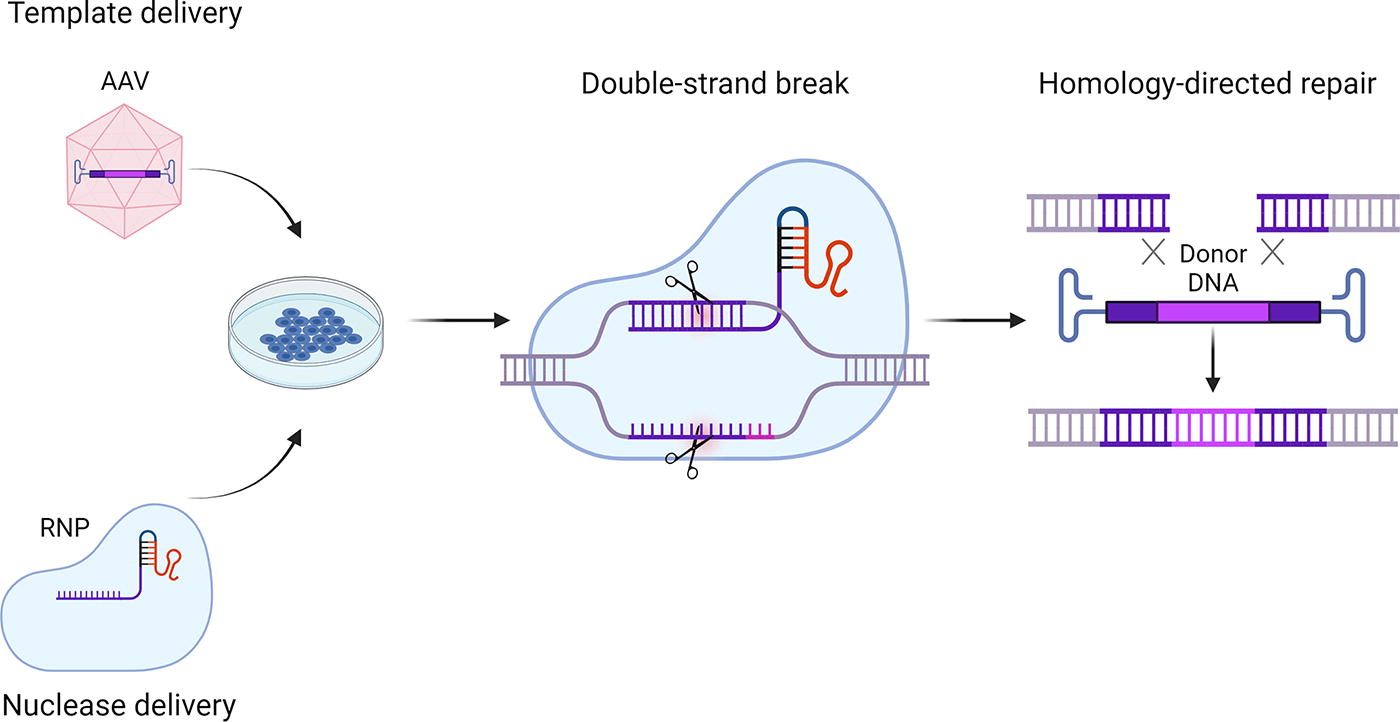
AAV vectors in gene editing. AAV vectors are used as a delivery system to transfer HDR templates in gene editing approaches. The HDR templates contain the sequences to be inserted, as well as homology arms to promote HDR. In order to introduce DSB in a target sequence selective manner, designer nucleases such as the here depicted CRISPR/Cas RNP complex are delivered to the cells either prior to or following AAV vector transduction, for example, by electroporation. HDR, homology-directed repair; RNP, ribonucleoprotein.
Despite this success, reports of AAV vectors causing the induction of a upregulated modulator of apoptosis (p53) dependent DNA damage response in HSPCs accompanied by a reduced cell engraftment call for comprehensive evaluation of the HSPC-first generation AAV vector interaction and subsequent changes in vector design and/or transfer protocols. 69 On a similar note, the same study also reported that the low transcriptional activity of the ITRs is also active in HSPCs and that ITR-containing fragments were found trapped at DSB. 69 Nevertheless, as of yet, combining designer nucleases with AAV6 vector-mediated HDR template delivery outperforms common lentiviral integration strategies with regard to safety profile. 70
A further change in strategy that is under development is the transition from ex vivo manipulation toward in vivo gene therapy of HSPCs. This will reduce the demand on specific infrastructure that is required for ex vivo HSPCs manipulation and transplantation. Moreover, a beneficial impact on cell viability and function is expected since HSPCs remain within the patient and are not cultured ex vivo within an artificial environment. First successful steps in this direction have already been reported. Andre Lieber and his team have developed a helper-dependent chimeric adenoviral (HDAd) vector, HDAd5/35++, targeting CD46, a receptor expressed across all HSPCs. 71 HSPCs mobilized from the bone marrow into the peripheral blood were transduced by intravenously applied HDAd5/35++ in a mouse model. Since adenoviral vectors—such as AAV vectors—are maintained as episomes, a combination with the Sleeping Beauty (SB) transposon system was explored to obtain stable transgene expression via random integration. Moreover, to increase the efficacy of transduction or to address the issue of seroprevalence, alternative HDAd chimera were investigated, such as HDAd5/3+, which targets the desmoglein 2 receptor 72 or HDAd6/35++, which showed lower off-target liver expression and a lower seroprevalence in the human population. 73 Yao and colleagues also focused on adenoviral vectors and reported on an engineered vector that, in addition to the hybrid fibers, contained mutations in adenoviral hexon proteins to reduce liver sequestration and a modified penton base in which the natural RGD loop had been replaced by the laminin-derived peptide SIKVAV to direct this novel adenoviral vector (termed AVID) toward α6β1, α6β4, α3β1, and α7β1 integrins. 74 Of importance in this regard is the α6 integrin (CD49f), which is expressed on hematopoietic cell with stem cell features. 74 Indeed, the authors reported targeted transduction of up to 20% of human CD34+CD38−CD45RA−HSPCs following intravenous administration in humanized mice. Moreover, modified particles showed a detargeting from liver, and mice showed no indications of a cytokine storm, hepatotoxicity, or thrombocytopenia. 74 Lipid nanoparticles (LNP) have also been explored as an alternative to viral vectors. Specifically, by conjugating an antibody directed against CD117 (c-Kit) to LNP, LNP were directed toward HSPCs to generate genetically modified HSPCs in mice without mobilization. 75,76 Beyond mRNA for Cre recombinase, 75,76 mRNA for base editing 75 or a pro-apoptotic factor (PUMA) 75 was successfully and efficiently delivered following intravenous administration of CD117-targeted LNP. While the base-editing approach was followed to show proof-of-concept for correction of sickle cell disease in HSPC samples, transfer of PUMA demonstrated targeted HSPC depletion as a nontoxic alternative to the conventional conditioning regimen. 75 However, the current system requires additional refinements to obtain LNP-retargeting, since CD117-targeted LNP and nontargeted LNP were detected with comparable efficiency in the liver due to binding of LNP to Apo-lipoprotein E and low-density lipoprotein. 75,76 Moreover, CD117-targeted LNP transduced lung tissue at a higher efficiency than nontargeted LNP. 75,76
Capsid-engineered AAV vectors might be a further alternative since they are significantly smaller compared with adenoviral vectors, less immunogenic, and strategies for liver detargeting and a redirected receptor-mediated transduction are in place (Fig. 3).
AAV VECTORS AND T LYMPHOCYTES
T lymphocytes are potent effector cells of the adaptive immune system and have been harnessed for therapeutic applications since the beginning of the age of gene therapy. Indeed, T cells were modified by gamma-retroviral vectors in an ex vivo gene therapy setting in the landmark clinical trial of Michael Blaese and French Anderson for adenosine deaminase deficiency (ADA-SCID), which is remembered as the first human gene therapy clinical trial. 77 The main focus when exploring T lymphocytes as targets in gene therapy is set on harnessing the target selective cytotoxic activity of CD8+ T lymphocytes in oncology and infectious diseases or the immune regulatory function of regulatory T cells (Tregs) in the context of autoimmune diseases. 1,78 For this purpose, T cells are genetically modified to respond with their target-selective activity to custom-defined target structures or antigens. 1 The most prominent examples in this regard are CAR T cells that are used as living drugs to treat B cell malignancies with currently six market-approved drugs available in the Western world (https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products). All market-approved CAR T cell GTMPs use either gamma-retroviral or lentiviral vectors to deliver the CAR coding sequence, which becomes integrated into the host cell genome in the process of T cell transduction. To advance this technology, gene editing strategies, among others exploring AAV vectors as the HDR template provider, are implemented to protect CAR T cells against fratricide, to knock out endogenous T-cell receptor (TCR) activity, to provide distinct cytokine stimuli, or to expand T cells from a personalized medicine into an “off-the-shelf” product. 1
One of the first reports on gene editing of both CD4+ and CD8+ T lymphocytes involving AAV vectors for HDR template delivery was reported by Wang and colleagues. 79 They used ZFN—provided as mRNA—to modify the chemokine (C-C motif) receptor 5 (CCR5) gene. 79 The CCR5 gene is classified as a safe harbor, based on the observation that humans with a mutation in CCR5 do not show any pathology. 80 Moreover, CCR5 is used as human immunodeficiency virus 1 (HIV-1) coreceptor, and thus, editing of this locus has also gained attraction among scientists aiming to develop gene therapy strategies to protect against HIV-1 infection. 80 Instead of ZFN, Sather and colleagues used megaTAL nucleases combined with AAV vector-mediated HDR delivery to modify the CCR5 locus. 81 Again, the coding information for the designer nuclease was provided as mRNA by electroporation, and AAV6 was used as the serotype capsid. This strategy yielded up to 60% modified T lymphocytes when a marker gene was used as template and was successfully expanded to anti-HIV therapeutics such as C46, an HIV gp41-derived peptide that blocks HIV-1 infection, or anti-HIV CARs. 81 In a subsequent work, the team developed a universal “off-the-shelf” CAR therapy using the TCR-α constant locus (TRAC) as target locus to avoid expression of the endogenous TCR. 82
A CreI homing endonuclease-based strategy was explored by MacLeod and colleagues. 83 The endonuclease targeting the TRAC locus was combined with AAV6 vector-mediated HDR template delivery for a CD19-directed CAR. These CAR T cells were functionally active in vitro as indicated by target cell driven cytolytic activity and release of pro-inflammatory cytokines, including IFN-γ. Moreover, in a murine B cell lymphoma model, CAR T cells eradicated CD19+ Raji cells. 83 The TRAC locus was also targeted through the CRISPR/Cas9 system using AAV6 vectors to deliver TCR2.5D6, a recombinant TCR that recognized a myeloperoxidase-derived peptide. 84 Of note, the construct was designed to place TCR2.5D6 under the control of the endogenous promoter and resulted in successful modification of 18% of the T cells. 84 Again, target selective activity of modified T lymphocytes was shown by ex vivo killing assays and following adoptive T cell transfer in a murine xenograft tumor model. 84
Of note, it has been shown that targeting the TRAC locus with a CAR construct designed to be endogenously regulated after integration results in a homogenous expression of the transgene product in different donors. 85 Moreover, these CAR T cells outperformed CAR T cells generated with conventional methods described in the literature, including lentiviral vector mediated delivery, in an in vivo mouse model of B cell leukemia. These enhancements depend on the reduced tonic signaling of the CAR when it is endogenously regulated, which subsequently reduces T lymphocyte differentiation and exhaustion in response to the antigen. 85
CRISPR/Cas9 RNP complexes—introduced by electroporation—were also used in combination with AAV6 vectors for HDR template delivery to modify the TCRαβ+/CD19+ cell fraction that is discharged during αβ-haplo-hematopoietic stem cell transplantation for the treatment of high-risk leukemia. 86 By inserting an anti-CD19 CAR into the TRAC locus of these cells, Wiebking and colleagues aimed to create an additional anti-leukemic therapy arm to enhance the effectiveness of the stem cell transplantation, without the risk of GvHD. 86 Dai and colleagues used, instead of Cas9, Cas12a/Cpf1 in combination with AAV6 vectors to deliver both the HDR template and the guide RNA. 87 Thereby, the programmed cell death protein 1 was knocked out, and an anti-CD22 CAR was delivered into the TRAC locus. 87 The same strategy was used to create double knock-in CAR T cells, expressing an anti-CD22 CAR in the TRAC locus and an anti-CD19 CAR in the programmed cell death protein 1 locus. 87 More recently, the T cells redirected toward universal cytokine-initiated killing (TRUCK) strategy was explored utilizing two different CRISPR/Cas9 RNP complexes and a single AAV6 vector containing both the HDR templates for a nectin cell adhesion molecule 4 (Nectin4) targeting CAR and for an internal ribosome entry site equipped IL-15. 88 The double knock-in cells, with CAR integrated in the TRAC locus and the internal ribosome entry site–IL-15 integrated in the 3′-ntranslated region (UTR) of the INF-γ gene, showed improved anti-tumoral activity both in vitro and in vivo in comparison with Nectin4 targeting CAR T cells. 88
While the nuclease is usually delivered by an mRNA or as an RNP complex, Moço and colleagues proposed a dual-AAV6 vector approach. One of the two AAV vector constructs encoded for the Staphylococcus aureus-derived SaCas9 and the corresponding guide RNA that targeted the TRAC locus, while the second AAV vector delivered the anti-CD19 CAR construct serving as the HDR template. However, vector doses around 10e6 were required to yield ∼10% correctly modified Jurkat cells, a T cell line. 89
Applications outside of the CAR T cell field have also explored gene editing with AAV vectors as the source of the HDR template. For example, Hunt and colleagues aimed to generate antigen-specific CD4+ Tregs. 90 They combined two different RNP complexes, targeting TRAC and FOXP3 loci, respectively, with two AAV6 vectors to deliver HDR templates. One AAV vector provided the template for a human islet-specific TCR (T1D4) to be inserted into the TRAC locus, while the second AAV vector was designed to provide a constitutively active promoter sequence to be inserted into the FOXP3 locus. With this dual-locus editing strategy, Hunt and colleagues were able to generate pancreatic islet antigen-specific Tregs that suppressed effector T cells directed against the islet specific antigens. Moreover, each AAV construct also contained a component of a heterodimeric chemically inducible signaling complex, created by fusing the γ and β subunits of IL-2 with the extracellular rapamycin binding domain. This construct was designed to activate the IL-2 signaling pathway in response to rapamycin, which enabled the selection and expansion of the double-edited cells. 90 Hubbard and colleagues used a targeted gene editing strategy to correct CD4+ T cells from patients with X-linked hyper-IgM syndrome, who suffered from loss of CD40 ligand (CD40L) expression. In that study, a TALEN-based approach was used to place the CD40L cDNA after the first start codon of the CD40L gene through HDR so that CD40L expression was under control of the endogenous promoter and regulatory elements, which restored CD40L expression to levels found in unaffected CD4+ cells. 91
Similarly, as for HSPCs, the SB system was also used to modify T lymphocytes. Specifically, electroporation of an mRNA encoding for the SB transposase and transduction by an AAV6 vector to deliver the therapeutic gene as an SB transposon, a strategy termed MAJESTIC (mRNA AAV-SB joint engineering of stable therapeutic immune cells), yielded CAR T cells directed against CD22, CD19/20, or human epidermal growth factor receptor 2 (HER2)/neu, as well as TCR-T lymphocytes. 92
To optimize a protocol for T cell modification using AAV6 vectors, Cannon and colleagues comprehensively studied conditions that influence transduction efficiency. A key finding of this study was the inhibitory effect of fetal bovine serum (FBS) in the medium. Moreover, they reported on how cell culture volume, time of transduction, and electroporation influence the outcome. Based on their findings, they proposed a protocol that combines a short transduction procedure with highly concentrated AAV6 vector preparations in the absence of FBS, followed by the electroporation step to introduce the nuclease. This protocol improved efficiency up to 7-fold compared with previously described protocols. 93 Moreover, treating the T lymphocytes with genistein, an isoflavone and natural tyrosine kinase inhibitor, prior to AAV6 vector administration appeared to improve the levels and durability of transcription, while the addition of OKT3 improved both transduction efficiency and level of transgene expression. 94 Moco and colleagues followed a different strategy by employing cell-penetrating peptides to enhance AAV vector-mediated cell transductions. While this led to a 10-fold increase in transduction of Jurkat cells, the effect was less pronounced on primary murine T lymphocytes. 95
Albeit AAV vectors based on naturally occurring serotypes display a broad tropism and—in general—transduce various cell types and tissues, there is a lack of variants that selectively and efficiently transduce T lymphocytes. Even with AAV6, a serotype considered to be the best suited for transduction of cells of the hematopoietic system, including T cells, vector doses of up to 10e6 per cell are required to achieve efficient transgene expression or an adequate number of gene edited cells. 79,96 These observations have fostered vector engineering approaches that aim to develop more efficient AAV capsid variants. The use of nanobodies (Nb) is one strategy that was applied to target T cells (Fig. 3). Hamann and colleagues, for example, used a capsid-engineered AAV2 variant as basis and modified either VP1 or VP2 proteins of the viral capsid by inserting Nb sequences. Two of the Nb-modified AAV capsids with Nb inserted into VP1 showed improved targeting of CD4+ cells in cell lines, mixed cultures, purified human CD4+ T cells, and human PBMC. 97
Other than human T cells, AAV variants that efficiently target murine T cells are also lacking, making it difficult to perform in vivo studies in syngeneic mouse models for preclinical studies. Michels and colleagues therefore selected a Design Ankyrin Repeat Protein (DARPin) that specifically targeted murine CD8. This DARPin, MSE10-LV, was inserted into VP1 capsid proteins using the GH2/3 loop, forming the highest peak of the capsid, as the target site and combined this with site-directed mutagenesis of the capsid to avoid natural primary receptor binding. 98 The resulting AAV capsid variant, mCD8-AAV, transduced murine T cells with a 10-fold increase in efficiency compared to the parental serotype and showed a strong preference for CD8+ cells. When used to transduce whole splenocyte cultures, mCD8-AAV was even more efficient, with a 26-fold increase in the percentage of EGFP+ cells and a specificity of >99%. 98 Instead of a DARPin library, Nyberg and colleagues screened an AAV capsid display library that was based on AAV6 on murine T cells in an ex vivo setting. 99 They thereby identified variant Ark313, which showed increased cell binding, uptake, and transduction efficiency in murine T lymphocytes. The variant was then used to generate murine anti-CD19 CAR T cells that showed anti-tumoral activity in vivo in an immunocompetent solid tumor mouse model. Of note, the group performed a CRISPR/Cas genome-wide knockout screen and identified the GPI-anchored protein H2-Q7 as an essential factor for the function of their Ark313 variant. 99
DARPins have also been used for in vivo retargeting of human T cells. The first approach was reported by Munch and colleagues using a CD4-directed DARPin to retarget AAV2. The respective targeting vector—displaying the DARPin as N-terminal fusion to VP2 proteins (also combined with site-directed mutagenesis to avoid natural primary receptor binding)—transduced human CD4+ lymphocytes following intravenous administration in mice transplanted with human PBMC. 100 Theuerkauf and colleagues modified the AAV2 capsid by inserting two DARPins specific for CD4 and for CD32a, respectively, connected by a linker into the GH2/3 loop of AAV2 VP1 proteins. This bispecific AAV variant showed superiority compared to AAV vectors that displayed only a single DARPin in ex vivo targeting and transduction of CD4-CD32a-double positive cells. This specificity was maintained in vivo when the vector was injected into immunodeficient mice engrafted with SupT1-CD4, SupT1-CD32a, and SupT1-CD4/CD32a cells. The bispecific vector was then used to deliver the coding information for CRISPR/Cas9 complex with guide RNAs directed against the tat and rev genes of HIV, as CD32a is described as a surface marker on HIV reservoir cells. This approach inhibited HIV replication in cultured T cells. 101 The DARPin strategy, however, at least when using the N-terminus of VP2 of AAV6 as the insertion site and the DARPin 55.2 to retarget the capsid was not effective in in vivo T lymphocyte transduction when administered subcutaneously in rhesus macaques. 102
Other strategies were also applied with the aim to transduce T lymphocytes in vivo by AAV vectors. Maguire and colleagues, who were the first to report on the use of AAV vectors associated with exosomes, 103 explored exosome-associated AAV8 and, for comparison, conventional AAV8 vectors for their ability to transduce CD4+ and CD8+ cells in C57BL/6 mice by introducing the human interleukin-2 receptor α (hIL-2Rα, hCD25) to the membrane of these immune cells. 104 In contrast, Nawaz and colleagues used a capsid variant derived from a shuffled capsid library screen on hepatocytes, 105 AAV-DJ, to transduce T cells with a CAR in a mouse model of T cell leukemia. A single AAV injection into humanized mice yielded sufficient CAR T cells for a potent anti-tumoral activity, with four out of six mice showing complete tumor remission. 106
In addition, local in vivo administration of AAV vectors with a naturally occurring capsid showed promise under distinct conditions. Specifically, Pouzolles and colleagues performed intrathymic injection for their approach to develop an AAV vector-based treatment for T cell immunodeficiency using AAV8 vectors encoding for the ZAP-70 gene and ZAP-70 deficient mice as a model system. In this setting, thymocytes are arrested in the double positive stage of development. Treated mice showed a significant development of medullar tissue and thymocyte reconstitution, as well as an unexpected long-term presence of corrected T cells. 107 The latter observation was followed by integration profile analyses that compared lymph nodes, spleen, and liver employing a new bioinformatic pipeline termed RAAVioli. While vector integration in the liver was limited and showed the expected random profile, vector genomes were found integrated in T cells with a clear preference for the TCR locus in the spleen and lymph nodes. Integration hot spots were near the RSS sequence, i.e., in a location where DSB is introduced into the cell genome through the activity of recombination activating genes during T cell maturation. Thus, these studies provide evidence that intrathymic AAV vector delivery can represent a new approach to obtain stable transgene expression from AAV vectors in T cells in a targeted manner. 108
AAV VECTORS AND B LYMPHOCYTES
B lymphocytes are antigen-presenting cells and at the same time the effector cells of the humoral arm of the immune system. 109,110 These unique features make B lymphocytes interesting targets in gene therapy. The most obvious function is using engineered B lymphocytes to produce and secrete therapeutic antibodies for the treatment of malignancies or infectious diseases. In the latter setting, even a prophylactic approach can be envisioned. The impressive capacity of B lymphocytes to produce and secrete proteins can also be harnessed to provide proteins other than antibodies, thereby addressing the patient needs in diseases caused by protein deficiencies. Indeed, these avenues have been followed using AAV vectors as engineering tools. To address the issue that episomal vectors are not maintained in proliferating cells, again, gene editing is applied using AAV vectors to deliver the HDR template. Initial tests by Johnson and colleagues to identify the most appropriate conditions for gene editing via the CRISPR/Cas9 system revealed highest efficiency and lowest impact on cell viability when the designer nuclease was introduced as RNP (instead of Cas9 mRNA) via electroporation into activated CD19+ B cells isolated from human PBMC. 110 This setting was next explored to move from gene knockout to HDR targeting the AAV integration site 1 as a safe harbor. While successful gene editing throughout a range of particle-per-cell ratios was reported, a peak in efficacy (∼18%) was reached at an AAV6 vector dose of 250,000 per cell. In a parallel study, Hung and colleagues confirmed AAV6 as a prime candidate for B cell engineering followed by AAV2, AAV2.5, and AAV-DJ. They also demonstrated that vector engineered B lymphocytes can differentiate into plasma cells that can serve as production plants to secrete factors other than antibodies. To improve the system by promoting survival of the modified B cells and sustaining transgene expression in vivo, they further engineered the cells to express B cell activating factor. 109
The study of Nahmad and colleagues is an example of developing engineered B cells for treating infectious diseases. 111 They focused on overexpression of broadly neutralizing anti-HIV antibodies using 3BNC117 as a model. To harness the native functions of B cells at their best, which includes antibody maturation, somatic antibody hypermutation, and affinity maturation, they decided for a vector design that fuses—following gene editing—the newly introduced antibody sequence with the endogenous constant part of the antibody. As a target locus, the authors decided to use the immunoglobulin (Ig) H locus. AAV6 vectors were used to modify human B cells, while AAV-DJ was used for work on murine B cells. In a set of comprehensive in vivo studies, Nahmad and colleagues demonstrated that their ex vivo engineered B cells home to germinal centers following adoptive B cell transfer into mice, are activated upon antigen encounter (authors used gp120 protein of HIV recognized by the expressed, membrane-localized 3BNC117), differentiate into memory and plasma cells, and perform antibody class switch, as well as somatic hypermutation and thus affinity maturation. Thus, they indeed engineered B cells as “living and evolving drugs”. 111
As mentioned, Breuer and colleagues explored exosome-associated AAV8 and AAV8 vectors in vivo in mice. 104 In addition to T cells, they also reported on B lymphocyte transduction, where peak transgene expression was reached on day 3 post vector administration. Expression was reported to remain stable for 14 days followed by a decline. Using a dual AAV vector approach, Nahmad and colleagues advanced the field a step further by demonstrating in vivo editing of B cells. 112 AAV-DJ vectors were used to deliver either the coding information for Cas9 and guide RNA specific to IgH locus or the donor template encoding for 3BNC117. An established prime-boost mechanism was followed in which mice were immunized with the gp120 HIV antigen. This resulted in antibody levels of up to 5 µg/mL of 3BNC117 in the blood, clonal expansion of engineered B cells in the germinal centers, as well as clonal selection. However, concerns have been raised regarding the high vector copy number detected in the liver and the detection of mutations in the antibody sequence.
AAV VECTORS AND DENDRITIC CELLS
DC are the most potent antigen presenting cells in the human body, 113 and play a pivotal role in mounting immune responses against infectious agents and aberrant cells. Numerous vaccine approaches based on DC have been developed during the last decades not only with a focus on cancer immunotherapy 114 but also for emerging infectious diseases. 115 Besides different strategies for DC-antigen loading, DC functions can be enhanced by ex vivo engineering. 116,117 Successful transduction of moDC by AAV vectors was reported as early as 2001 using ssAAV2 coding for EGFP as a transgene. 118 Changing the vector genome configuration to an sc design increased transduction efficiency. 119 Importantly, no cytotoxicity was observed, and scAAV-transduced DC showed no changes in surface marker profiles or functional properties, such as antigen uptake. 119 Ussher and colleagues in particular focused on AAV6 and showed that a single amino acid substitution in the capsid proteins (lysine residue at position 531 to glutamic acid, i.e., K531E), which is known to inhibit binding to the extracellular matrix component heparan sulfate proteoglycan, abolishes DC transduction. This finding is in line with a report by Rossi and colleagues who observed that AAV2-mediated moDC transduction is dependent upon binding to heparan sulfate proteoglycan. 14 In contrast, exchanging the tyrosine residue at position 731 for phenylalanine (i.e., Y731F) resulted in a modest increase (2-fold) in transduction efficiency, 120 possibly due to the avoidance of proteasomal degradation as outlined above.
Several studies demonstrated that introduction of the coding sequence for antigens (i.e., hepatitis B virus antigen, cytomegalovirus antigens, papillomavirus type 16 E7 antigen, or the tumor associated antigen HER2/neu) through an ex vivo AAV2 vector-mediated transduction of moDC results in strong antigen-specific T cell responses, which were sufficient to kill target cells in vitro. 121 –124 Concordantly, AAV2 vector-mediated transduction of moDC with both carcinoembryonic antigen as an antigen and IL-12 as an immune stimulator enhanced T cell proliferation, cytotoxic T cell formation, and target cell killing. 125 Interestingly, the use of AAV vectors appears to be more effective in pulsing DC than pulsing with a cancer cell lysate. 126
With the aim to improve transduction of immature moDC, Rossi and colleagues screened an AAV2 peptide display library. Thereby, the capsid variant AAV-VSSTSPR was identified to transduce DC with higher efficiencies compared to the parental serotype. This property correlated with an improved vector uncoating, and intramuscular injection of AAV-VSSTSPR coding for the model antigen ovalbumin (ova) resulted in an improved anti-ova T cell response compared with AAV2. 14
Site-directed mutagenesis of distinct residues was also explored for AAV2, and, in line with the above-mentioned study of Ussher and colleagues, 120 two AAV2 capsid variants that transduced moDC with higher efficacy were discovered. 127 Initially, a serine residue at position 662 was substituted by valine. This S662V variant coding for hTERT as an antigen was able to induce a potent CTL response and led to specific lysis of K562 target cells. 127 Later, the same group used an AAV6 capsid variant that was modified at the threonine residue at position 492 in addition to S663 (i.e., T492V+S663), to drive prostate specific antigen expression, which again induced cytotoxic T cell responses and target cell lysis. 128
Krotova and colleagues further improved the vector transgene cassette regarding antigen presentation in DC and - when delivered through the AAV6-S663V capsid variant - empowered DC in lymph nodes adjacent to the injection site to mediate a strong in vivo T cell, as well as humoral immune response in mice against the model antigen ova. The vaccination delayed solid tumor growth significantly, but did not completely prevent tumor formation. 129 As a further development of this concept, the same group showed that this AAV6 variant could be used in a vaccination setting in the aggressive B16F10 mouse melanoma model by delivery of melanoma antigens (i.e., glycoprotein 100 [gp100], tyrosine related protein 1 [TRP1], TRP2, or tyrosinase), which required breaking self-tolerance in order to work. 130
AAV VECTORS AND NK CELLS
NK cells are part of the innate immune system and have important functions such as recognition and elimination of virus-infected cells and tumor cells. In contrast to T cells, NK cells identify target cells in a MHC-independent manner and the decision to kill the target cell is based on the balance of activating and inactivating cellular signals. Upon activation, NK cells can kill target cells via release of perforins and granzymes, as well as through death receptor-induced apoptosis. Recognition of the MHC-I acts as an inactivating signal to prevent NK cell-mediated destruction of “self” cells. These control mechanisms likely contribute to the relative lack of GvHD observed upon infusion of allogeneic NK cells, even with mismatched unrelated donors. Adoptive cell therapy using NK cells is developed for the treatment of cancer, a strategy that is inspired by their natural cytotoxicity, which allows tumor cell killing without prior antigen-specific activation. To optimize this procedure with regard to target cell recognition and cytotoxic capacities, genetic engineering of NK cells has become a very active field in cellular immunotherapy mainly exploiting lentiviral vectors as a delivery tool. 131 Another important asset is the possibility of an “off-the-shelf production.” As a clinical example, CAR-NK cells targeting CD19 have shown impressive results in clinical trials for treatment of patients with relapsed/refractory B cell lymphomas. 132 Advances in gene editing technologies further expand the possibilities for modifying NK cells for cancer therapy.
However, manipulation of primary NK cells has been challenging because of a broad resistance to genetic modification strategies that work well in T cells. Thus, Buchholz et al. used a DARPin library-based screening approach that identified an NK cell marker NKp46-directed DARPin. 133 AAV vectors displaying the NKp46-directed DARPin specifically transduced NKp46 expressing HT1080 cells, but not the parental NKp46 negative cell line. 133 This demonstrates the potential of this principle, but it remains to be proven if this targeting approach will work in primary NK cells or NK cell lines. Interestingly, however, analyses of cell types that were transduced by the AAV-DJ vector to deliver the CD4-CAR expression cassette following intravenous administration in humanized mice with the intention to generate CAR T cells in vivo (see above), showed that 1.5% of CD56+ cells were positive for the transgene CD4-CAR. 106
The transduction of primary human NK cells was also tested with a set of naturally occurring AAV serotypes (AAV4, AAV6, AAV8, and AAV9) at a vector dose of 300,000 per cell using EGFP as a reporter gene under control of a ubiquitously active promoter. 134 Of note, scAAV6 vectors were the only serotype to show transgene expression, which was, however, below 1% despite the confirmed presence of intracellular vector genomes. 134 This argues for a post-entry barrier toward AAV vector-mediated transduction. 135 In contrast, use of AAV6 vectors to deliver the same expression cassette as template for HDR in combination with CRISPR/Cas9 RNP complexes resulted in over 80% transgene (EGFP) expressing NK cells, which indicates that this method for integration of a transgene can overcome post-entry barriers to AAV. The system was further developed using two different CAR constructs that contained the same CD33 targeting domain, but differed in the transmembrane and signaling domains. 135 Also in this case, around 60% CAR+ cells were detected when a vector dose of 300,000 per cell was used. These CAR-NK cells showed an enhanced anti-tumor activity against two CD33+ Acute myeloid leukemia (AML) cell lines (Kasumi-1 and HL60) and one patient-derived AML cell sample. 135
As an alternative to CRISPR/Cas-mediated vector genome integration, Ye and colleagues used their SB MAJESTIC system to introduce HER2/neu-directed CARs into the NK-92 cell line. Stable expression of the HER2.CAR was reported with efficacies of ∼44% of CAR+ cells, which showed an enhanced HER2/neu target-specific killing activity compared with unmodified NK-92 cells. 92
SUMMARY AND OUTLOOK
In vivo gene therapy is the main workspace of AAV vectors with a focus on tissues with nondividing or slowly dividing target cell types since long-term therapeutic transgene expression can only be conferred under these conditions. This restriction is based on the nonintegrative nature of AAV vectors, which lack—in contrast to gamma-retroviral and lentiviral vectors—an integrase and instead use vector genome episomes as a maintenance format. Success of this vector system is indicated by the so far eight AAV vector-based GTMPs that have received market approval and the impressive number of human clinical trials that employ AAV vectors as delivery tools. Some main challenges are—in addition to the here discussed immune responses directed against the vector or the transgene product—the general broad tropism of AAV vectors based on naturally-occurring serotypes and the high vector doses that are required for some of the applications and that pose the risk of severe side effects. As briefly mentioned, engineering of AAV capsids, as well as vector genomes, is explored as a response to these challenges with encouraging results and approaches to translate these next generation AAV vectors into the clinics.
In the past, HSPCs, T cells, B cells, DC, or NK cells have not been considered as target cells for AAV vector-mediated transduction because of their proliferative nature and the low efficacy of transduction. Moreover, T and B cells, as well as DC and NK cells, are key components of the host immune response involved in “fighting” AAV vector-mediated transductions. With the advent of gene editing, these cell types have become target cells of the AAV vector system, enabling HDR-mediated genome modification with impressive efficiency. Interestingly, transduction efficiencies as measured by transgene expression products are higher following vector genome integration, which might be due to post-entry barriers toward this viral vector system. Furthermore, the intracellular sensing of AAV vectors might impact cell viability and engraftment. 69 In addition, with regard to the use of AAV vectors in conjunction with DSB-introducing gene editing strategies, integration of vector genomes needs to be addressed. 69,136 Nevertheless, tailoring AAV vectors by capsid and/or genome engineering is expected to improve the efficacy of transduction of these new target cells and, in combination with advanced gene editing strategies, sets the stage to translate current ex vivo approaches into in vivo applications.
Footnotes
ACKNOWLEDGMENT
Figures were created with BioRender.com.
AUTHORS’ CONTRIBUTIONS
A.E.A., M.B., X.P.G., A.K., C.F., U.T.H.: literature research and writing—parts of initial draft. M.M.: writing—reviewing and editing. U.T.H.: conceptualization (supporting). H.B.: conceptualization (lead), literature research, writing—original article draft (lead), and funding. All authors reviewed and edited the final article.
AUTHOR DISCLOSURE
H.B. is an inventor on patent applications focusing on AAV capsid engineering and vector development. The remaining authors declare no conflict of interests.
FUNDING INFORMATION
This work was supported by academic third-party funding from German Research Foundation (DFG) (